
MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
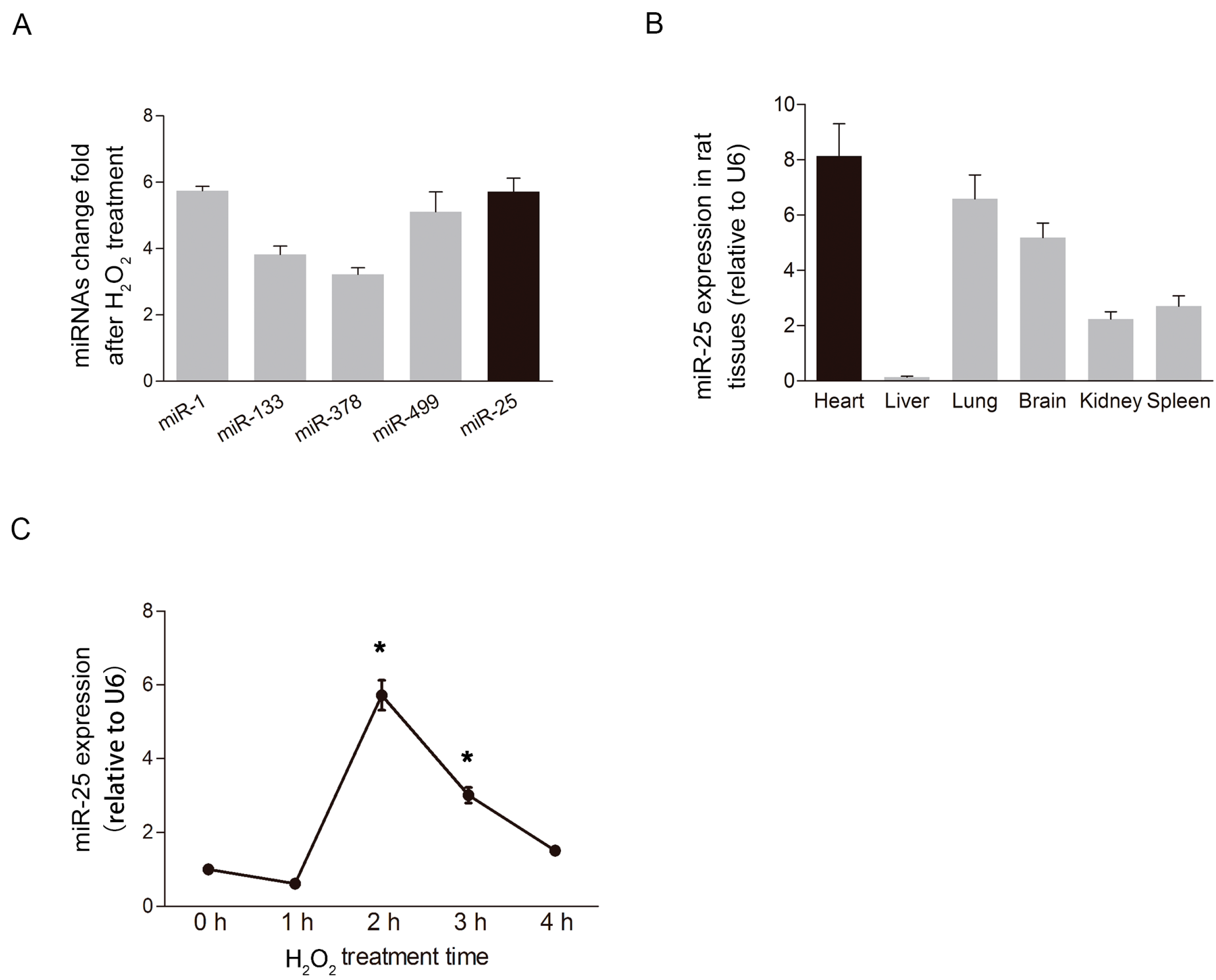
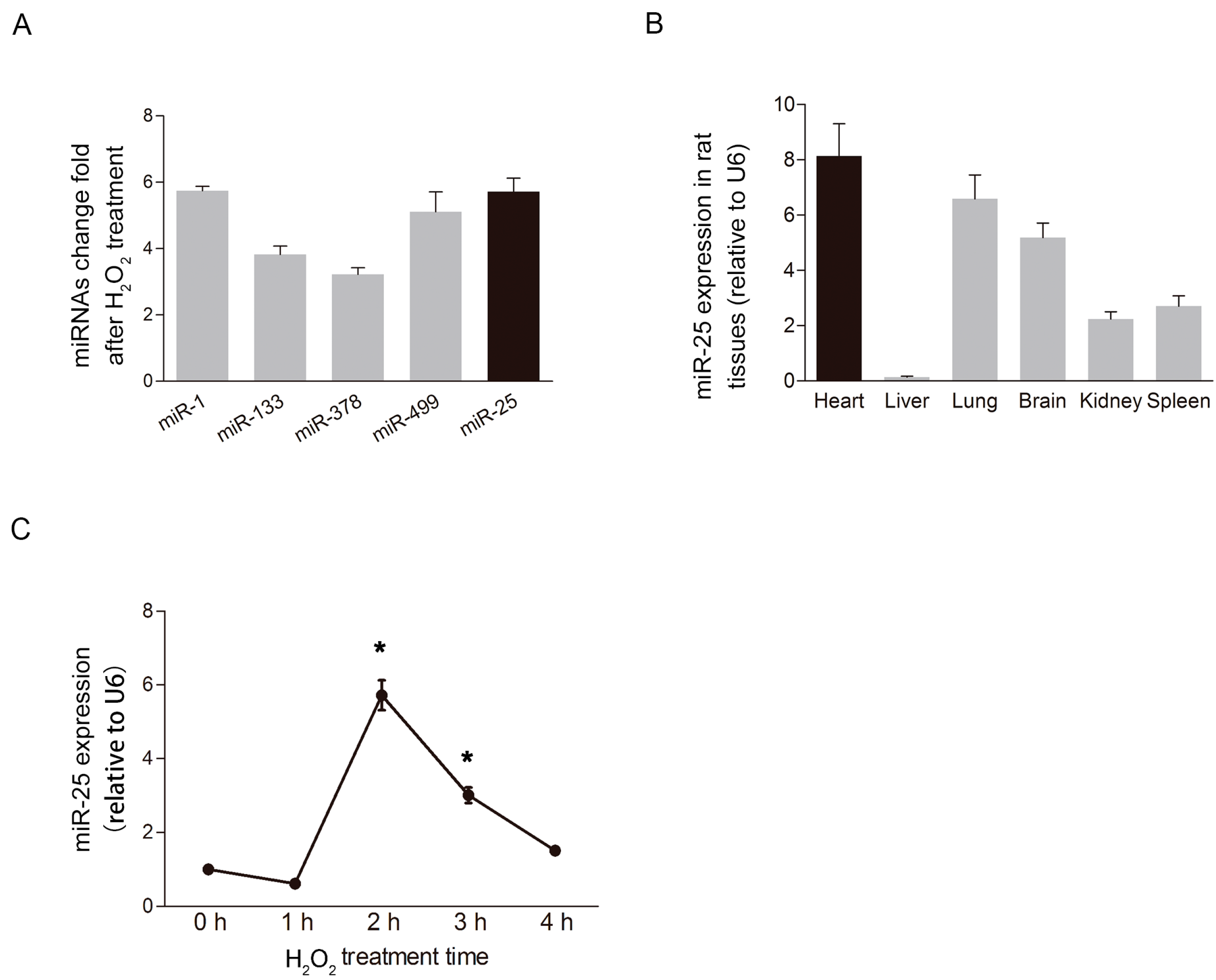
2.1. MiR-25 Was Remarkably Elevated under Oxidative Stress
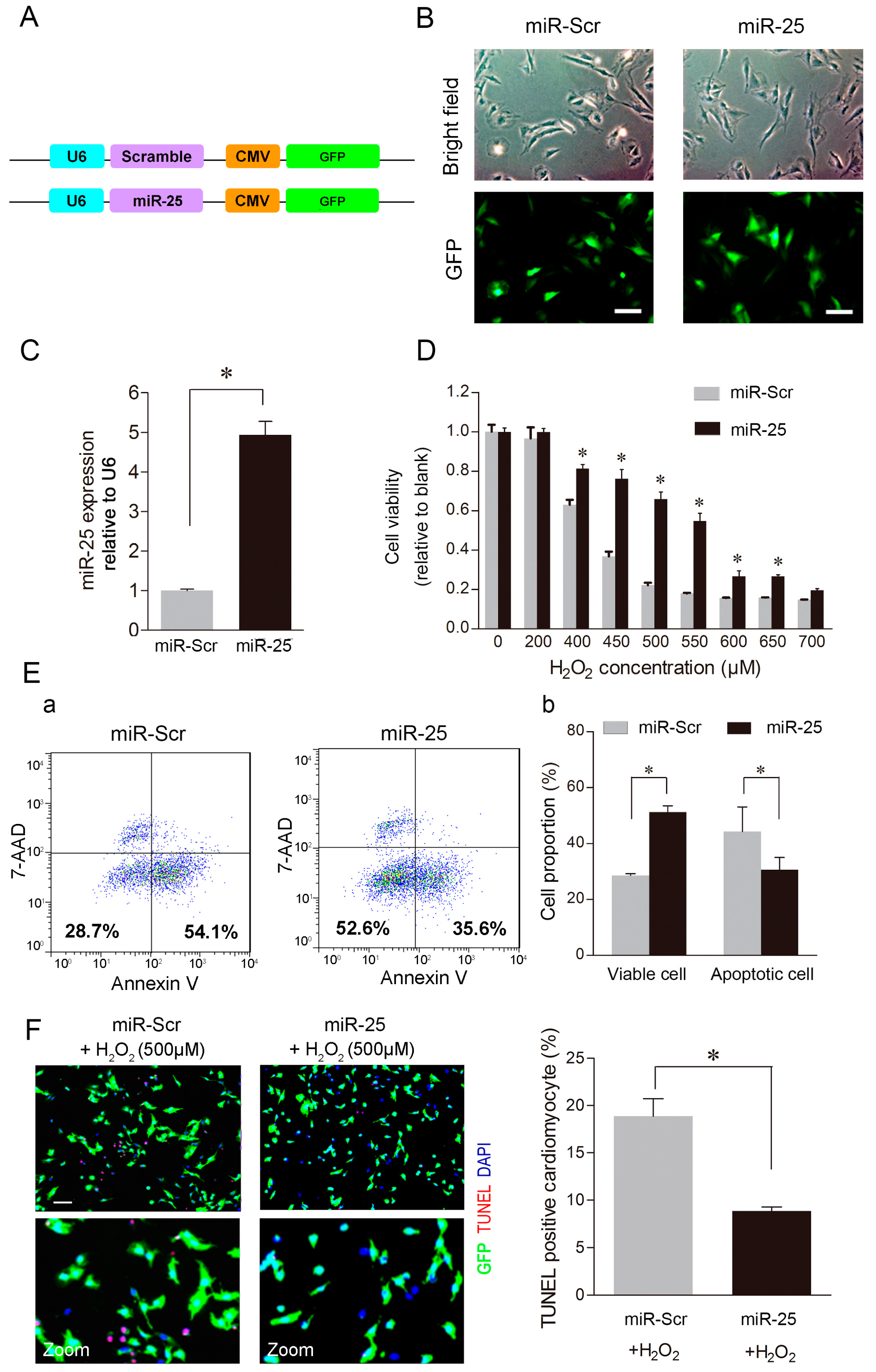
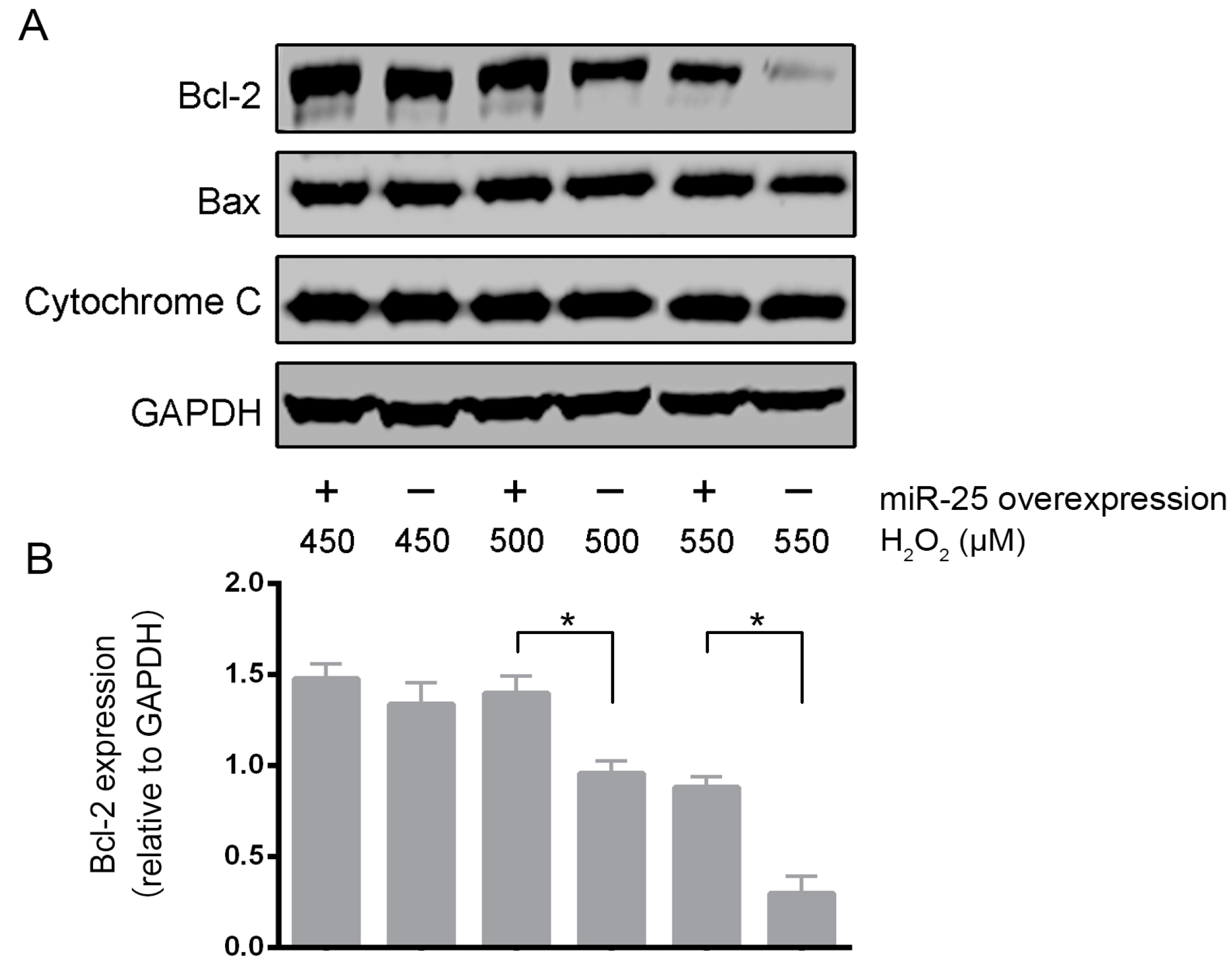
2.2. Over-Expression of MiR-25 Protected Cardiomyocytes against Oxidative Damage by Inactivating the Mitochondrial Apoptosis Pathway
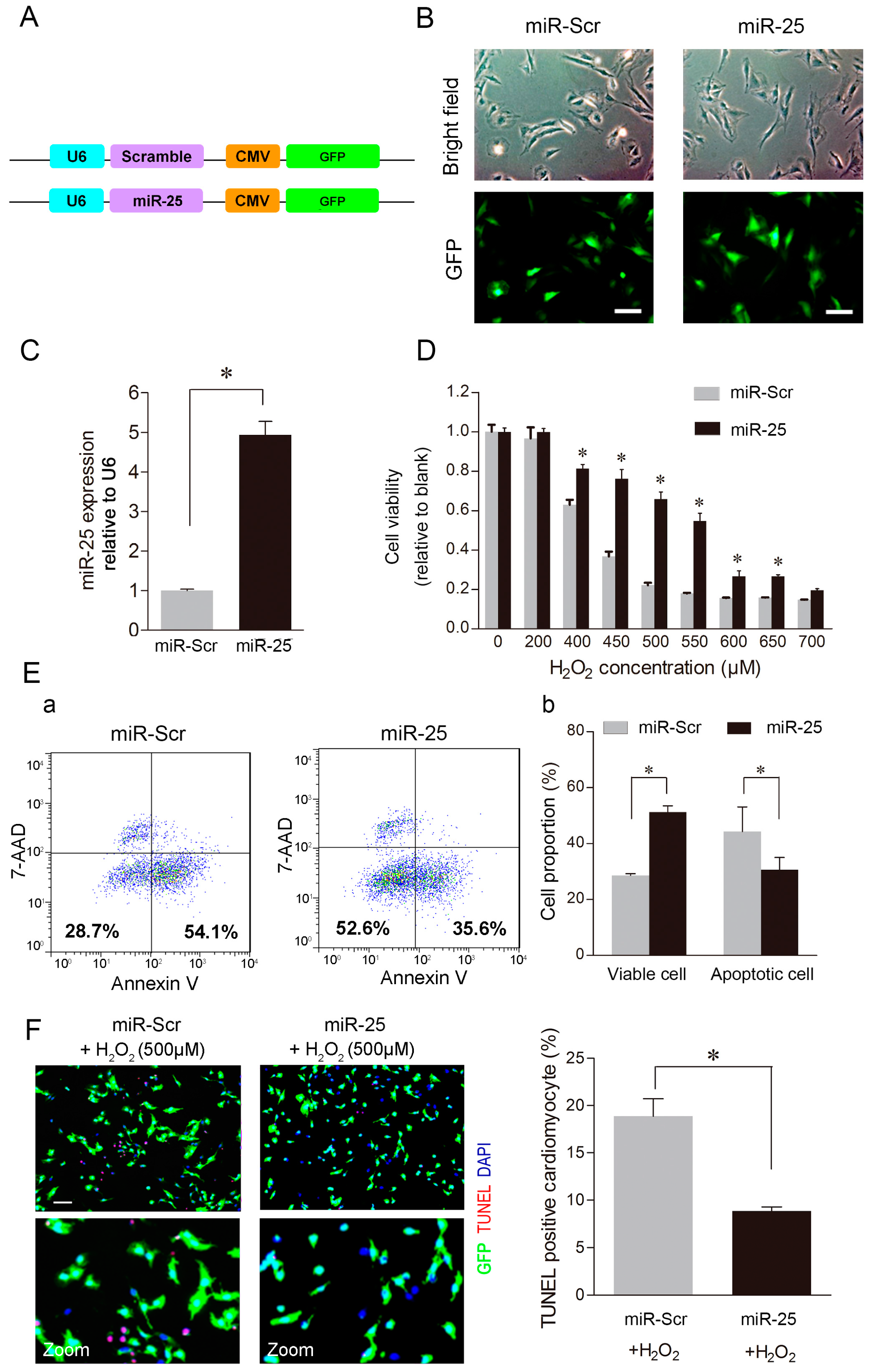
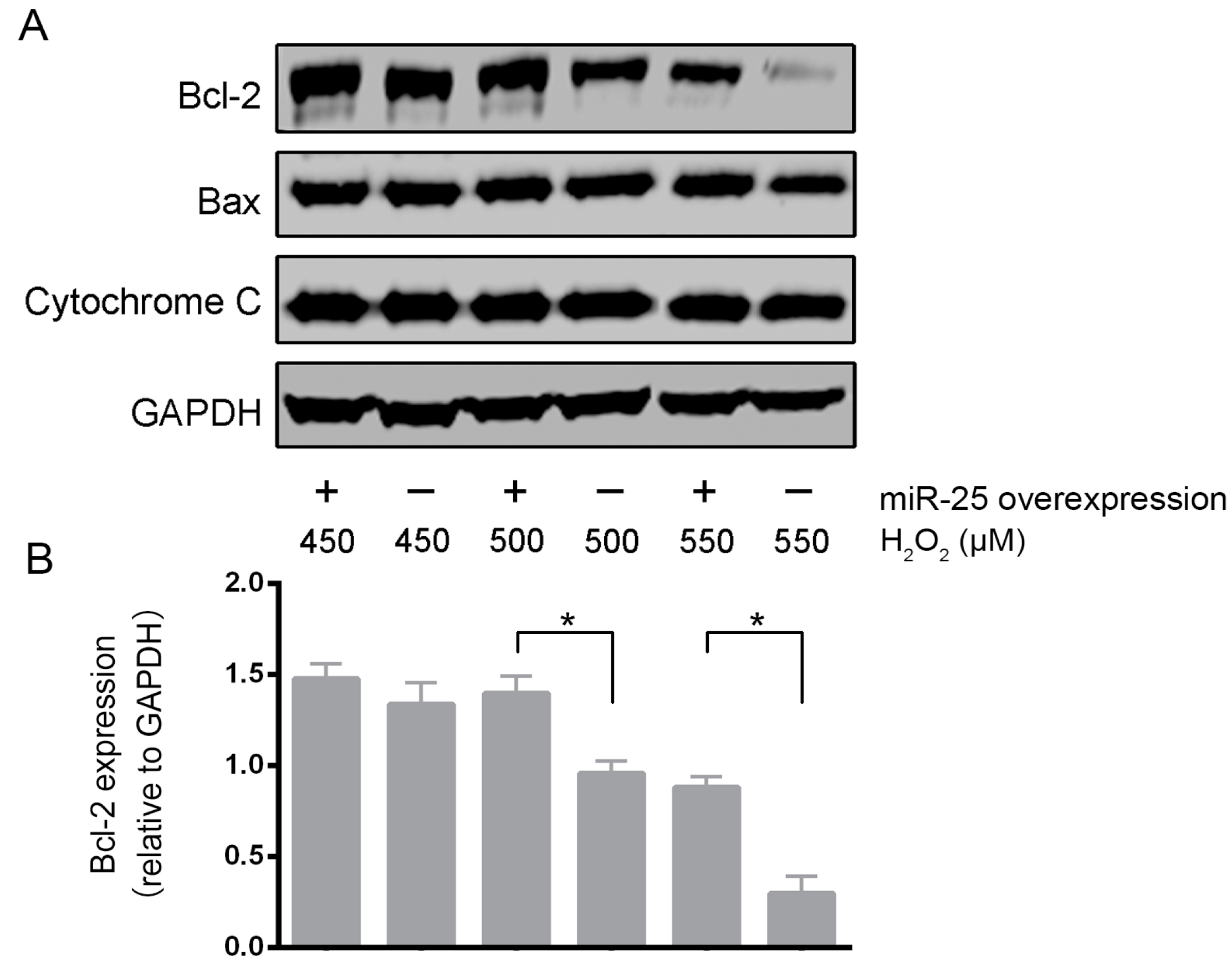
2.3. MCU Was a Direct Target of MiR-25

2.4. MiR-25 Significantly Decreased H2O2-Induced Elevation of Mitochondrial Ca2+ Concentration
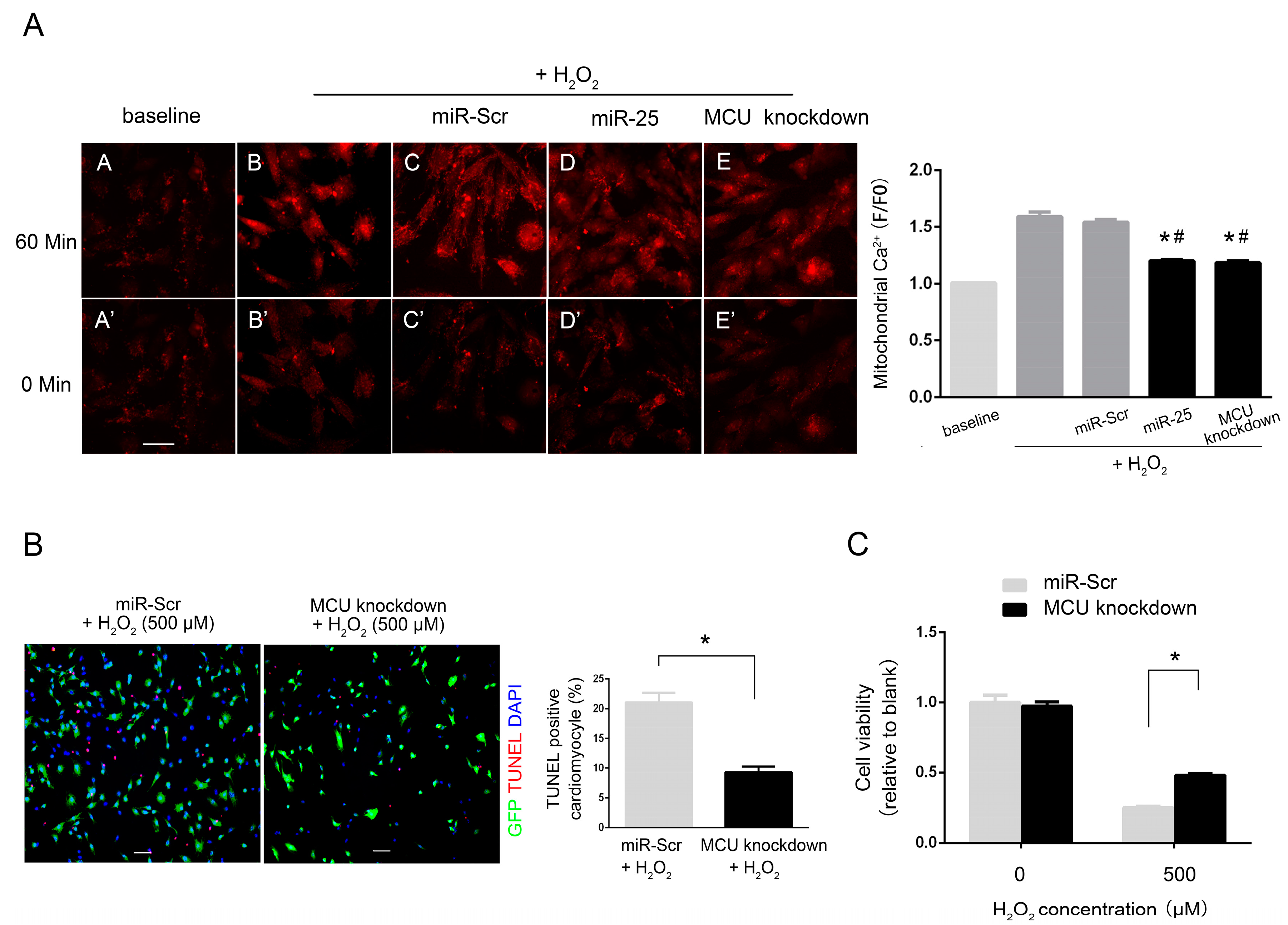
3. Discussion
4. Methods and Materials
4.1. Cell Culture and Viral Infection
4.2. Quantitative Reverse Transcription-Polymerase Chain Reaction
4.3. Fluorescence-Activated Cell Sorting (FACS)
