
Aβ1-25-Derived Sphingolipid-Domain Tracer Peptide SBD Interacts with Membrane Ganglioside Clusters via a Coil-Helix-Coil Motif



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
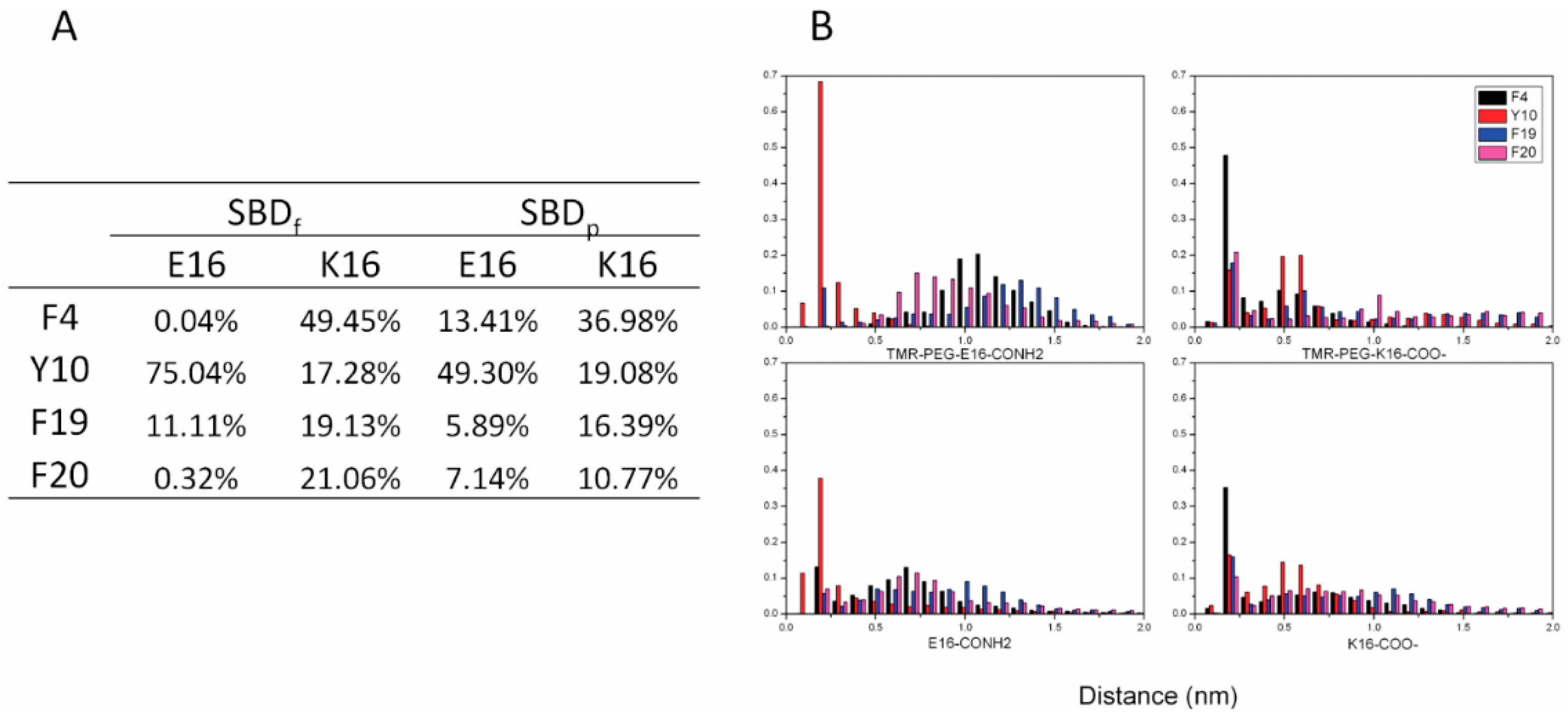
2. Results
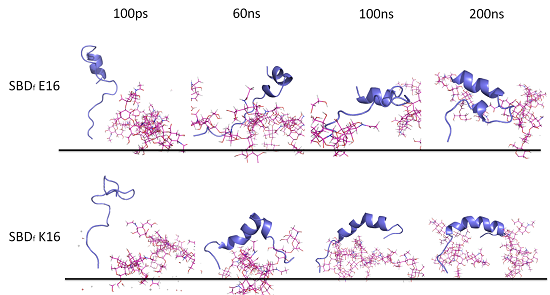
