
Cell-Penetrating Ability of Peptide Hormones: Key Role of Glycosaminoglycans Clustering

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence a,b | Charge c |
|---|---|---|
| PACAP 38 | HSDGIFTDSYSRYRKQMAVKKYLAAVLGKRYKQRVKNK | 10 |
| PACAP 27 | HSDGIFTDSYSRYRKQMAVKKYLAAVL | 4 |
| VIP | HSDAVFTDNYTRLRKQMAVKKYLNSILN | 4 |
| Secretin | HSDGTFTSELSRLLREGARLQRLQGLV | 2 |
| Glucagon | HSQGTFTSDYSKYLDSRRAQDFVQWLMNT | 1 |
| GLP-1 | HDEFERHAEGTFTSDVSSYLEGQAAQGFIAWLVKGRG | −2 |
| Calcitonin | CGNLSTCMLGTYTQDFNKFHTFPQTAIGVGAP | 1 |
| TAT(48–60) | GRKKRRQRRRPPQ | 9 |
2. Results and Discussion
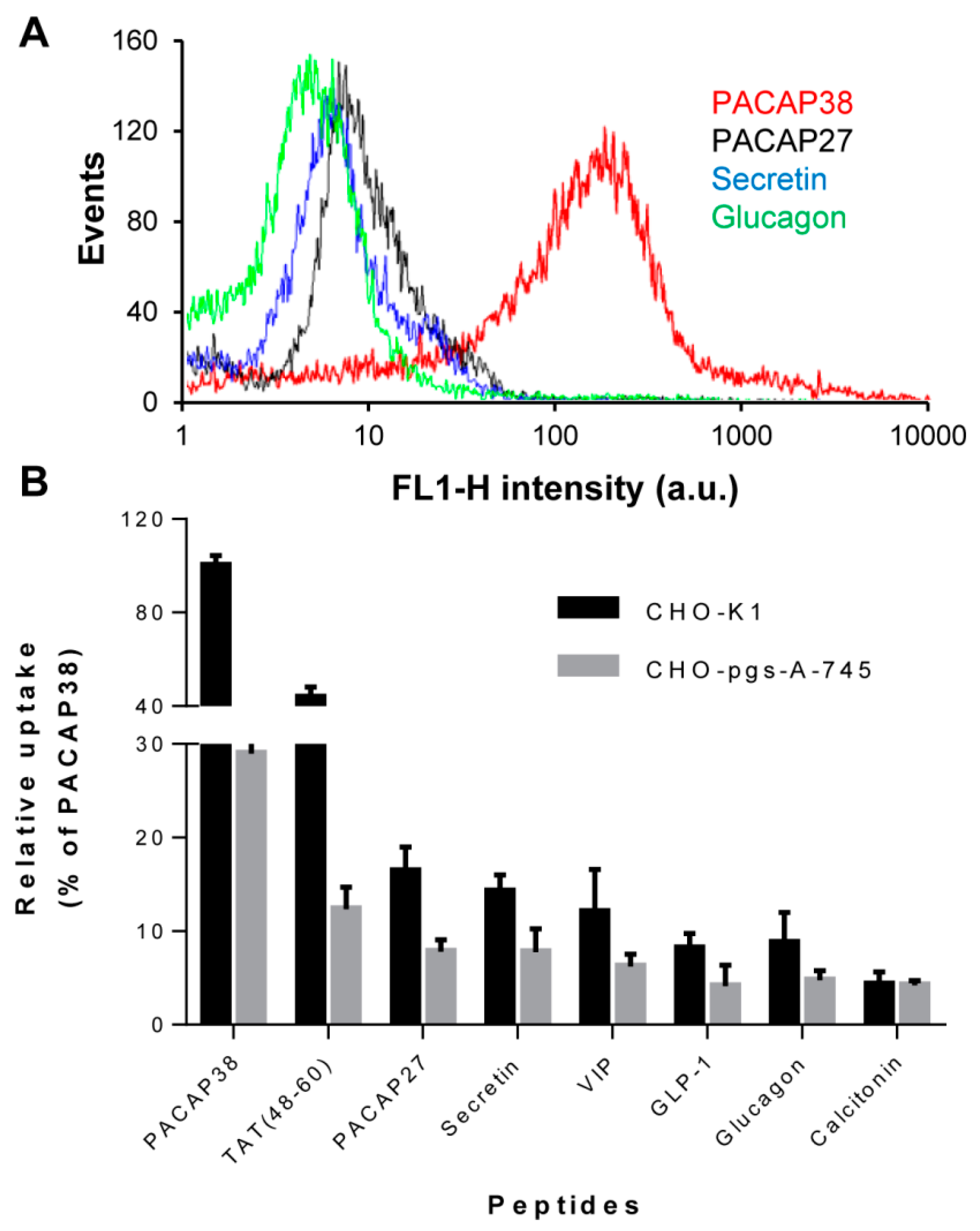
2.1. Cell Surface Glycosaminoglycans Promote Cellular Uptake of Class B GPCR Ligands
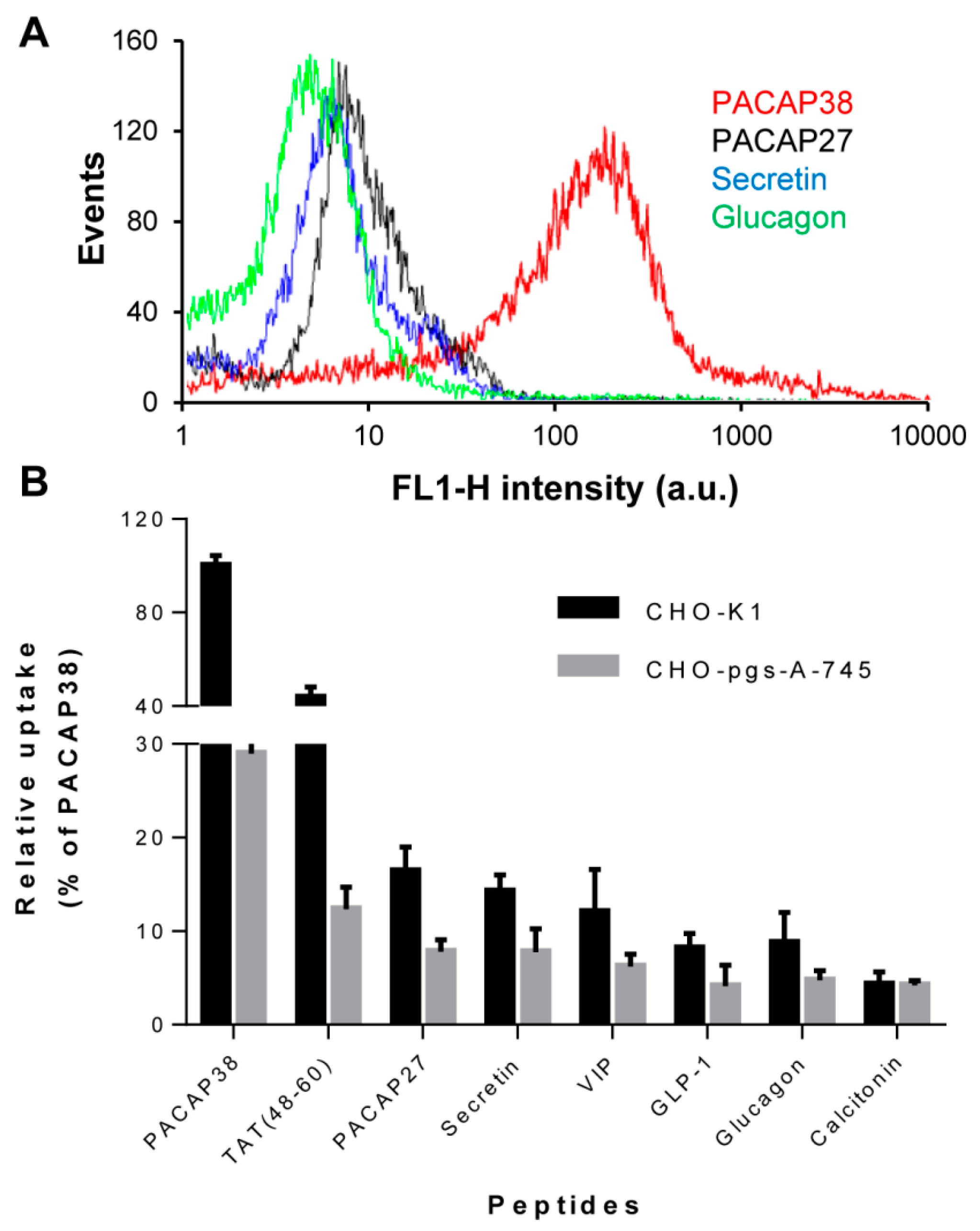
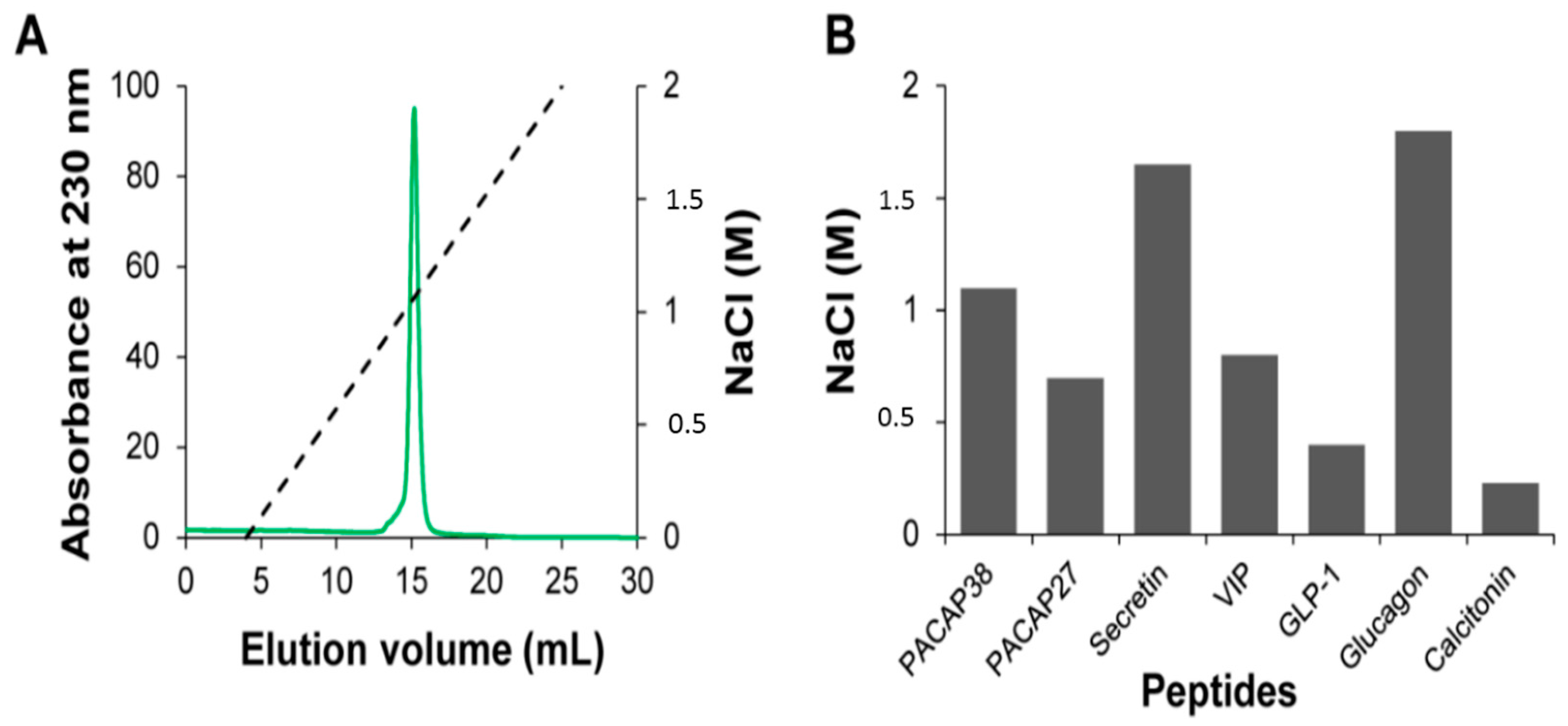
2.2. Relative Affinity for Sulfated Glycosaminoglycans

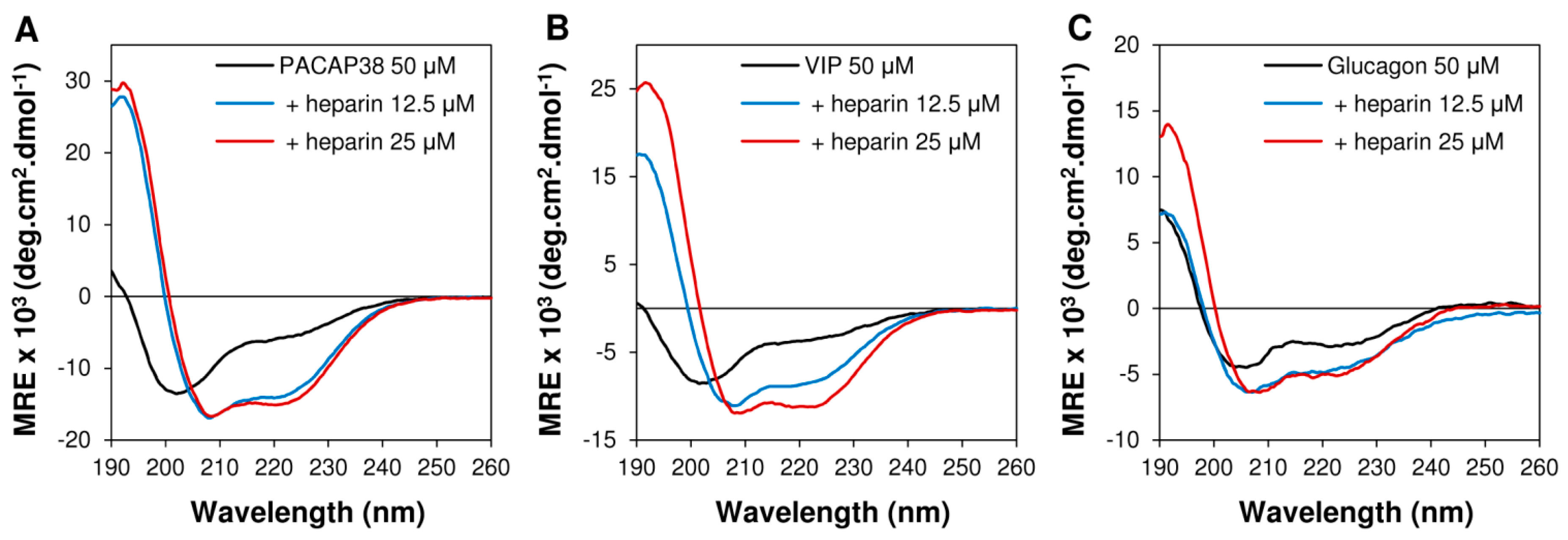
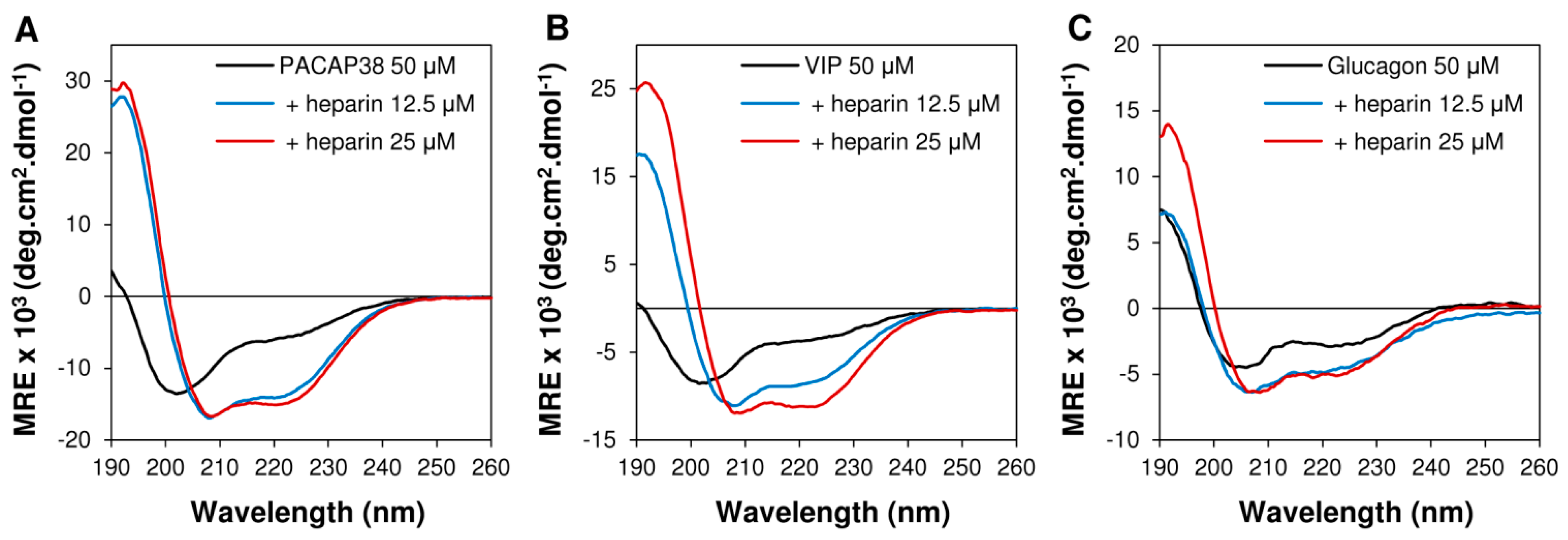
2.3. Heparin Binding Induced Conformational Conversion of Peptide Hormones
