
mTOR Inhibition Induces EGFR Feedback Activation in Association with Its Resistance to Human Pancreatic Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. AZD8055 Transiently Inhibits Pancreatic Cancer Cell Growth
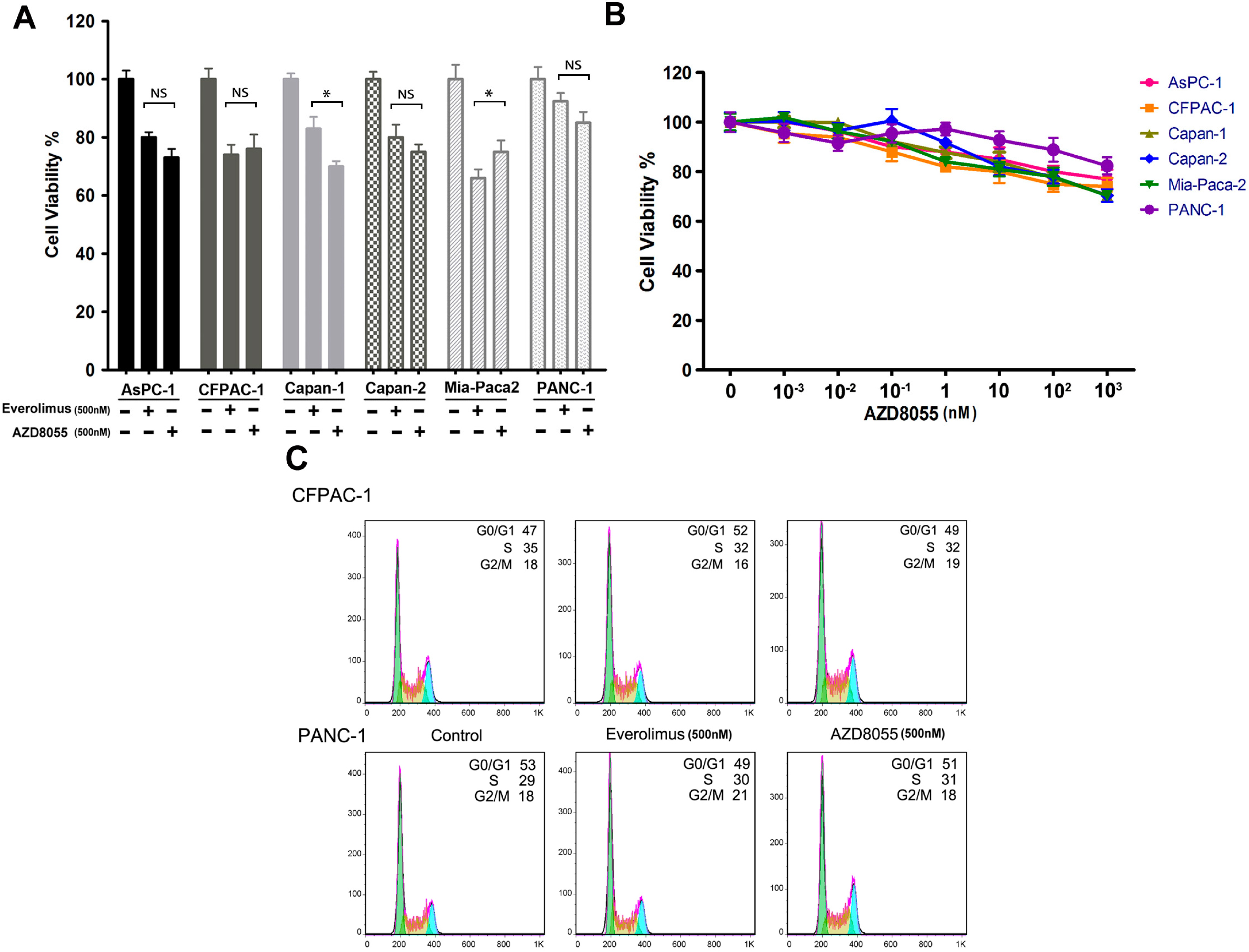
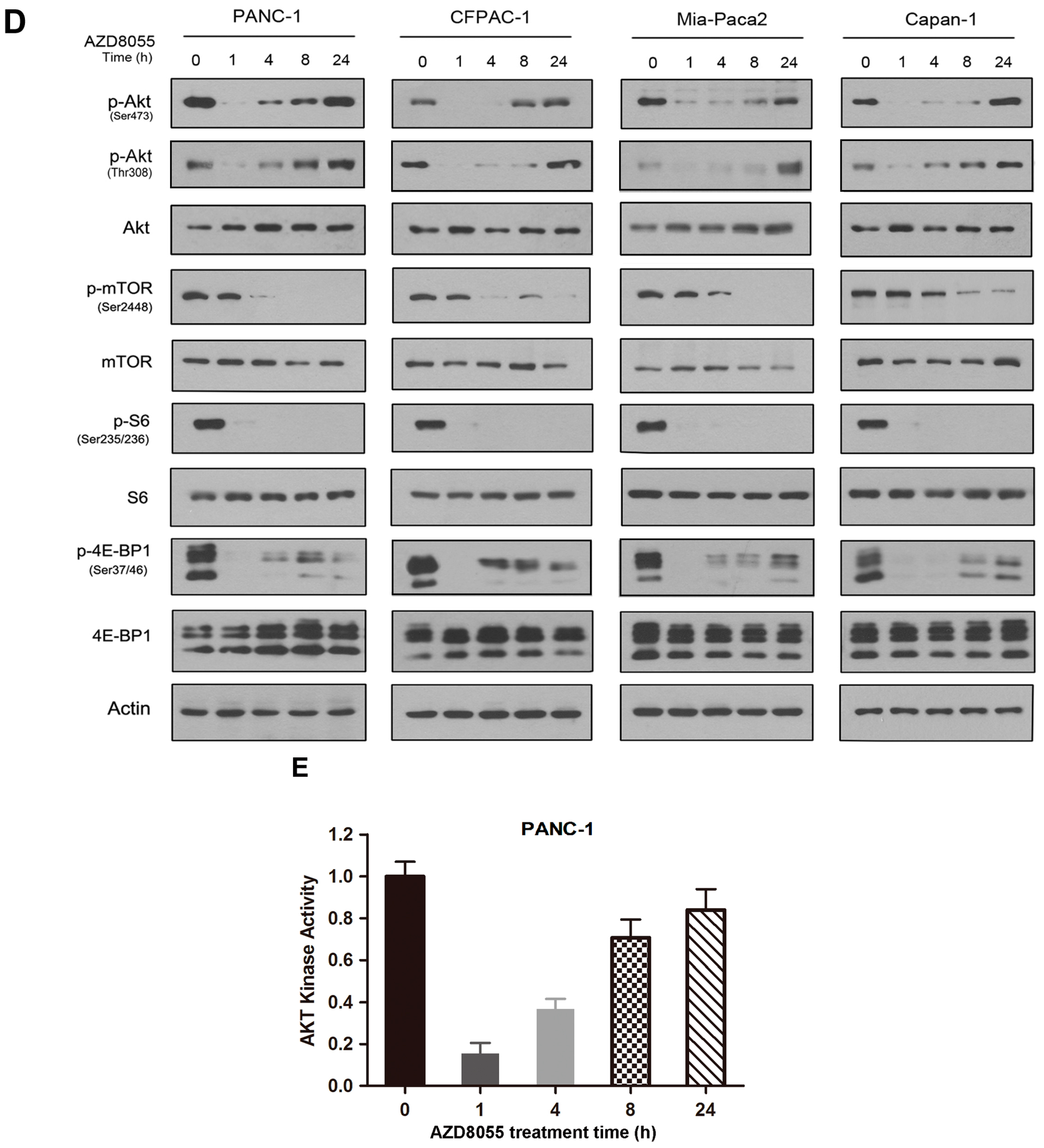
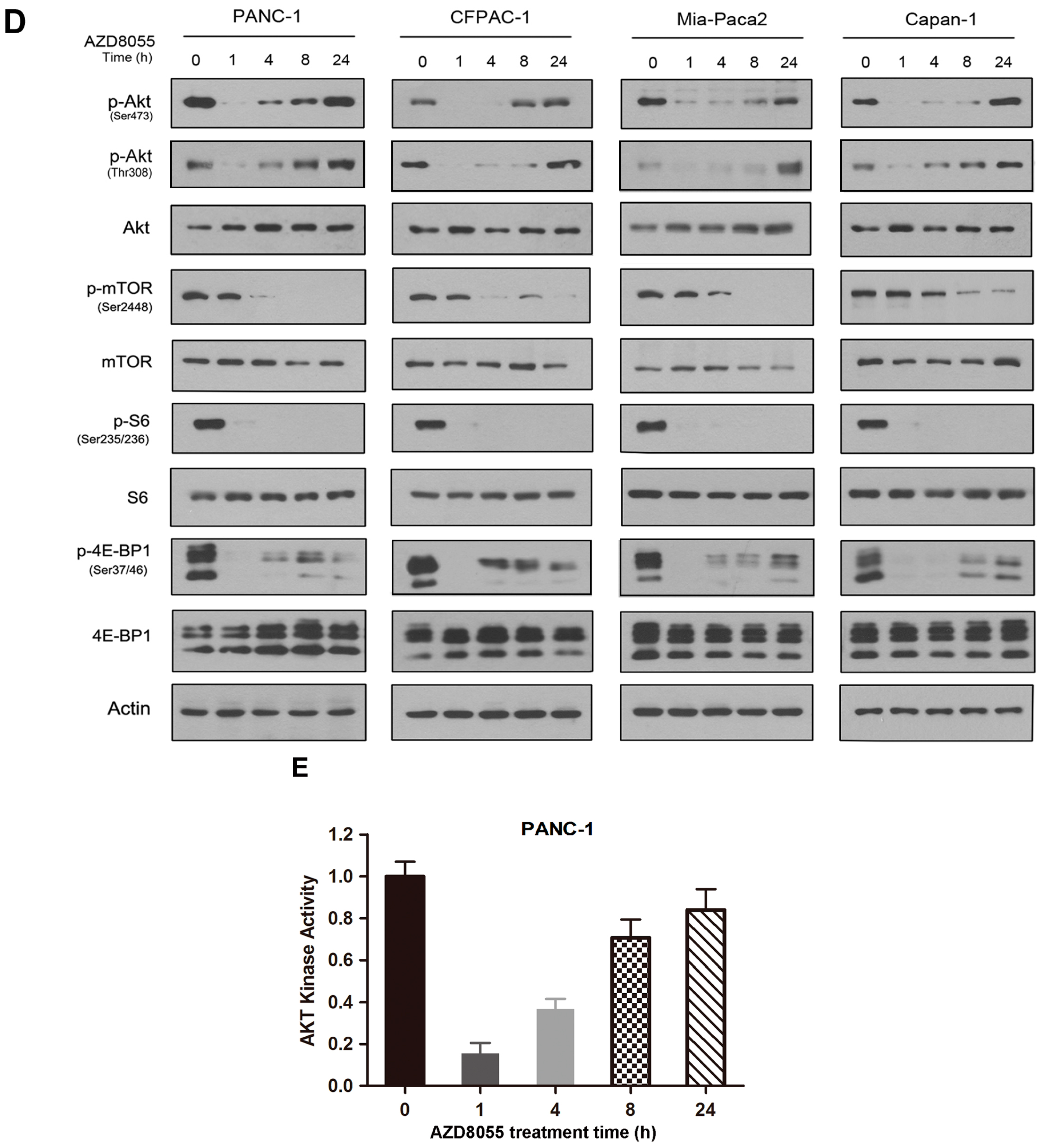
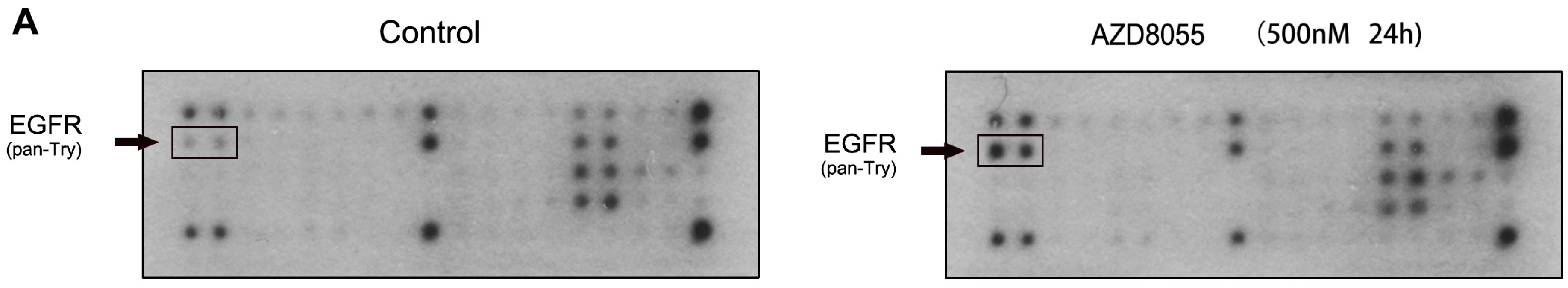
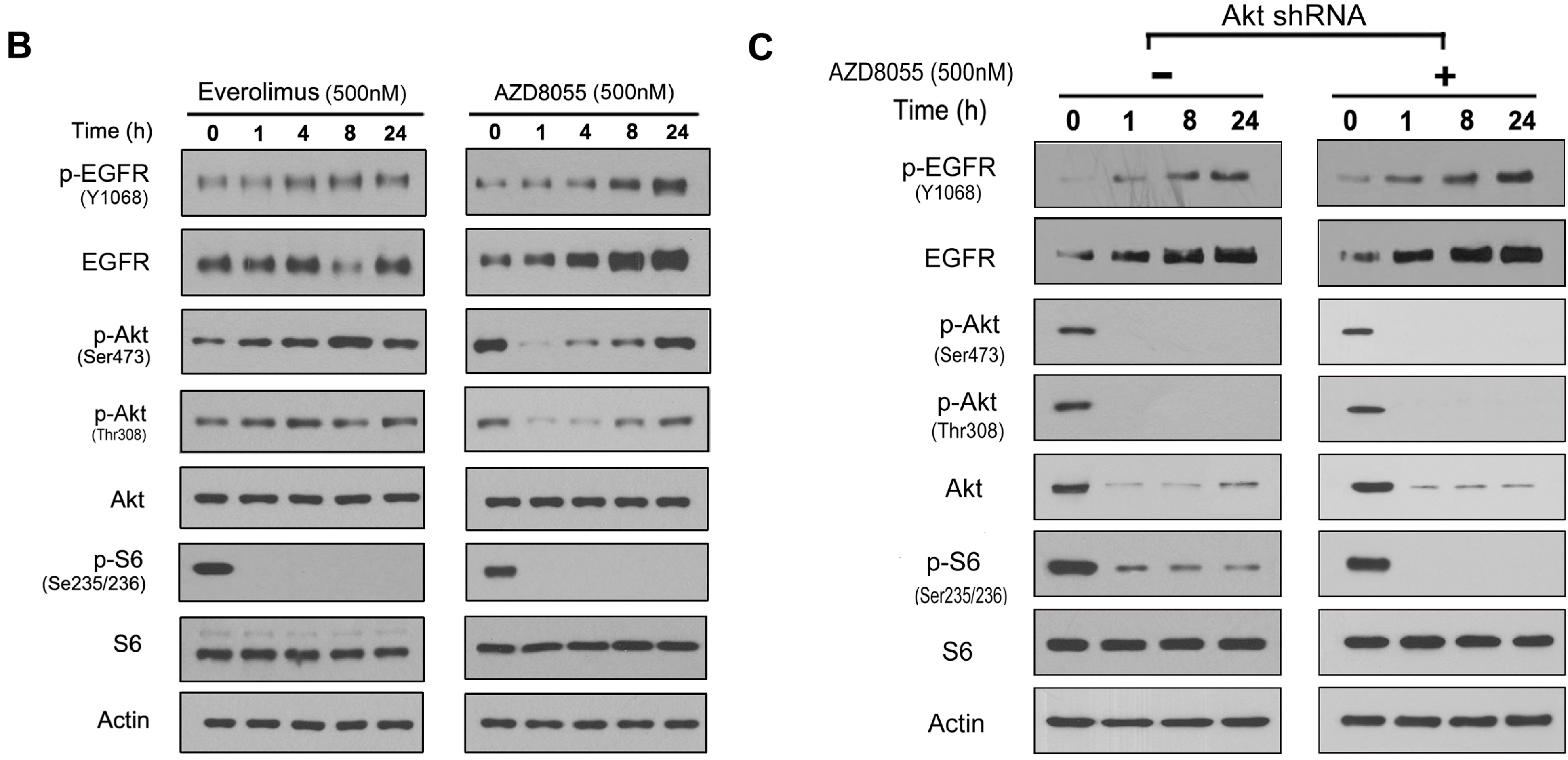
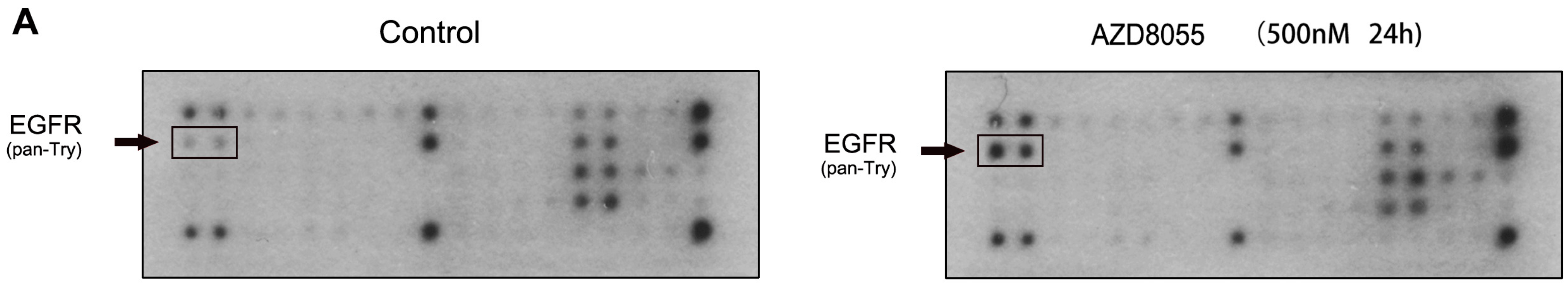
2.2. AZD8055 Induces Epidermal Growth Factor Receptor (EGFR) Up-Regulation in an AKT-Dependent Manner in Pancreatic Cancer Cells
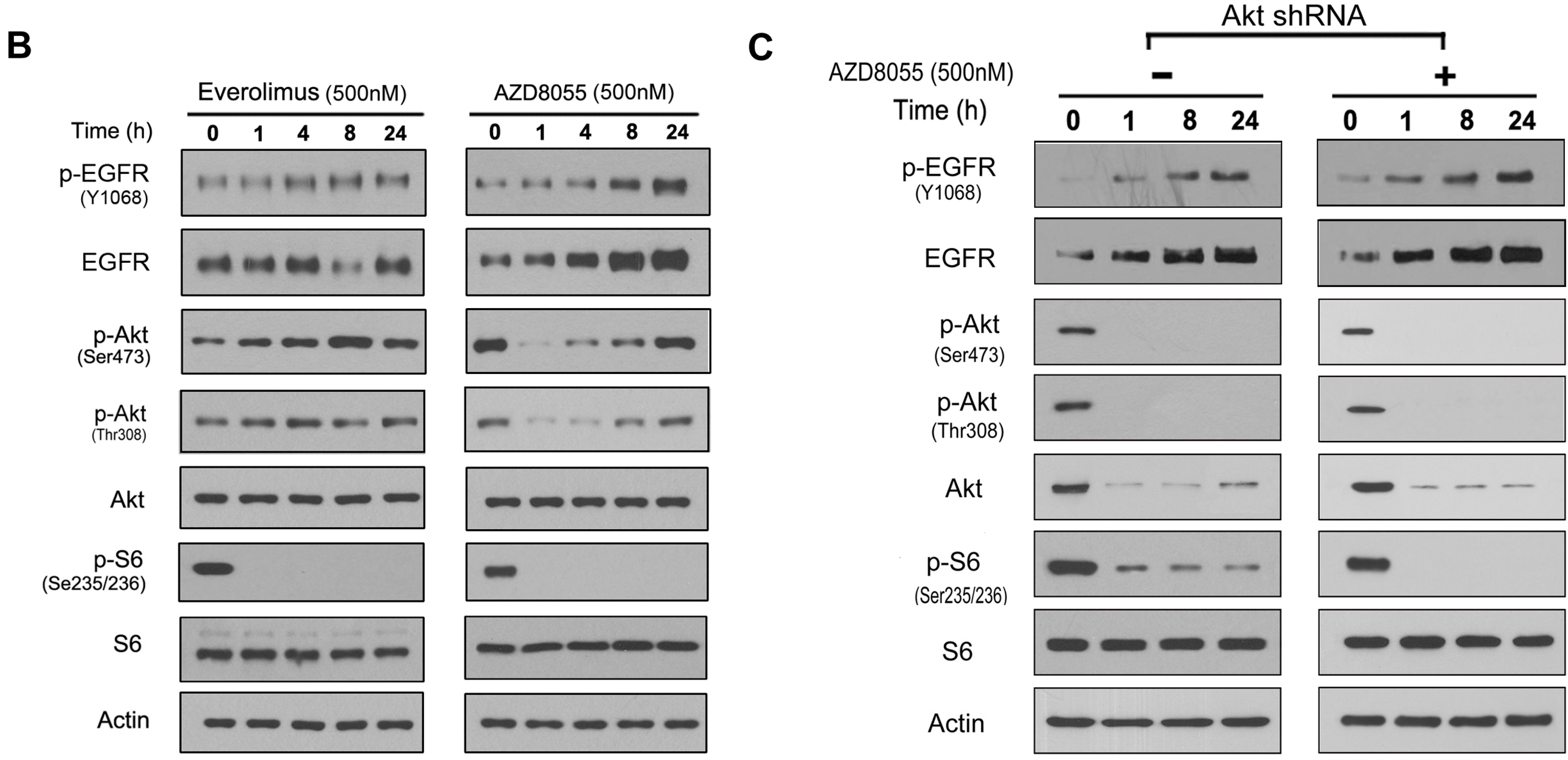
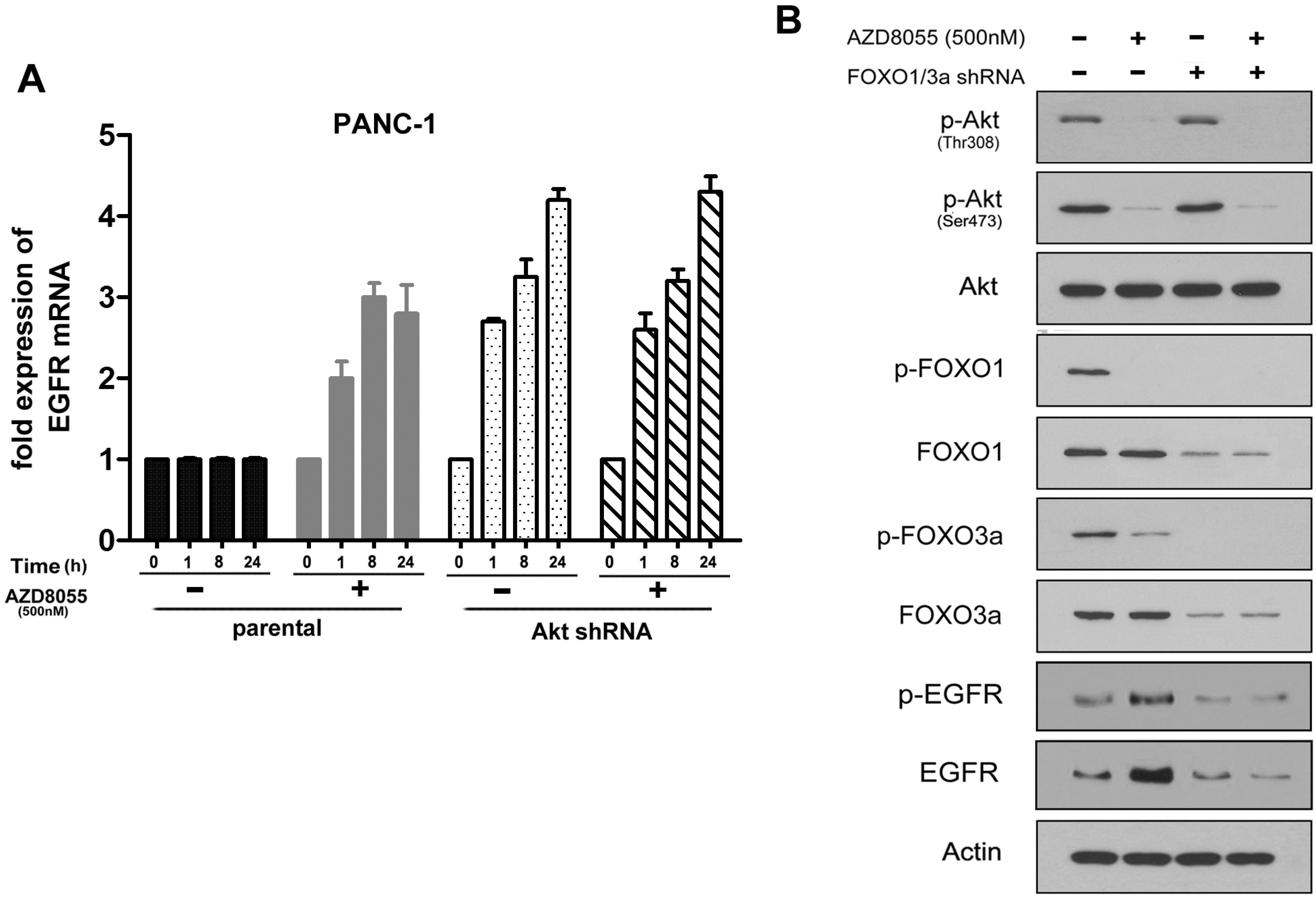
2.3. AKT Inhibition Releases Fork-Head Box O (FoxO), Contributing to AZD8055-Induced EGFR Up-Regulation
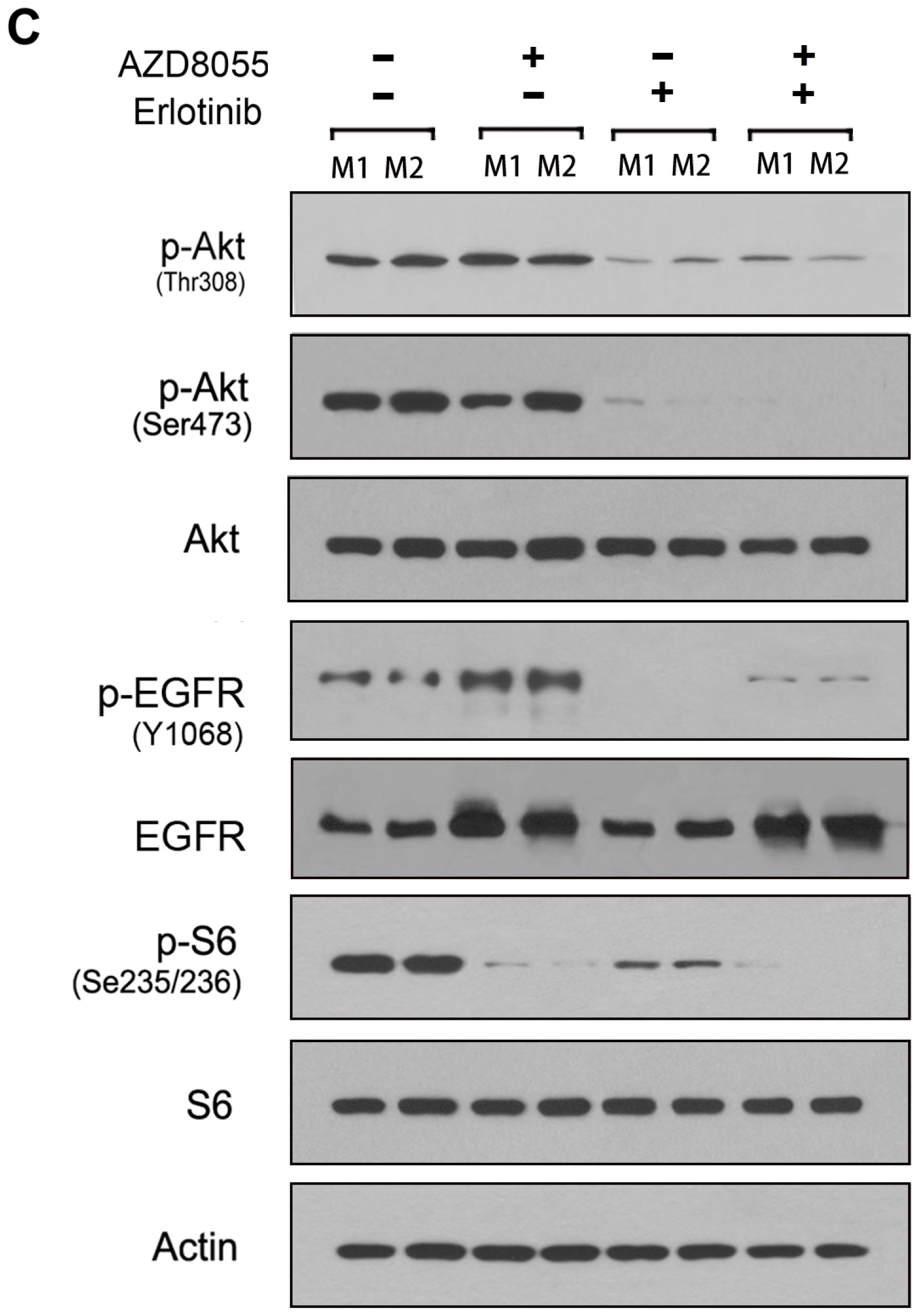
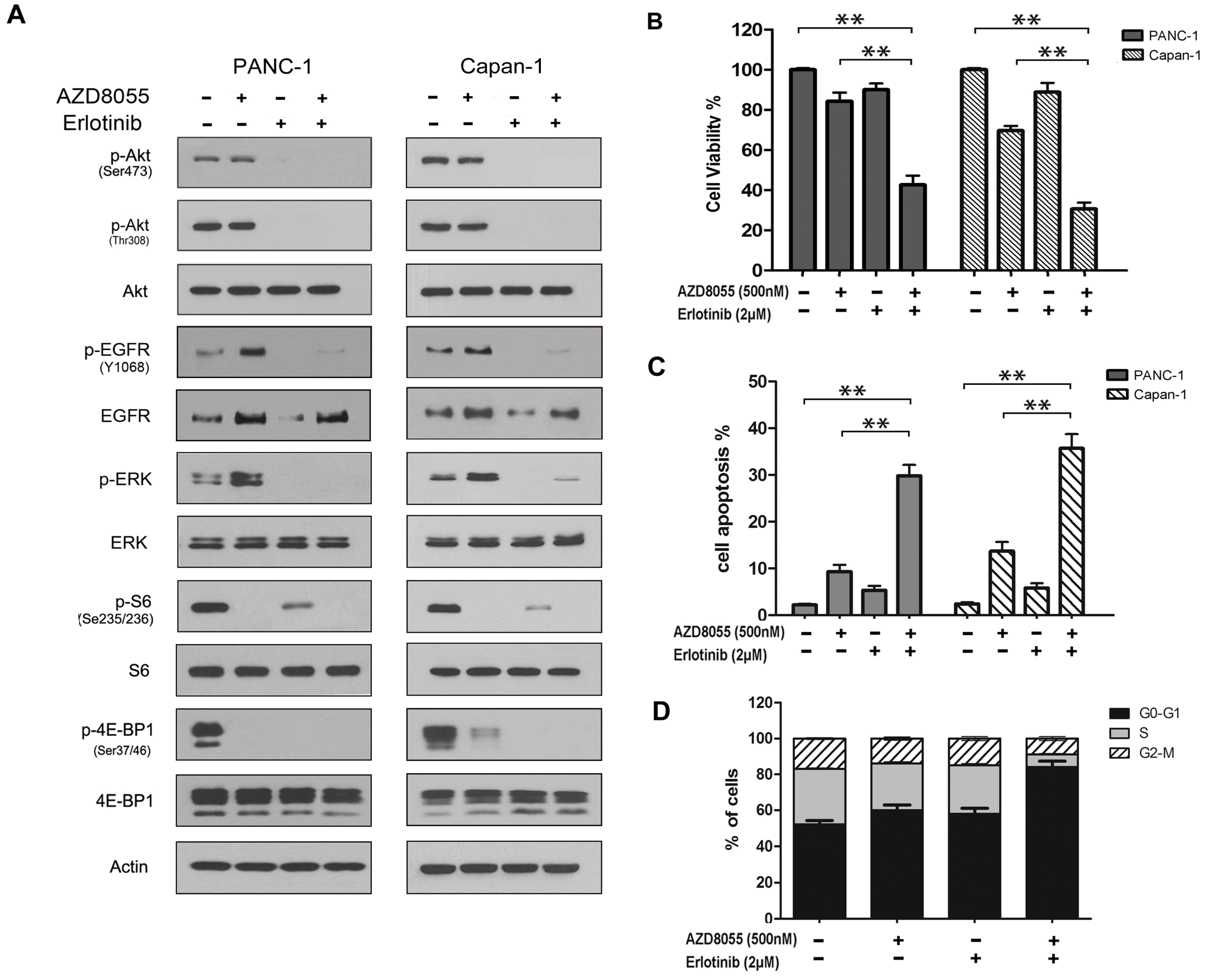
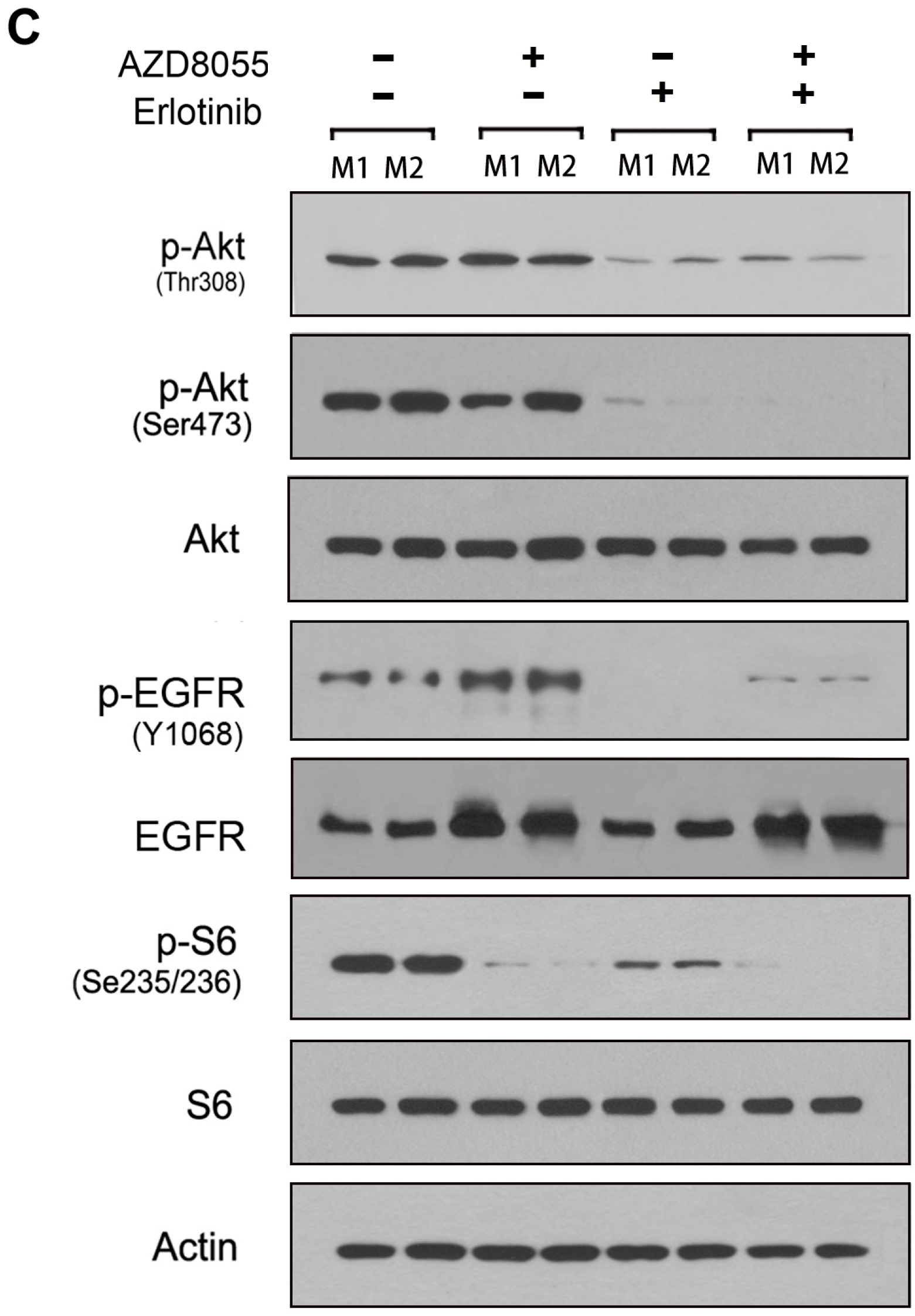
2.4. Combined Inhibition of the Mammalian Target of Rapamycin (mTOR) and EGFR Persistently Inhibits AKT Activation and Synergistically Inhibits Cell Growth in Vitro
