
Species-Dependent Splice Recognition of a Cryptic Exon Resulting from a Recurrent Intronic CEP290 Mutation that Causes Congenital Blindness




Abstract
:1. Introduction
2. Results
2.1. Generation and Validation of CEP290 Minigenes
2.2. Assessment of Cryptic Splice Events in Different Species
2.3. Identification of a Suitable Sequence to Increase the Recognition of Exon X in Mouse
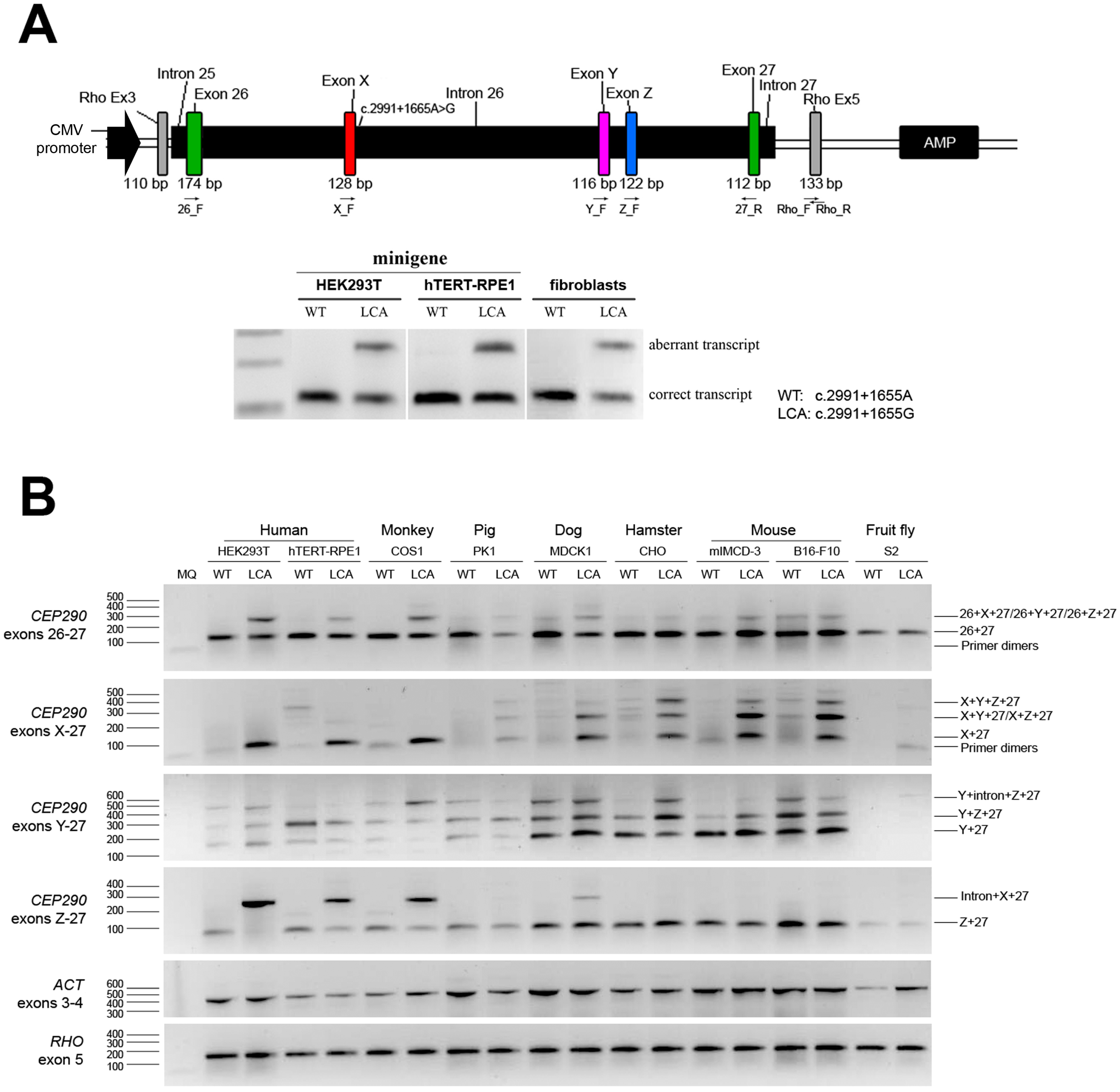

{kind=link}
{kind=link}
| Name | Forward Primers (Sequence 5'–3') |
|---|---|
| Act_F | ACTGGGACGACATGGAGAAG |
| Rho_F | ATCTGCTGCGGCAAGAAC |
| CEP290_attB1_i25_F | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGGCCGCTCTTTCTCAAAAGTGGC |
| CEP290_m1_F | GCCCGGCTAATTTTTTGTATTTTCAGGAGAGATGGGGTTTCACCTTG |
| CEP290_m2_F | CACCTGGCCCCAGTTGTAATGGTGAGTATCTCATACCTATCCC |
| CEP290__F | TGCTAAGTACAGGGACATCTTGC |
| CEP290_X_F | GCACCTGGCCCCAGTTG |
| CEP290_Y_F | CATAGCTCATTGCAGCCTTG |
| CEP290_Z_F | TGCCTCAGTCTCCTGAGTAG |
| Reverse Primers (Sequence 5'–3') | |
| Act_R | TCTCAGCTGTGGTGGTGAAG |
| Rho_R | AGGTGTAGGGGATGGGAGAC |
| CEP290_attB2_i27_R | GGGGACCACTTTGTACAAGAAAGCTGGGTGCTTGGTGGGGTTAAGTACAGG |
| CEP290_m1_R | CAAGGTGAAACCCCATCTCTCCTGAAAATACAAAAAATTAGCCGGGC |
| CEP290_m2_R | GGGATAGGTATGAGATACTCACCATTACAACTGGGGCCAGGTG |
| CEP290_27_R | AGACTCCACTTGTTCTTTTAAGGAG |
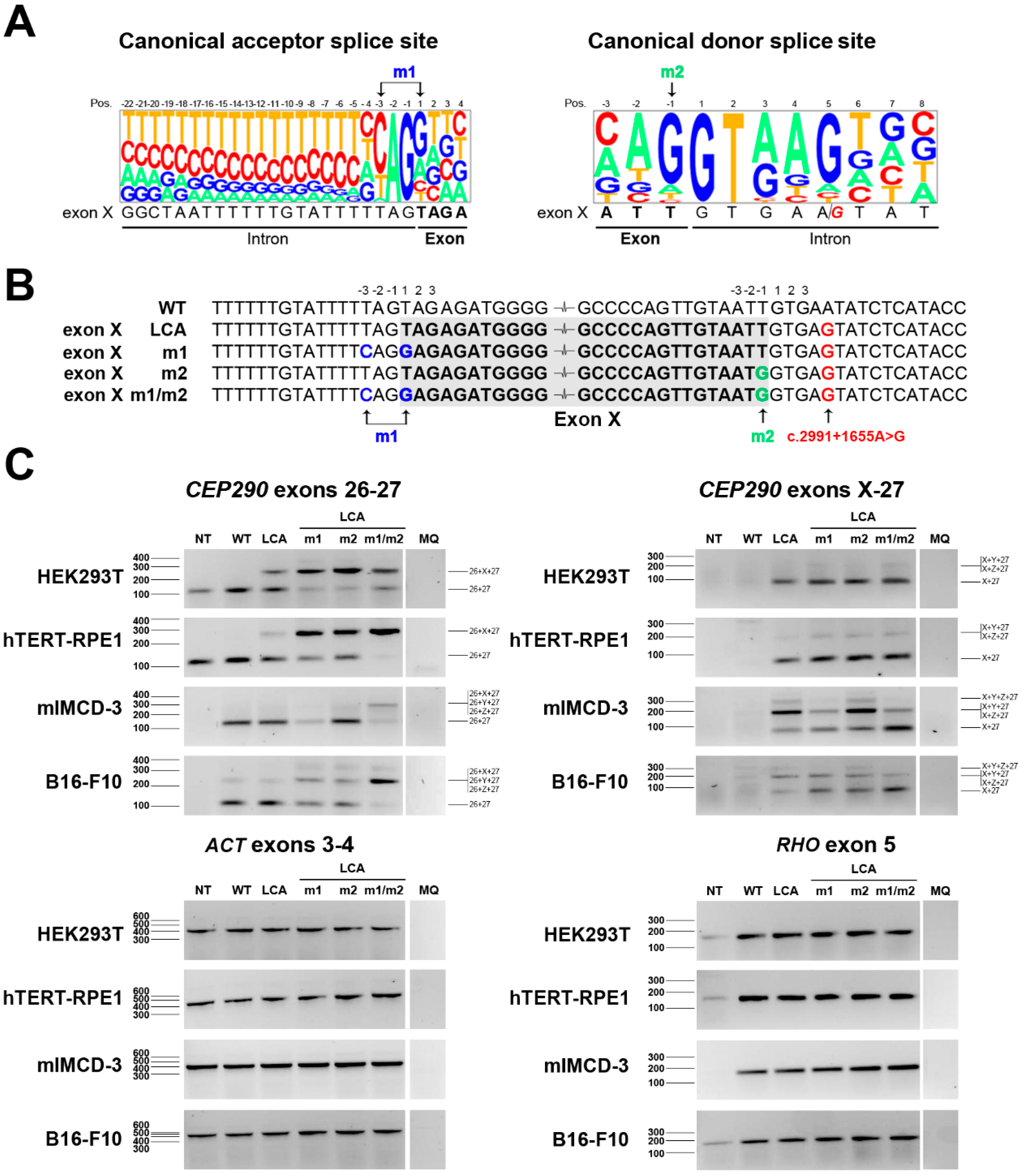
3. Discussion
4. Experimental Section
4.1. Minigene Generation
4.2. Site-Directed Mutagenesis
4.3. Cell Lines
4.4. Transfection
4.5. RT-PCR and Transcriptional Analysis
| Cell Line (Source) | Animal | Tissue of Origin | Culture Medium | Temp. |
|---|---|---|---|---|
| Fibroblast (skin biopsy) | Human | Skin | DMEM supplemented with 20% Fetal Calf Serum (FCS), 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| HEK293T (ATCC® CRL-3216™) | Human | Embryonic kidney | DMEM supplemented with 10% Fetal Calf Serum (FCS), 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| hTERT-RPE1 (ATCC® CRL-4000™) | Human | Eye | DMEM:F10 (1:1) supplemented with 10% FCS, 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| COS1 (ATCC® CRL-1650™) | Monkey | Kidney | DMEM supplemented with 10% FCS, 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| PK1 (ATCC® CRL-101™) | Pig | Kidney | DMEM supplemented with 5% FCS, 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| MDCK (ATCC® CRL-2935™) | Dog | Kidney | DMEM supplemented with 5% FCS, 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| CHO (ATCC® CRL-61™) | Hamster | Ovary | DMEM supplemented with 10% FCS, 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| mIMCD-3 (ATCC® CRL-2123™) | Mouse | Kidney | DMEM:F10 (1:1) supplemented with 10% FCS, 1% NaPyr, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| B16-F10 (ATCC® CRL-6475™) | Mouse | Skin | MEM supplemented with 5% FCS, 1% Non-essential amino acid (NEAA), 1% NaPyr, 1.5% MEM vitamins, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| N2A (ATCC® CCL-131™) | Mouse | Brain | DMEM supplemented with 10% FCS, 1% l-Glutamine, 1% NaPyr, 1% NEAA, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| ATT20 (ATCC® CCL-89™) | Mouse | Pituitary | DMEM supplemented with 7% FCS, 7% HS, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| EL4 (ATCC® TIB-39™) | Mouse | Lymphocyte | Iscove’s medium supplemented with 5% FCS, 100 U/mL penicillin and 100 μg/mL streptomycin | 37 °C |
| S2 (ATCC® CRL-1963™) | Fly | Embryo | Schneider’s Drosophila medium supplemented with 10% heat-inactivated FCS, 50 U/mL penicillin and 50 μg/mL streptomycin | 25 °C |
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chang, B.; Khanna, H.; Hawes, N.; Jimeno, D.; He, S.; Lillo, C.; Parapuram, S.K.; Cheng, H.; Scott, A.; Hurd, R.E.; et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 2006, 15, 1847–1857. [Google Scholar]
- Craige, B.; Tsao, C.C.; Diener, D.R.; Hou, Y.; Lechtreck, K.F.; Rosenbaum, J.L.; Witman, G.B. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; de Baere, E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar]
- Den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P.M. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef] [PubMed]
- Frank, V.; den Hollander, A.I.; Bruchle, N.O.; Zonneveld, M.N.; Nurnberg, G.; Becker, C.; Du Bois, G.; Kendziorra, H.; Roosing, S.; Senderek, J.; et al. Mutations of the CEP290 gene encoding a centrosomal protein cause meckel-gruber syndrome. Hum. Mutat. 2008, 29, 45–52. [Google Scholar]