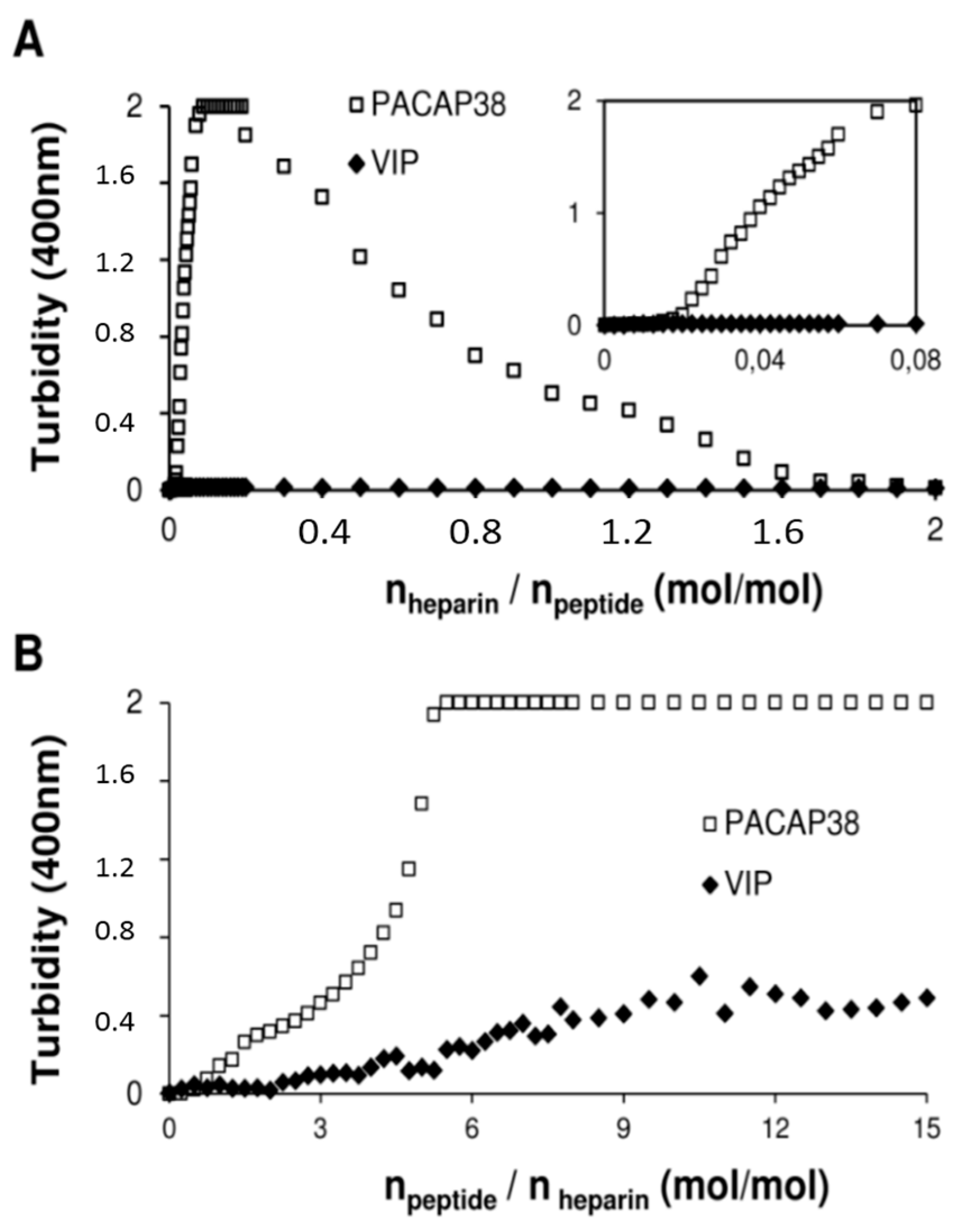
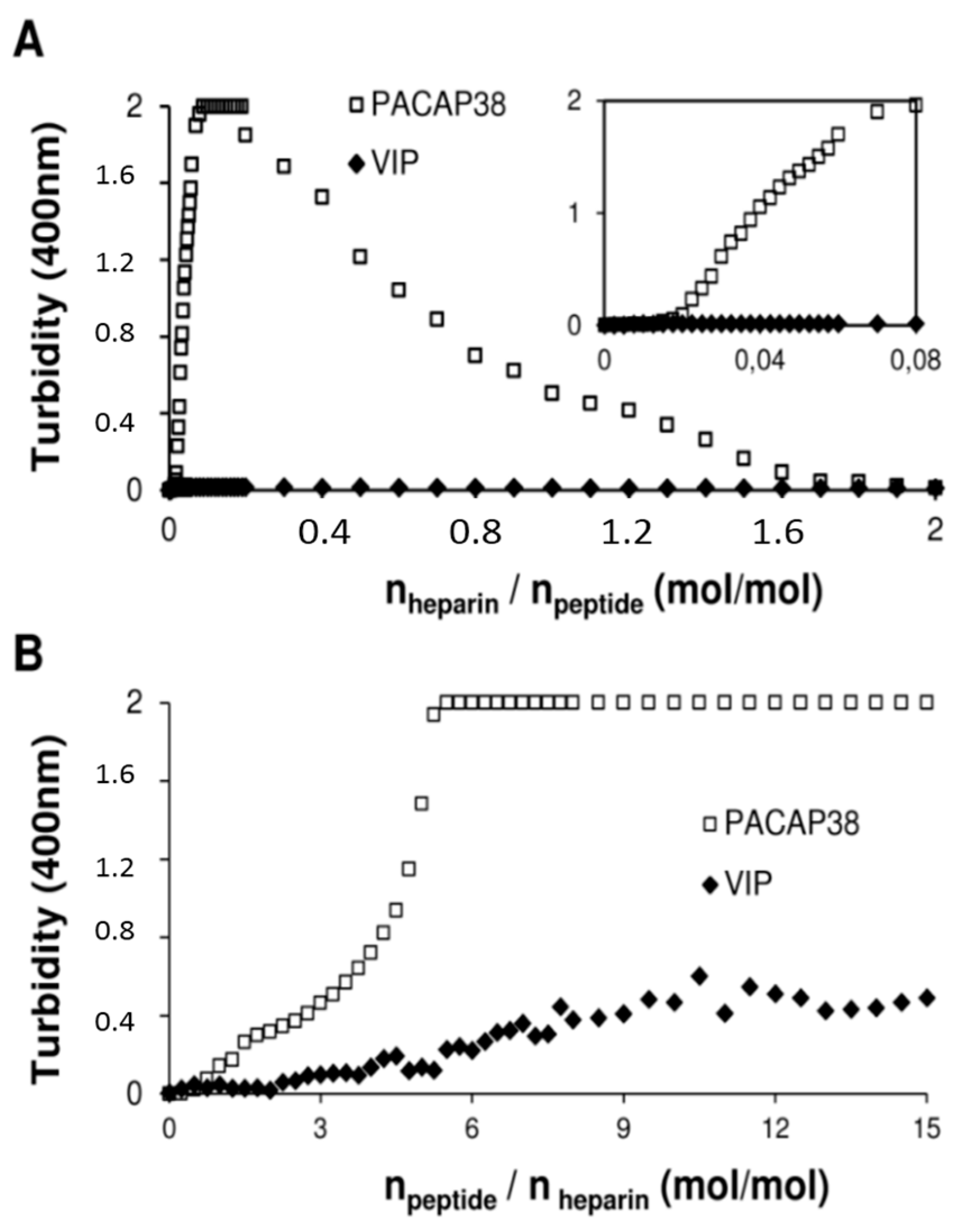
2.4. PACAP38 Is Unique to Induce Heparin Clustering
3. Experimental Section
3.1. Peptide Synthesis, Purification and Characterization
3.2. Cell Culture
3.3. Evaluation of Peptide Uptake by Flow Cytometry
3.4. Affinity Chromatography
3.5. Circular Dichroism Spectroscopy
3.6. Clustering Measured by Turbidity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Controll. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Schmidt, J.; Pelegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Ishino, M.; Loewenstein, P.M. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress hiv-ltr-driven gene expression. Cell 1989, 58, 215–223. [Google Scholar] [CrossRef]
- Doan, N.D.; Chatenet, D.; Letourneau, M.; Vaudry, H.; Vaudry, D.; Fournier, A. Receptor-independent cellular uptake of pituitary adenylate cyclase-activating polypeptide. Biochim. Biophys. Acta 2012, 1823, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.D.; Letourneau, M.; Vaudry, D.; Doucet, N.; Folch, B.; Vaudry, H.; Fournier, A.; Chatenet, D. Design