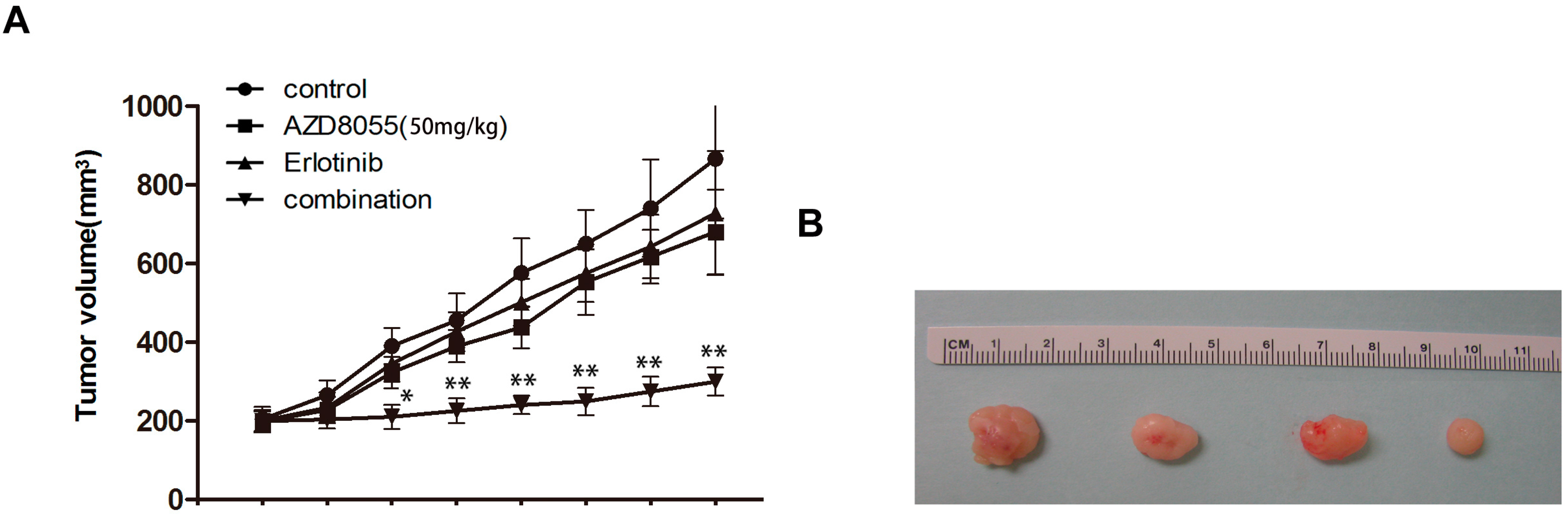
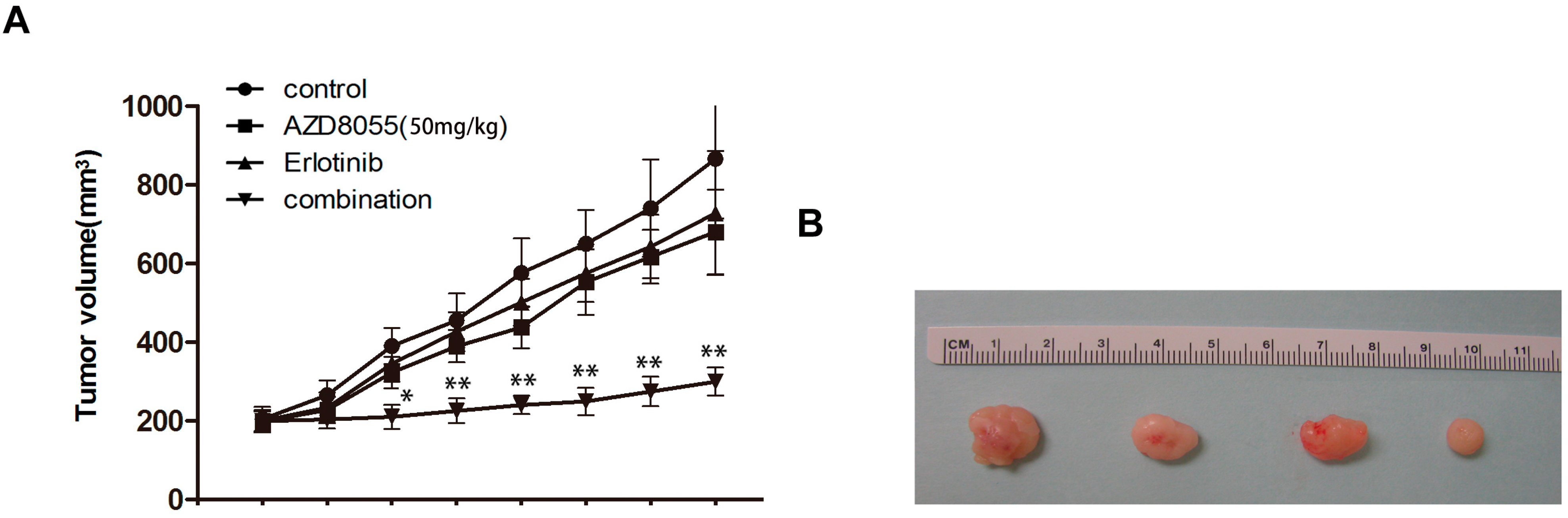
2.5. Combined Erlotinib and AZD8055 Treatment Leads to the Effective Suppression of Pancreatic Cancer Xenografts
3. Experimental Section
3.1. Materials
3.2. Cell Lines, Cell Culture and Transfection
3.3. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Assay
3.4. Cell Lysate and Western Blot Assay
3.5. RNA Isolation and Quantitative Real-Time PCR
3.6. Cell Cycle and Cell Apoptosis Analysis
3.7. Phosphorylated Receptor Tyrosine Kinase (RTK) Array
3.8. Animal Studies
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA 2011, 61, 69–90. [Google Scholar] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Liu, Y.; Guo, Y.; Xiang, A.; Wang, G.; Xue, X.; Lu, Z. Mir-99b-targeted mTOR induction contributes to irradiation resistance in pancreatic cancer. Mol. Cancer 2013, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Phan, A.T.; Jehl, V.; Shah, G.; Meric-Bernstam, F. Everolimus in advanced pancreatic neuroendocrine tumors: The clinical experience. Cancer Res. 2013, 73, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013, 340, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Asahina, H.