4.4. Cell Vitality Analysis
4.5. TUNEL Assay
4.6. Immunoblotting
4.7. Luciferase Reporter Assay
4.8. Mitochondrial Calcium Recordings and Analyses
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tamargo, J.; Lopez-Sendon, J. Novel therapeutic targets for the treatment of heart failure. Nat. Rev. Drug Discov. 2011, 10, 536–555. [Google Scholar] [CrossRef] [PubMed]
- Ansley, D.M.; Wang, B. Oxidative stress and myocardial injury in the diabetic heart. J. Pathol. 2013, 229, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Walters, A.M.; Porter, G.A., Jr.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Olson, E.N. MicroRNA regulatory networks in cardiovascular development. Dev. Cell 2010, 18, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Srivastava, D. MicroRNA regulation of cardiovascular development. Circ. Res. 2009, 104, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; Von, D.M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Matkovich, S.J.; Hecker, P.A.; Zhang, Y.; Edwards, J.R.; Dorn, G.W., 2nd. Epitranscriptional orchestration of genetic reprogramming is an emergent property of stress-regulated cardiac microRNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19864–19869. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fuscoe, J.C.; Zhao, C.; Guo, C.; Jia, M.; Qing, T.; Bannon, D.I.; Lancashire, L.; Bao, W.; Du, T.; et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 2014, 5, 3230. [Google Scholar] [PubMed]
- Ku, H.C.; Chen, W.P.; Su, M.J. DPP4 deficiency exerts protective effect against H2O2 induced oxidative stress in isolated cardiomyocytes. PLOS One 2013, 8, e54518. [Google Scholar] [CrossRef] [PubMed]