and characterization of novel cell-penetrating peptides from pituitary adenylate cyclase-activating polypeptide. J. Controll. Release 2012, 163, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Vaudry, D.; Dejda, A.; Doan, N.D.; Vaudry, H.; Fournier, A. Pituitary adenylate cyclase-activating polypeptide: Focus on structure-activity relationships of a neuroprotective peptide. Curr. Med. Chem. 2009, 16, 4462–4480. [Google Scholar] [CrossRef] [PubMed]
- Hoare, S.R. Mechanisms of peptide and nonpeptide ligand binding to class B G-protein-coupled receptors. Drug Discov. Today 2005, 10, 417–427. [Google Scholar] [CrossRef]
- Bourgault, S.; Vaudry, D.; Guilhaudis, L.; Raoult, E.; Couvineau, A.; Laburthe, M.; Segalas-Milazzo, I.; Vaudry, H.; Fournier, A. Biological and structural analysis of truncated analogs of pacap27. J. Mol. Neurosci. 2008, 36, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Vaudry, D.; Segalas-Milazzo, I.; Guilhaudis, L.; Couvineau, A.; Laburthe, M.; Vaudry, H.; Fournier, A. Molecular and conformational determinants of pituitary adenylate cyclase-activating polypeptide (PACAP) for activation of the pac1 receptor. J. Med. Chem. 2009, 52, 3308–3316. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Chatenet, D.; Wurtz, O.; Doan, N.D.; Leprince, J.; Vaudry, H.; Fournier, A.; Vaudry, D. Strategies to convert pacap from a hypophysiotropic neurohormone into a neuroprotective drug. Curr. Pharm. Des. 2011, 17, 1002–1024. [Google Scholar] [CrossRef] [PubMed]
- Tchoumi Neree, A.; Nguyen, P.T.; Chatenet, D.; Fournier, A.; Bourgault, S. Secondary conformational conversion is involved in glycosaminoglycans-mediated cellular uptake of the cationic cell-penetrating peptide pacap. FEBS Lett. 2014, 588, 4590–4596. [Google Scholar] [CrossRef] [PubMed]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.; Neundorf, I.; Beck-Sickinger, A.G. Calcitonin-derived peptide carriers: Mechanisms and application. Adv. Drug Deliv. Rev. 2008, 60, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.C.; Rothen-Rutishauser, B.; Rist, B.; Beck-Sickinger, A.; Wunderli-Allenspach, H.; Rubas, W.; Sadee, W.; Merkle, H.P. Translocation of human calcitonin in respiratory nasal epithelium is associated with self-assembly in lipid membrane. Biochemistry 1998, 37, 16582–16590. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Stewart, T.E.; Taylor, W.H. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 1985, 82, 3197–3201. [Google Scholar] [CrossRef] [PubMed]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of protein function by glycosaminoglycans—As exemplified by chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef] [PubMed]
- Amand, H.L.; Rydberg, H.A.; Fornander, L.H.; Lincoln, P.; Norden, B.; Esbjorner, E.K. Cell surface binding and uptake of arginine- and lysine-rich penetratin peptides in absence and presence of proteoglycans. Biochim. Biophys. Acta 2012, 1818, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Rullo, A.; Qian, J.; Nitz, M. Peptide-glycosaminoglycan cluster formation involving cell penetrating peptides. Biopolymers 2011, 95, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Fromm, J.R.; Hileman, R.E.; Caldwell, E.E.; Weiler, J.M.; Linhardt, R.J. Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. Arch. Biochem. Biophys. 1995, 323, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Pallerla, M.; Zaltsman, Y.; Burlina, F.; Alves, I.D.; Lequin, O.; Sagan, S. Tryptophan within basic peptide sequences triggers glycosaminoglycan-dependent endocytosis. FASEB J. 2013, 27, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Seelig, J. Contributions of glycosaminoglycan binding and clustering to the biological uptake of the nonamphipathic cell-penetrating peptide wr9. Biochemistry 2011, 50, 4650–4664. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic tat peptide transduction domain enters cells by macropinocytosis. J. Controll. Release 2005, 102, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Al-Taei, S.; Penning, N.A.; Simpson, J.C.; Futaki, S.; Takeuchi, T.; Nakase, I.; Jones, A.T. Intracellular