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Tamura, Y.; Honda, K.; Seki, Y.; Tanabe, Y.; Shimada, H.; Shi, X.; et al. Safety and tolerability of AZD8055 in Japanese patients with advanced solid tumors: A dose-finding phase I study. Investig. New Drugs 2013, 31, 677–684. [Google Scholar]
- Jordan, N.J.; Dutkowski, C.M.; Barrow, D.; Mottram, H.J.; Hutcheson, I.R.; Nicholson, R.I.; Guichard, S.M.; Gee, J.M. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014, 16, R12. [Google Scholar] [CrossRef] [PubMed]
- Preuss, E.; Hugle, M.; Reimann, R.; Schlecht, M.; Fulda, S. Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J. Biol. Chem. 2013, 288, 35287–35296. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Coffee, E.M.; Costa, C.; Dastur, A.; Ebi, H.; Hata, A.N.; Yeo, A.T.; Edelman, E.J.; Song, Y.; Tam, A.T.; et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014, 4, 42–52. [Google Scholar]
- Holt, S.V.; Logie, A.; Davies, B.R.; Alferez, D.; Runswick, S.; Fenton, S.; Chresta, C.M.; Gu, Y.; Zhang, J.; Wu, Y.L.; et al. Enhanced apoptosis and tumor growth suppression elicited by combination of MEK (selumetinib) and mTOR kinase inhibitors (AZD8055). Cancer Res. 2012, 72, 1804–1813. [Google Scholar]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of Akt signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.; Parsons, R. mTOR inhibition, the second generation: ATP-competitive mTOR inhibitor initiates unexpected receptor tyrosine kinase-driven feedback loop. Cancer Discov. 2011, 1, 203–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ke, L.D.; Adler-Storthz, K.; Clayman, G.L.; Yung, A.W.; Chen, Z. Differential expression of epidermal growth factor receptor in human head and neck cancers. Head Neck 1998, 20, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.B.; Harris, A.L. The epidermal growth factor receptor in breast cancer. J. Mammary Gland Biol. Neoplasia 1997, 2, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Faivre, S.; Armand, J.P. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs 2000, 60, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.E.; Murren, J.R. Erlotinib Osi/Roche/Genentech. Curr. Opin. Investig. Drugs 2002, 3, 1385–1395. [Google Scholar] [PubMed]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. Akt inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-over-expressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar]
- Dansen, T.B.; Burgering, B.M. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008, 18, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (FOX) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.; Eyzaguirre, A.; Haley, J.D.; Gibson, N.W.; Cagnoni, P.; Iwata, K.K. Inactivation of Akt by the epidermal growth factor receptor inhibitor erlotinib is mediated by HER-3 in pancreatic and colorectal tumor cell lines and contributes to erlotinib sensitivity. Mol. Cancer Ther. 2006, 5, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Nyakern, M.; Cappellini, A.; Mantovani, I.; Martelli, A.M. Synergistic induction of apoptosis in human leukemia t cells by the Akt inhibitor perifosine and etoposide through activation of intrinsic and Fas-mediated extrinsic cell death pathways. Mol. Cancer Ther. 2006, 5, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, S.Y.; Owonikoko, T.K.; Sica, G.L.; Curran, W.J.; Khuri, F.R.; Deng, X. Rapamycin induces bad phosphorylation in association with its resistance to human lung cancer cells. Mol. Cancer Ther. 2012, 11, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Sirotnak, F.M.; Zakowski, M.F.; Miller, V.A.; Scher, H.I.; Kris, M.G. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin. Cancer Res. 2000, 6, 4885–4892. [Google Scholar] [PubMed]
- Wang, X.; Hawk, N.; Yue, P.; Kauh, J.; Ramalingam, S.S.; Fu, H.; Khuri, F.R.; Sun, S.Y. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors’ anticancer efficacy. Cancer Biol. Ther. 2008, 7, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA 2013, 63, 11–30. [Google Scholar] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA 2008, 58, 71–96. [Google Scholar]