- Coppieters, F.; Casteels, I.; Meire, F.; de Jaegere, S.; Hooghe, S.; van Regemorter, N.; van Esch, H.; Matuleviciene, A.; Nunes, L.; Meersschaut, V.; et al. Genetic screening of LCA in Belgium: Predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum. Mutat. 2010, 31, E1709–E1766. [Google Scholar]
- Collin, R.W.; den Hollander, A.I.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Thanaraj, T.A.; Clark, F. Human GC-AG alternative intron isoforms with weak donor sites show enhanced consensus at acceptor exon positions. Nucleic Acids Res. 2001, 29, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Grodecka, L.; Lockerova, P.; Ravcukova, B.; Buratti, E.; Baralle, F.E.; Dusek, L.; Freiberger, T. Exon first nucleotide mutations in splicing: Evaluation of in silico prediction tools. PLoS One 2014, 9, e89570. [Google Scholar] [CrossRef] [PubMed]
- Ratsch, G.; Sonnenburg, S.; Scholkopf, B. Rase: Recognition of alternatively spliced exons in C. Elegans. Bioinformatics 2005, 21, i369–i377. [Google Scholar] [CrossRef]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick one, but be quick: 5' Splice sites and the problems of too many choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; van Beersum, S.E.; Peters, T.A.; Roepman, R.; Cremers, F.P.; Collin, R.W. Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for leber congenital amaurosis. PLoS One 2013, 8, e79369. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Riera, M.; Pomares, E.; Permanyer, J.; de Castro-Miro, M.; Sava, F.; Abril, J.F.; Marfany, G.; Gonzalez-Duarte, R. High transcriptional complexity of the retinitis pigmentosa CERKl gene in human and mouse. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5202–5214. [Google Scholar] [CrossRef]
- Lu, X.; Ferreira, P.A. Identification of novel murine- and human-specific RPGRIP1 splice variants with distinct expression profiles and subcellular localization. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1882–1890. [Google Scholar] [CrossRef]
- Tian, X.L.; Paul, M. Species-specific splicing and expression of angiotensin converting enzyme. Biochem. Pharmacol. 2003, 66, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics 2013. [Google Scholar] [CrossRef]