- Dulchavsky, S.A.; Davidson, S.B.; Cullen, W.J.; Devasagayam, T.P.; Diebel, L.N.; Dutta, S. Effects of deferoxamine on H2O2-induced oxidative stress in isolated rat heart. Basic Res. Cardiol. 1996, 91, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, S.; Rieger, G.; Amberger, A.; Kuznetsov, A.V.; Margreiter, R.; Gnaiger, E. H2O2-mediated oxidative stress versus cold ischemia-reperfusion: Mitochondrial respiratory defects in cultured human endothelial cells. Transplantation 2002, 74, 1800–1803. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Sharp, P.A. MicroRNA functions in stress responses. Mol. Cell 2010, 40, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Sun, X.; Ren, J.; Li, X.; Gao, X.; Lu, C.; Zhang, Y.; Sun, H.; Wang, Y.; Wang, H.; et al. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS One 2012, 7, e50515. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.X.; Lin, Q.X.; Fu, Y.H.; Deng, C.Y.; Zhou, Z.L.; Zhu, J.N.; Liu, X.Y.; Zhang, Y.Y.; Li, Y.; Lin, S.G.; et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem. Biophys. Res. Commun. 2009, 381, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, H.; Shi, C.; Liu, K.; Liu, M.; Wang, N.; Wang, K.; Zhang, H.; Wang, G.; Xiao, X. ZNF667/Mipu1 is a novel anti-apoptotic factor that directly regulates the expression of the rat Bax gene in H9c2 cells. PLoS One 2014, 9, e111653. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Maack, C. SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013, 55, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Varnai, P.; Golenar, T.; Sheu, S.S.; Hajnoczky, G. Calcium transport across the inner mitochondrial membrane: Molecular mechanisms and pharmacology. Mol. Cell. Endocrinol. 