traffic and fate of protein transduction domains HIV-1 Tat peptide and octaarginine. Implications for their utilization as drug delivery vectors. Bioconjug. Chem. 2006, 17, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Solomon, J.P.; Reixach, N.; Kelly, J.W. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry 2011, 50, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Elimova, E.; Kisilevsky, R.; Ancsin, J.B. Heparan sulfate promotes the aggregation of HDL-associated serum amyloid A: Evidence for a proamyloidogenic histidine molecular switch. FASEB J. 2009, 23, 3436–3448. [Google Scholar] [CrossRef] [PubMed]
- Wallbrecher, R.; Verdurmen, W.P.; Schmidt, S.; Bovee-Geurts, P.H.; Broecker, F.; Reinhardt, A.; van Kuppevelt, T.H.; Seeberger, P.H.; Brock, R. The stoichiometry of peptide-heparan sulfate binding as a determinant of uptake efficiency of cell-penetrating peptides. Cell. Mol. Life Sci. 2014, 71, 2717–2729. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchoumi Neree, A.; Nguyen, P.T.; Bourgault, S. Cell-Penetrating Ability of Peptide Hormones: Key Role of Glycosaminoglycans Clustering. Int. J. Mol. Sci. 2015, 16, 27391-27400. https://doi.org/10.3390/ijms161126025
Tchoumi Neree A, Nguyen PT, Bourgault S. Cell-Penetrating Ability of Peptide Hormones: Key Role of Glycosaminoglycans Clustering. International Journal of Molecular Sciences. 2015; 16(11):27391-27400. https://doi.org/10.3390/ijms161126025
Chicago/Turabian StyleTchoumi Neree, Armelle, Phuong Trang Nguyen, and Steve Bourgault. 2015. "Cell-Penetrating Ability of Peptide Hormones: Key Role of Glycosaminoglycans Clustering" International Journal of Molecular Sciences 16, no. 11: 27391-27400. https://doi.org/10.3390/ijms161126025
APA StyleTchoumi Neree, A., Nguyen, P. T., & Bourgault, S. (2015). Cell-Penetrating Ability of Peptide Hormones: Key Role of Glycosaminoglycans Clustering. International Journal of Molecular Sciences, 16(11), 27391-27400. https://doi.org/10.3390/ijms161126025