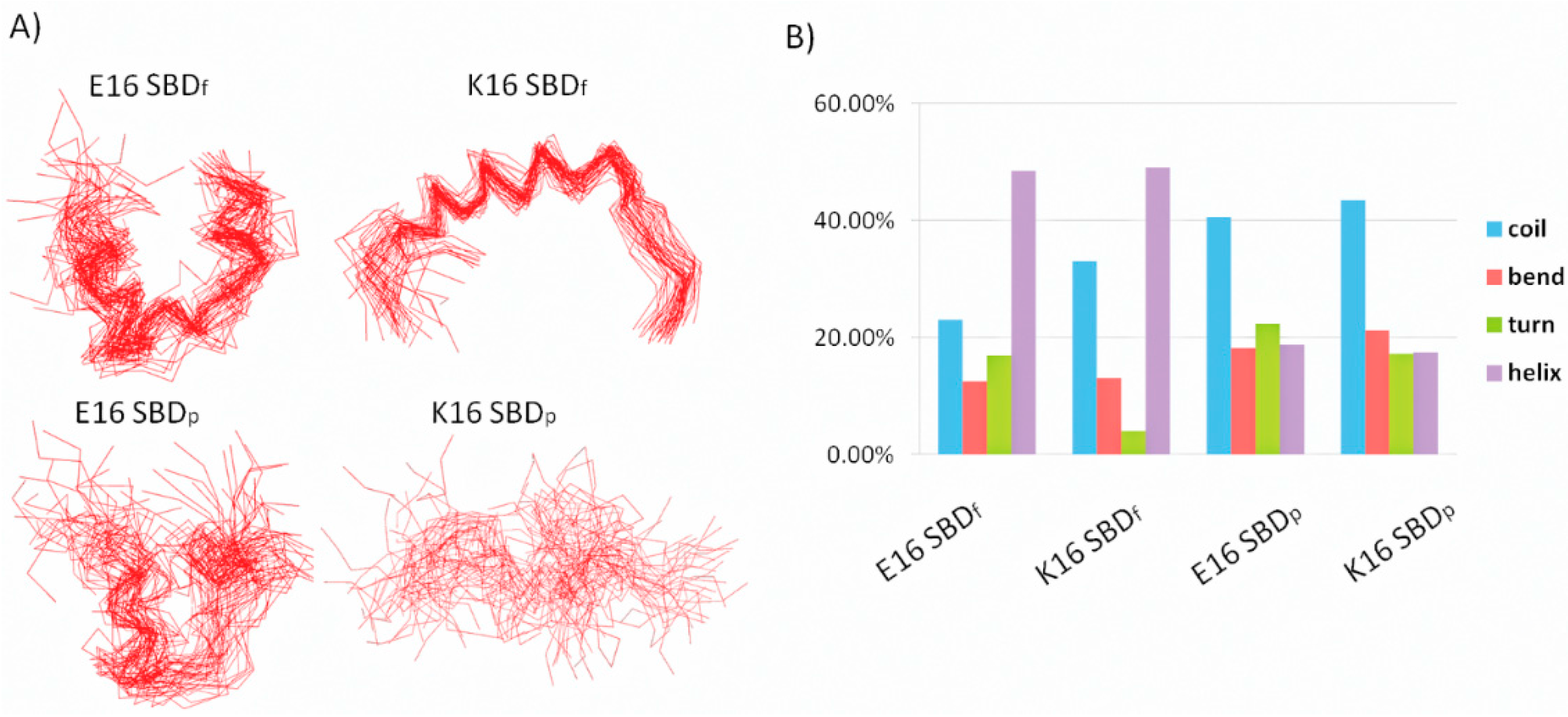
2.1. E16/K16 Sphingolipid Binding Domain (SBD) Variants Adopt Different Conformations