- Zhang, Y.J.; Dai, Q.; Sun, D.F.; Xiong, H.; Tian, X.Q.; Gao, F.H.; Xu, M.H.; Chen, G.Q.; Han, Z.G.; Fang, J.Y. mTOR signaling pathway is a target for the treatment of colorectal cancer. Ann. Surg. Oncol. 2009, 16, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.T.; Chakrabarty, A.; Arteaga, C.L. Will PI3K pathway inhibitors be effective as single agents in patients with cancer? Oncotarget 2011, 2, 1314–1321. [Google Scholar] [PubMed]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R.A.C. Elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; de Ruiter, N.D.; de Vries-Smits, A.M.; Powell, D.R.; Bos, J.L.; Burgering, B.M. Direct control of the forkhead transcription factor AFX by protein kinase B. Nature 1999, 398, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Kurten, R.C.; Cadena, D.L.; Gill, G.N. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science 1996, 272, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Bae, G.Y.; Choi, S.J.; Lee, J.S.; Jo, J.; Lee, J.; Kim, J.; Cha, H.J. Loss of e-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget 2013, 4, 2512–2522. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, F.; Zhang, Y.; Geng, L.; Zhang, P.; Wang, G.; Liu, Y. mTOR Inhibition Induces EGFR Feedback Activation in Association with Its Resistance to Human Pancreatic Cancer. Int. J. Mol. Sci. 2015, 16, 3267-3282. https://doi.org/10.3390/ijms16023267
Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. mTOR Inhibition Induces EGFR Feedback Activation in Association with Its Resistance to Human Pancreatic Cancer. International Journal of Molecular Sciences. 2015; 16(2):3267-3282. https://doi.org/10.3390/ijms16023267
Chicago/Turabian StyleWei, Feng, Yandong Zhang, Li Geng, Ping Zhang, Guangyi Wang, and Yan Liu. 2015. "mTOR Inhibition Induces EGFR Feedback Activation in Association with Its Resistance to Human Pancreatic Cancer" International Journal of Molecular Sciences 16, no. 2: 3267-3282. https://doi.org/10.3390/ijms16023267
APA StyleWei, F., Zhang, Y., Geng, L., Zhang, P., Wang, G., & Liu, Y. (2015). mTOR Inhibition Induces EGFR Feedback Activation in Association with Its Resistance to Human Pancreatic Cancer. International Journal of Molecular Sciences, 16(2), 3267-3282. https://doi.org/10.3390/ijms16023267