- Neidhardt, J.; Glaus, E.; Barthelmes, D.; Zeitz, C.; Fleischhauer, J.; Berger, W. Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 2007, 28, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Cooper, N.G. Characterization of the transcripts and protein isoforms for cytoplasmic polyadenylation element binding protein-3 (CPEB3) in the mouse retina. BMC Mol. Biol. 2009. [Google Scholar] [CrossRef]
- Gamsiz, E.D.; Ouyang, Q.; Schmidt, M.; Nagpal, S.; Morrow, E.M. Genome-wide transcriptome analysis in murine neural retina using high-throughput RNA sequencing. Genomics 2012, 99, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Bush, S.J.; Tovar-Corona, J.M.; Castillo-Morales, A.; Urrutia, A.O. Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol. Biol. Evol. 2014, 31, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Florea, L. Bioinformatics of alternative splicing and its regulation. Brief. Bioinform. 2006, 7, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Xie, J. Differential evolution of signal-responsive RNA elements and upstream factors that control alternative splicing. Cell Mol. Life Sci. 2014, 71, 4347–4360. [Google Scholar] [CrossRef] [PubMed]
- Kol, G.; Lev-Maor, G.; Ast, G. Human-mouse comparative analysis reveals that branch-site plasticity contributes to splicing regulation. Hum. Mol. Genet. 2005, 14, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Gladman, J.T.; Bebee, T.W.; Edwards, C.; Wang, X.; Sahenk, Z.; Rich, M.M.; Chandler, D.S. A humanized SMN gene containing the SMN2 nucleotide alteration in exon 7 mimics SMN2 splicing and the SMA disease phenotype. Hum. Mol. Genet. 2010, 19, 4239–4252. [Google Scholar] [CrossRef] [PubMed]
- Hims, M.M.; Shetty, R.S.; Pickel, J.; Mull, J.; Leyne, M.; Liu, L.; Gusella, J.F.; Slaugenhaupt, S.A. A humanized IKBKAP transgenic mouse models a tissue-specific human splicing defect. Genomics 2007, 90, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Vadolas, J.; Nefedov, M.; Wardan, H.; Mansooriderakshan, S.; Voullaire, L.; Jamsai, D.; Williamson, R.; Ioannou, P.A. Humanized â-thalassemia mouse model containing the common IVSI-110 splicing mutation. J. Biol. Chem. 2006, 281, 7399–7405. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Swaminathan, S.; Martin, B.K.; Sharan, S.K. Aberrant splicing induced by missense mutations in BRCA1: Clues from a humanized mouse model. Hum. Mol. Genet. 2003, 12, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Bochner, R.; Ziv, Y.; Zeevi, D.; Donyo, M.; Abraham, L.; Ashery-Padan, R.; Ast, G. Phosphatidylserine increases IKBKAP levels in a humanized knock-in IKBKAP mouse model. Hum. Mol. Genet. 2013, 22, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. Aon-mediated exon skipping restores ciliation in fibroblasts harboring the common leber congenital amaurosis CEP290 mutation. Mol. Ther. Nucleic Acids 2012. [Google Scholar] [CrossRef]
- Shafique, S.; Siddiqi, S.; Schraders, M.; Oostrik, J.; Ayub, H.; Bilal, A.; Ajmal, M.; Seco, C.Z.; Strom, T.M.; Mansoor, A.; et al. Genetic spectrum of autosomal recessive non-syndromic hearing loss in pakistani families. PLoS One 2014, 9, e100146. [Google Scholar]
- Rasband, W.S. Image J software. Available online: http://imagej.nih.gov/ij/ (accessed on 25 January 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garanto, A.; Duijkers, L.; Collin, R.W.J. Species-Dependent Splice Recognition of a Cryptic Exon Resulting from a Recurrent Intronic CEP290 Mutation that Causes Congenital Blindness. Int. J. Mol. Sci. 2015, 16, 5285-5298. https://doi.org/10.3390/ijms16035285
Garanto A, Duijkers L, Collin RWJ. Species-Dependent Splice Recognition of a Cryptic Exon Resulting from a Recurrent Intronic CEP290 Mutation that Causes Congenital Blindness. International Journal of Molecular Sciences. 2015; 16(3):5285-5298. https://doi.org/10.3390/ijms16035285
Chicago/Turabian StyleGaranto, Alejandro, Lonneke Duijkers, and Rob W. J. Collin. 2015. "Species-Dependent Splice Recognition of a Cryptic Exon Resulting from a Recurrent Intronic CEP290 Mutation that Causes Congenital Blindness" International Journal of Molecular Sciences 16, no. 3: 5285-5298. https://doi.org/10.3390/ijms16035285
APA StyleGaranto, A., Duijkers, L., & Collin, R. W. J. (2015). Species-Dependent Splice Recognition of a Cryptic Exon Resulting from a Recurrent Intronic CEP290 Mutation that Causes Congenital Blindness. International Journal of Molecular Sciences, 16(3), 5285-5298. https://doi.org/10.3390/ijms16035285