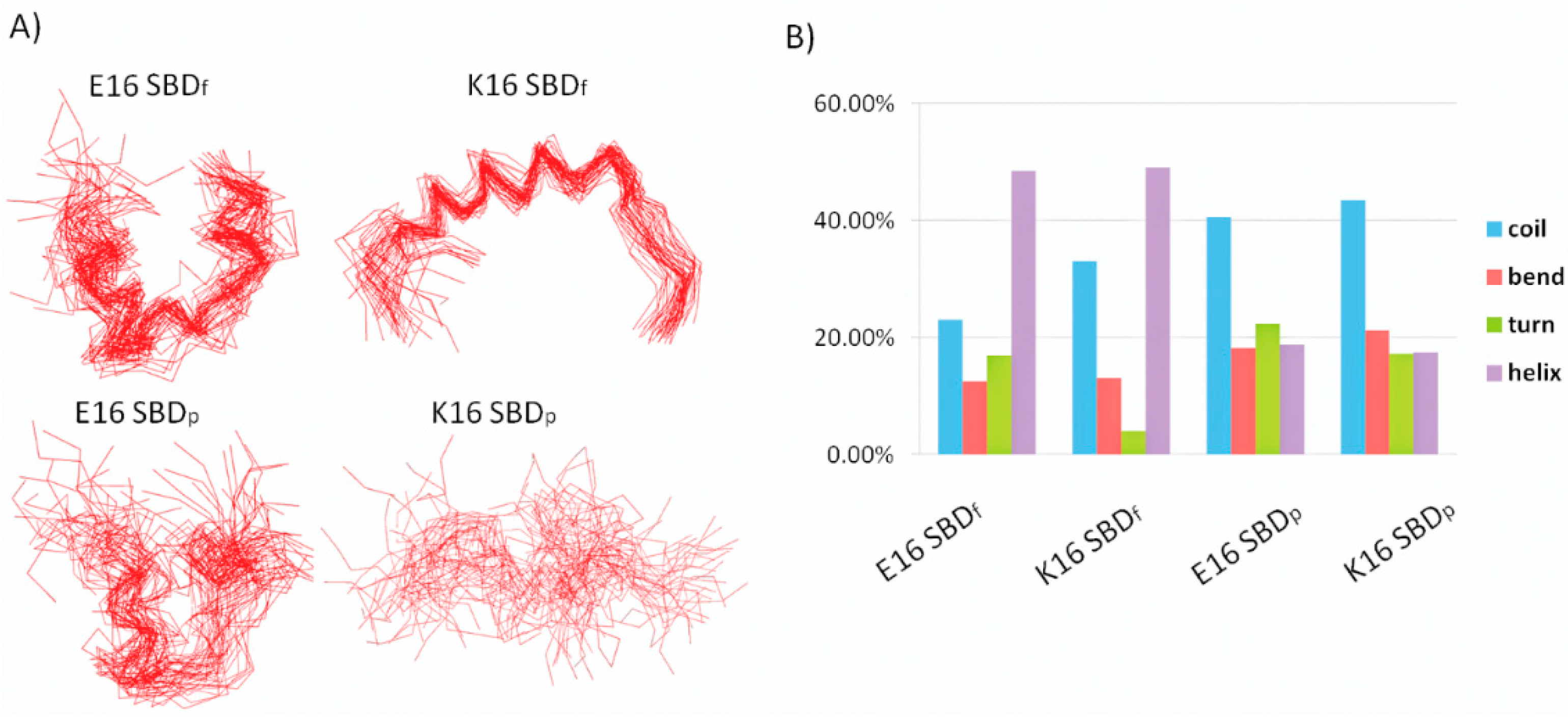
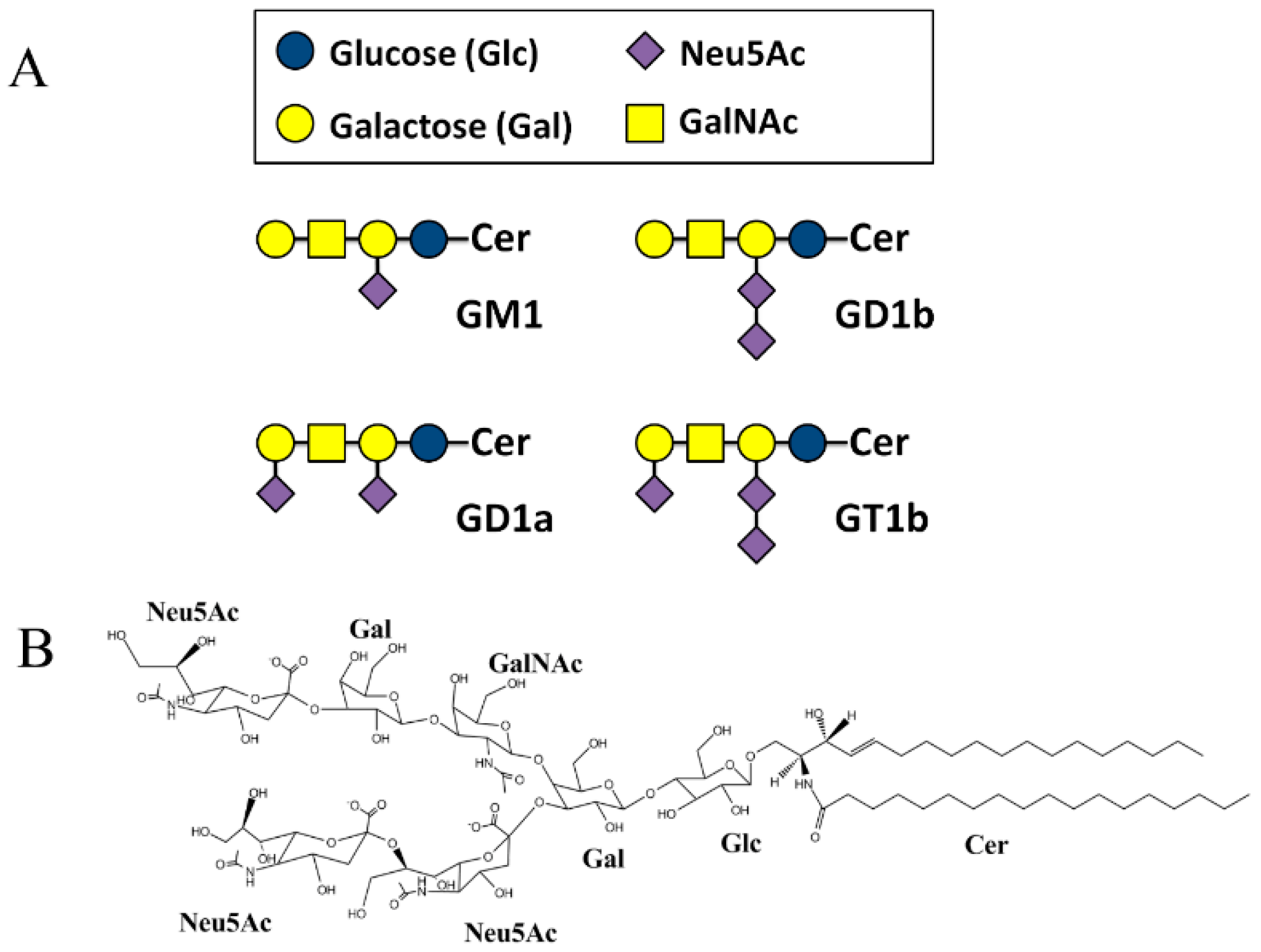
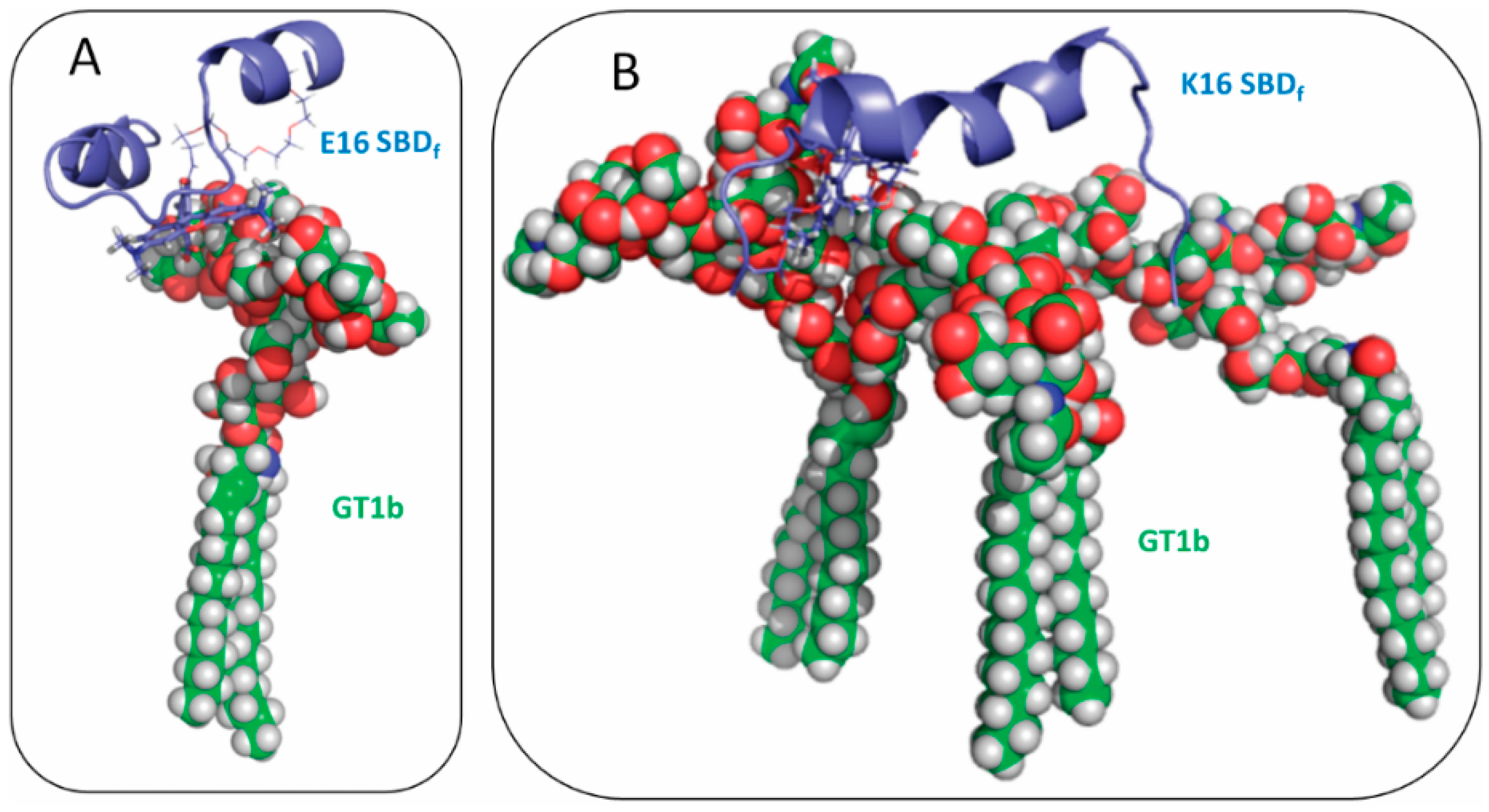
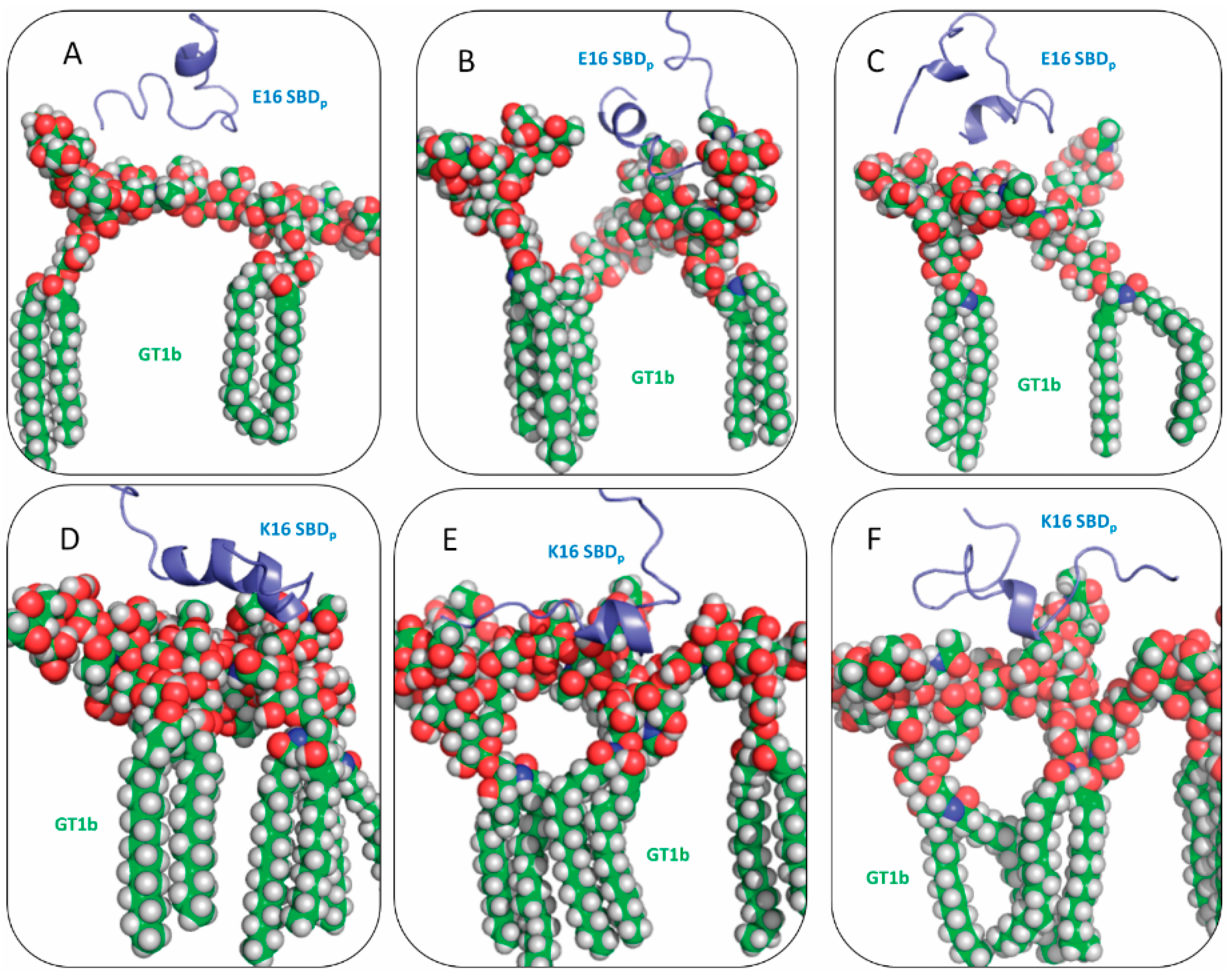