2012, 353, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, EJ. Mitochondrial calcium transport in the heart: Physiological and pathological roles. J. Mol. Cell. Cardiol. 2009, 46, 789–803. [Google Scholar]
- Drago, I.; de Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed]
- Santo-Domingo, J.; Vay, L.; Hernandez-Sanmiguel, E.; Lobaton, C.D.; Moreno, A.; Montero, M.; Alvarez, J. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br. J. Pharmacol. 2007, 151, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corra, F.; Giorgi, C.; de Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Wahlquist, C.; Jeong, D.; Rojas-Munoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.; Doevendans, P.A.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, D.G.; Doseff, A.; Chau, B.N.; Lim, D.S.; de Souza-Pinto, N.C.; Hansford, R.; Kastan, M.B.; Lazebnik, Y.A.; Hardwick, J.M. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 1999, 274, 21155–21161. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.T.; Fonseca, E.A.; Melo, F.M.; Andrade, V.A.; Aguiar, C.J.; Andrade, L.M.; Pinheiro, A.C.; Casteluber, M.C.; Resende, R.R.; Pinto, M.C.; et al. Mitochondrial calcium regulates rat liver regeneration through the modulation of apoptosis. Hepatology 2011, 54, 296–306. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, L.; Huang, B.-J.; Ma, X.-E.; Wang, S.-Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.-M.; Liang, D.-D.; et al. MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter. Int. J. Mol. Sci. 2015, 16, 5420-5433. https://doi.org/10.3390/ijms16035420
Pan L, Huang B-J, Ma X-E, Wang S-Y, Feng J, Lv F, Liu Y, Liu Y, Li C-M, Liang D-D, et al. MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter. International Journal of Molecular Sciences. 2015; 16(3):5420-5433. https://doi.org/10.3390/ijms16035420
Chicago/Turabian StylePan, Lei, Bi-Jun Huang, Xiu-E Ma, Shi-Yi Wang, Jing Feng, Fei Lv, Yuan Liu, Yi Liu, Chang-Ming Li, Dan-Dan Liang, and et al. 2015. "MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter" International Journal of Molecular Sciences 16, no. 3: 5420-5433. https://doi.org/10.3390/ijms16035420
APA StylePan, L., Huang, B.-J., Ma, X.-E., Wang, S.-Y., Feng, J., Lv, F., Liu, Y., Liu, Y., Li, C.-M., Liang, D.-D., Li, J., Xu, L., & Chen, Y.-H. (2015). MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter. International Journal of Molecular Sciences, 16(3), 5420-5433. https://doi.org/10.3390/ijms16035420