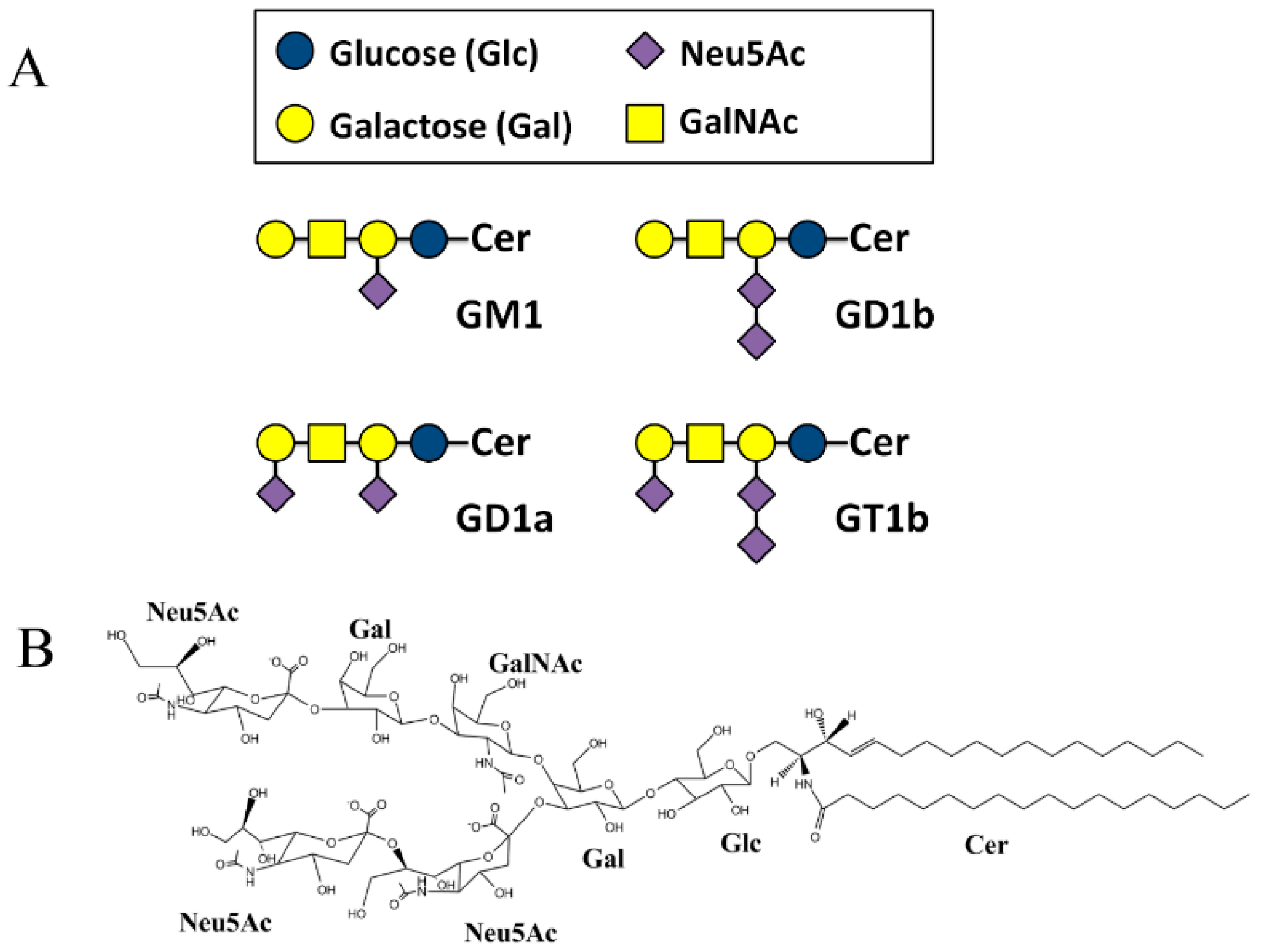
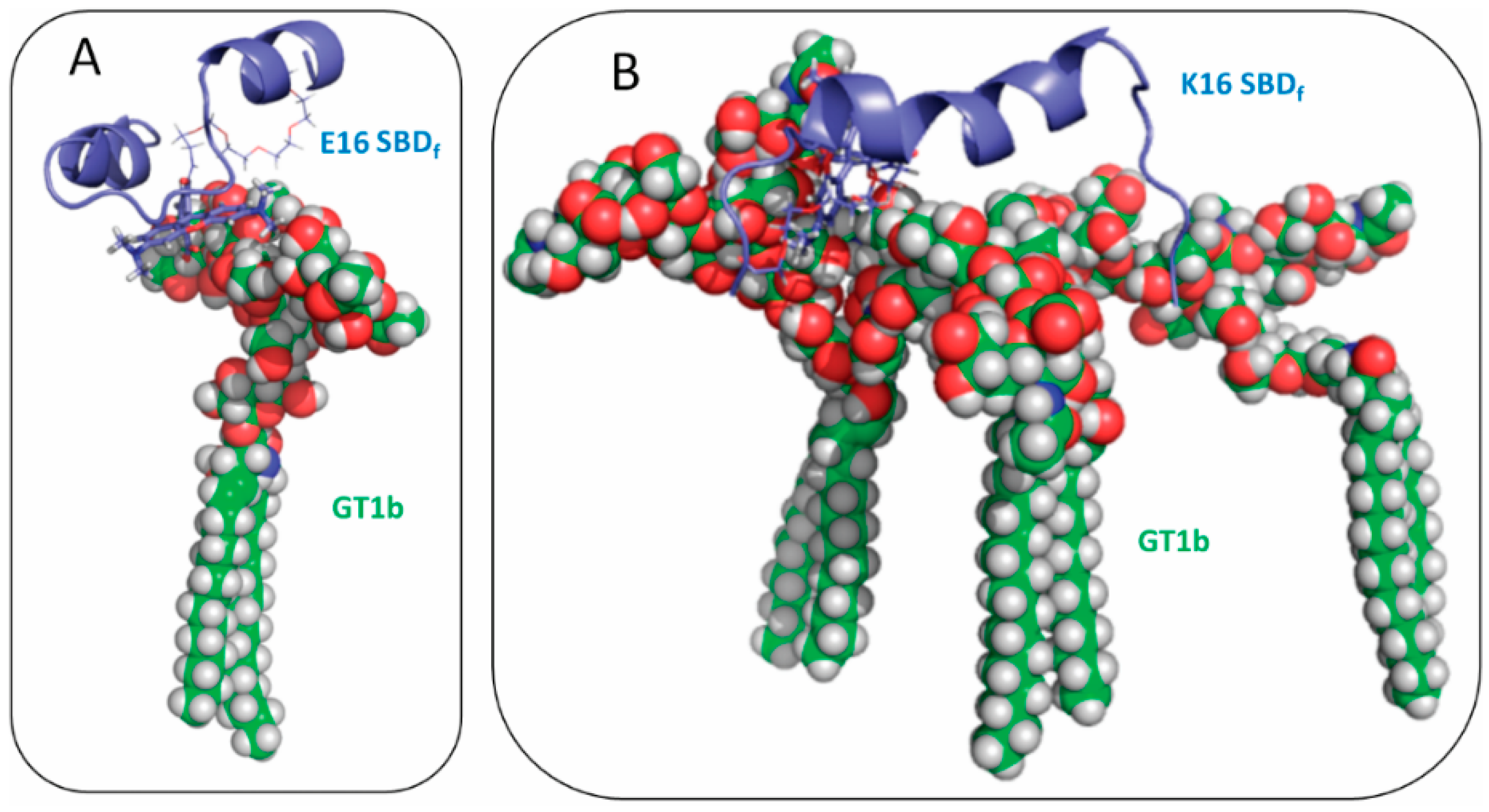
2.2. SBDp Peptides Are Capable of Binding to GT1b Clusters in Distinct Modes
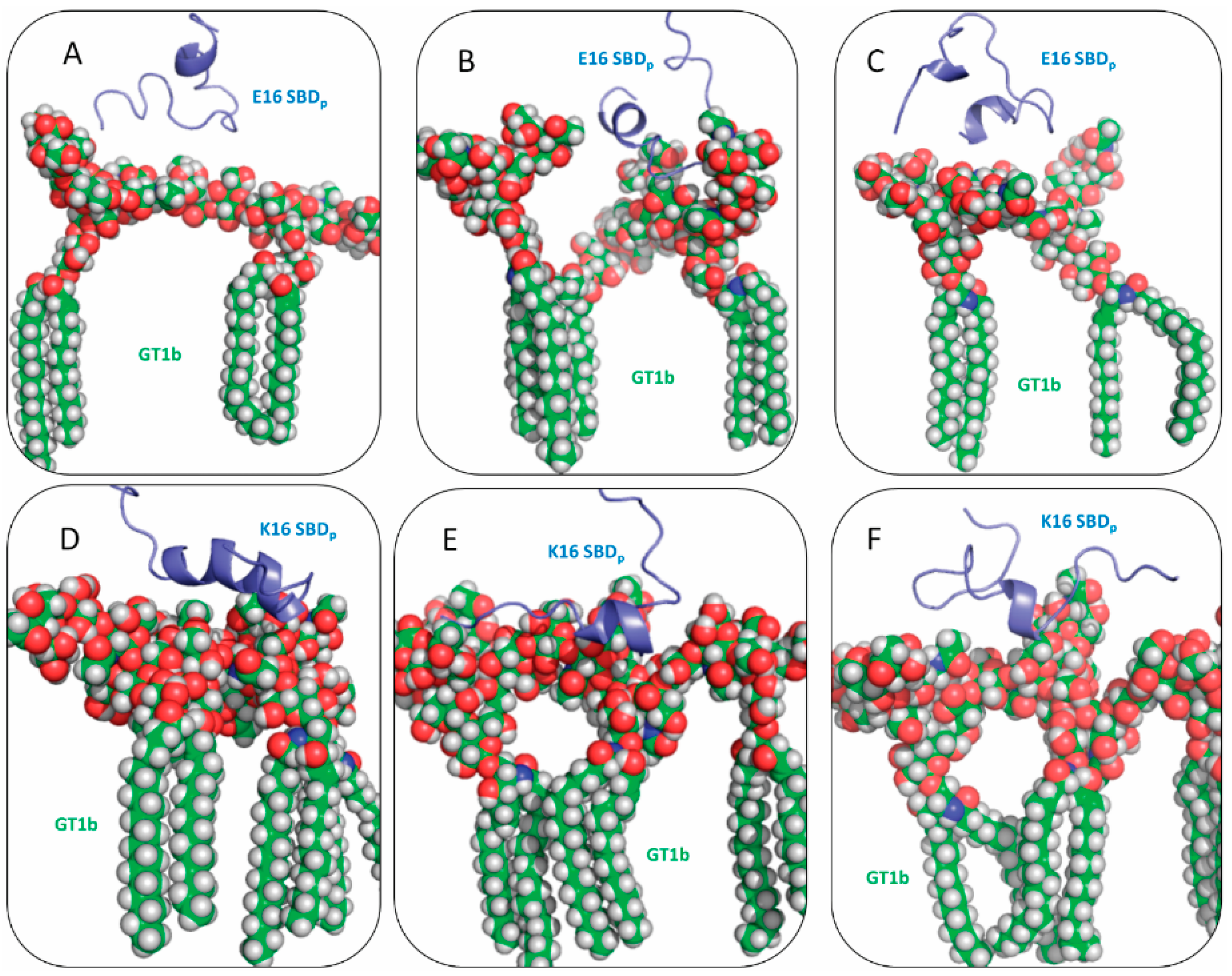
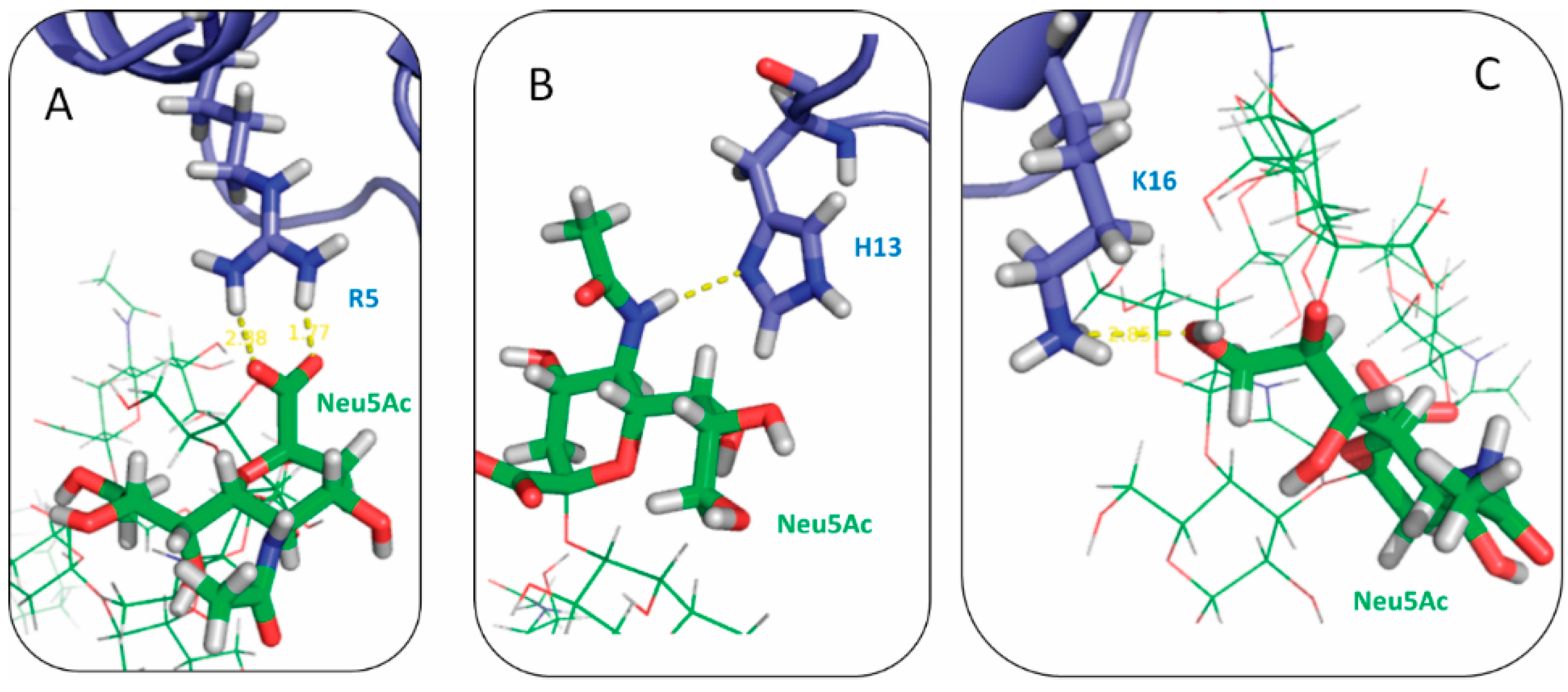
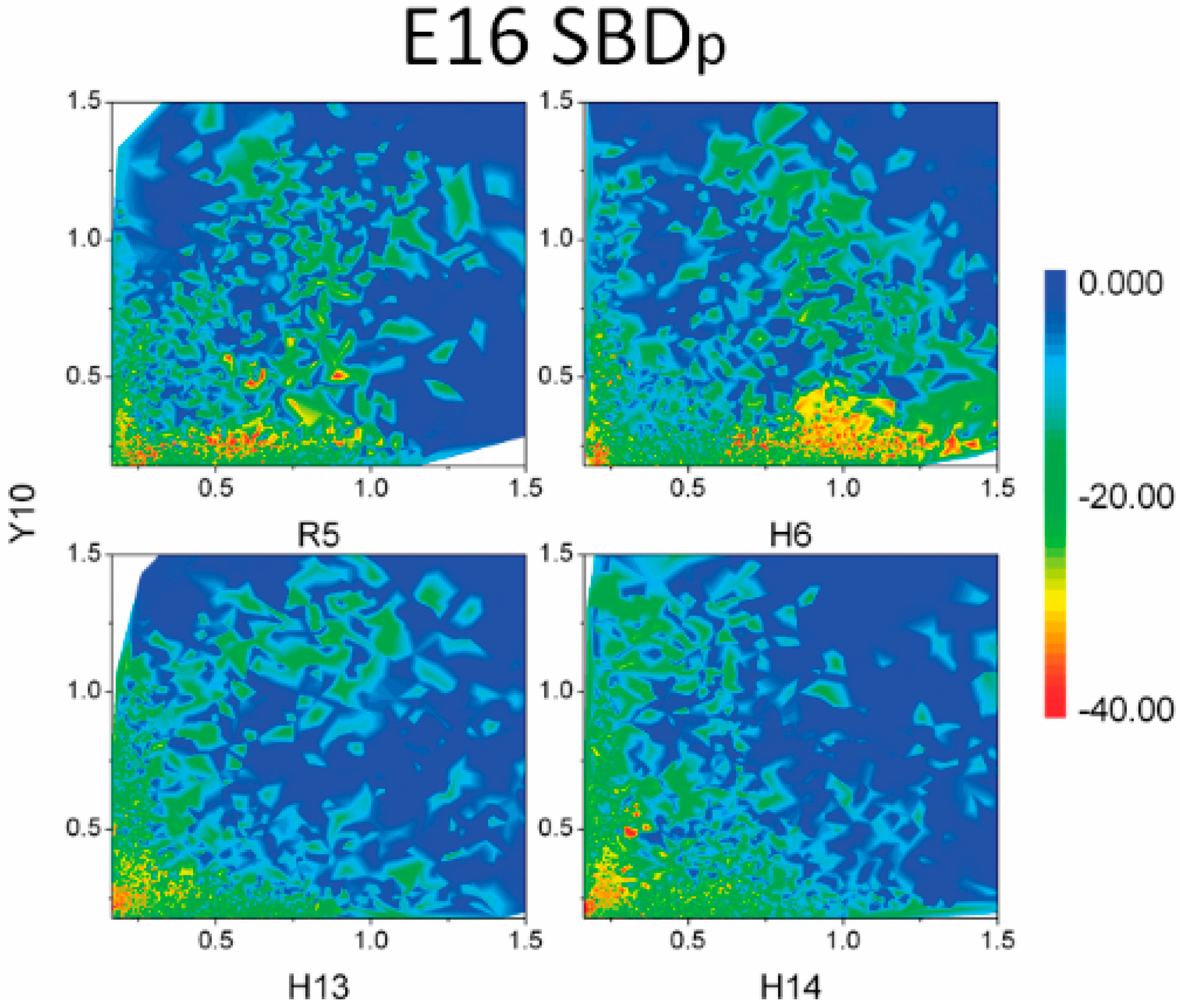
2.3. Residues R5, H13 and K16 Interact Electrostatically with Ganglioside Sialic Acid Groups
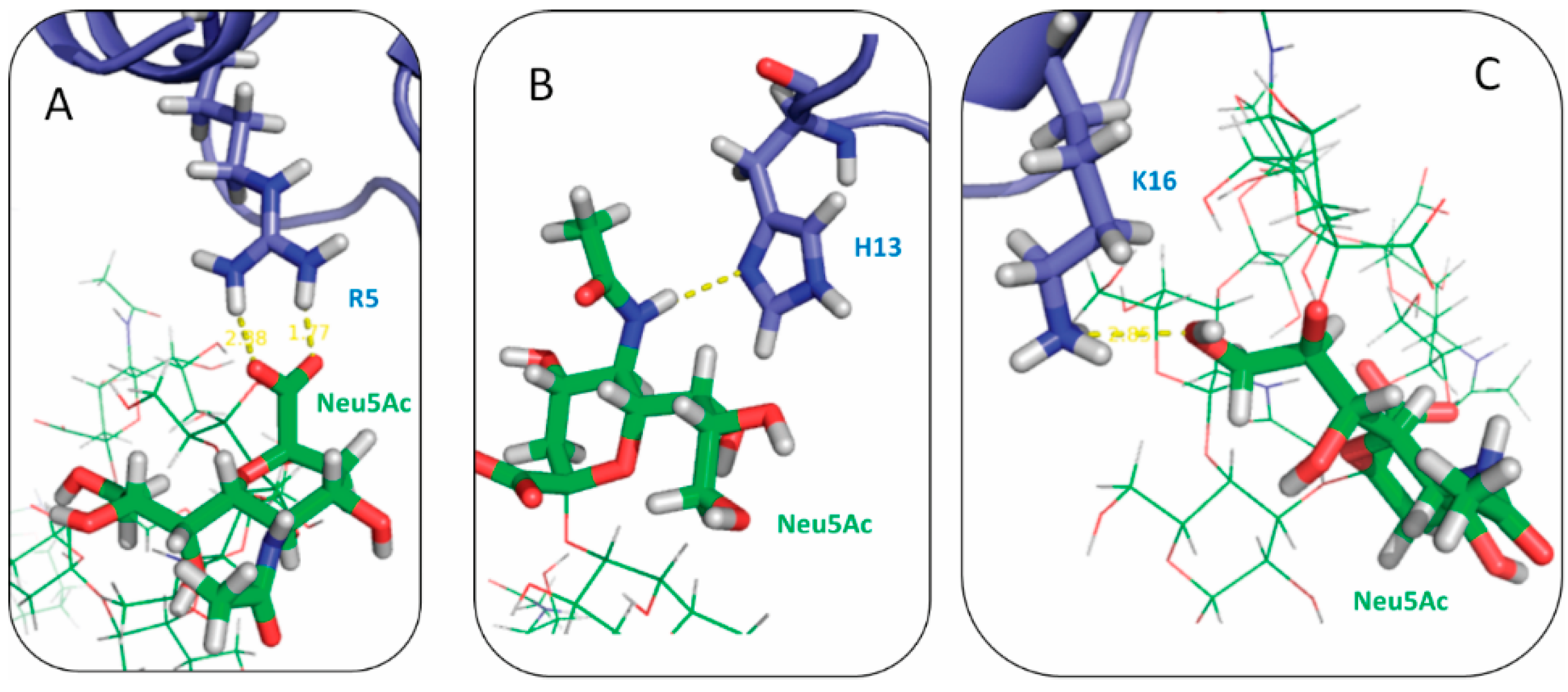
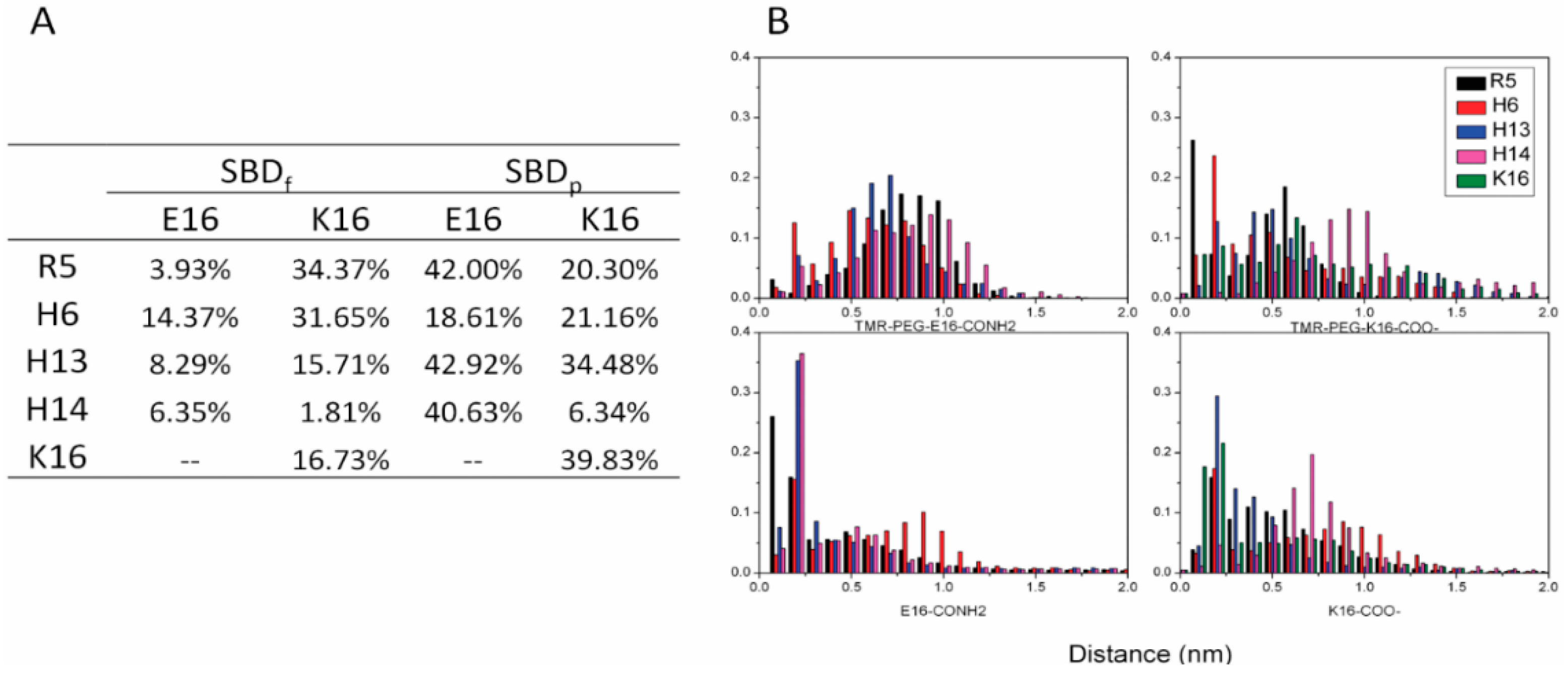
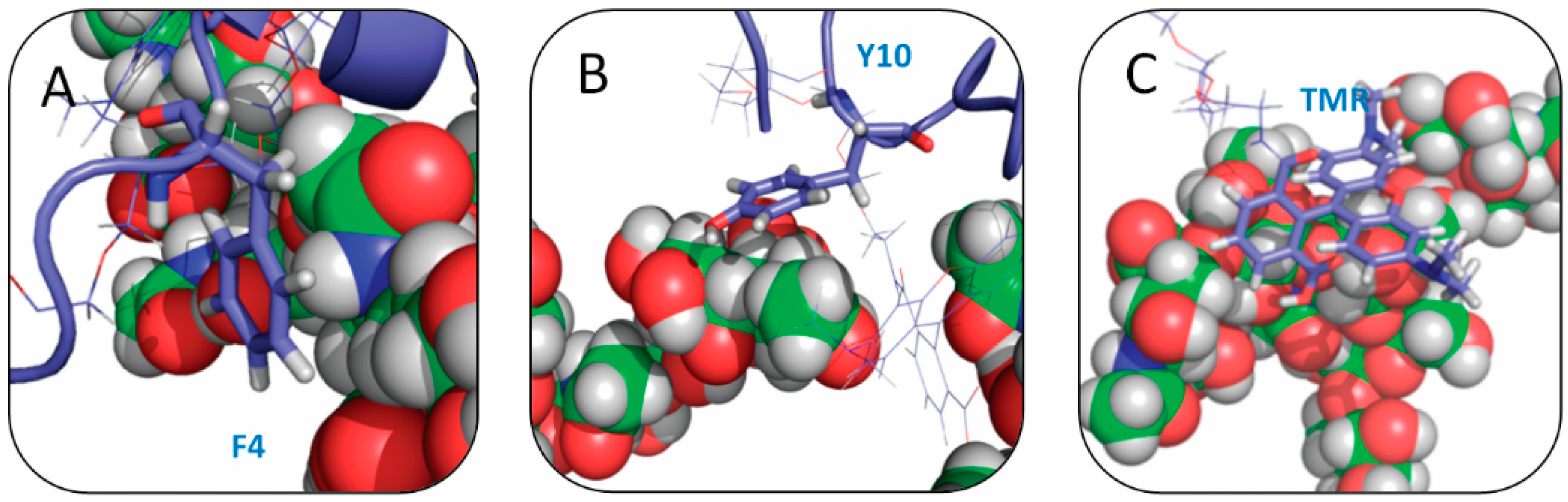
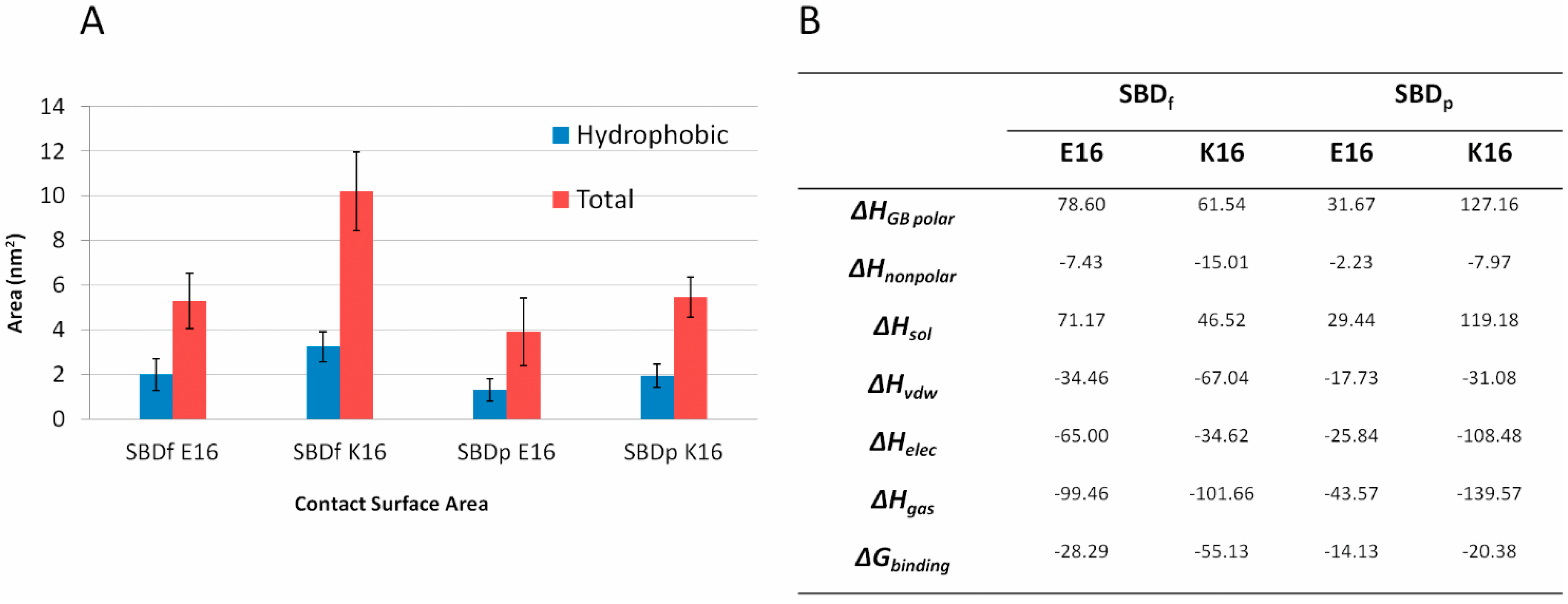
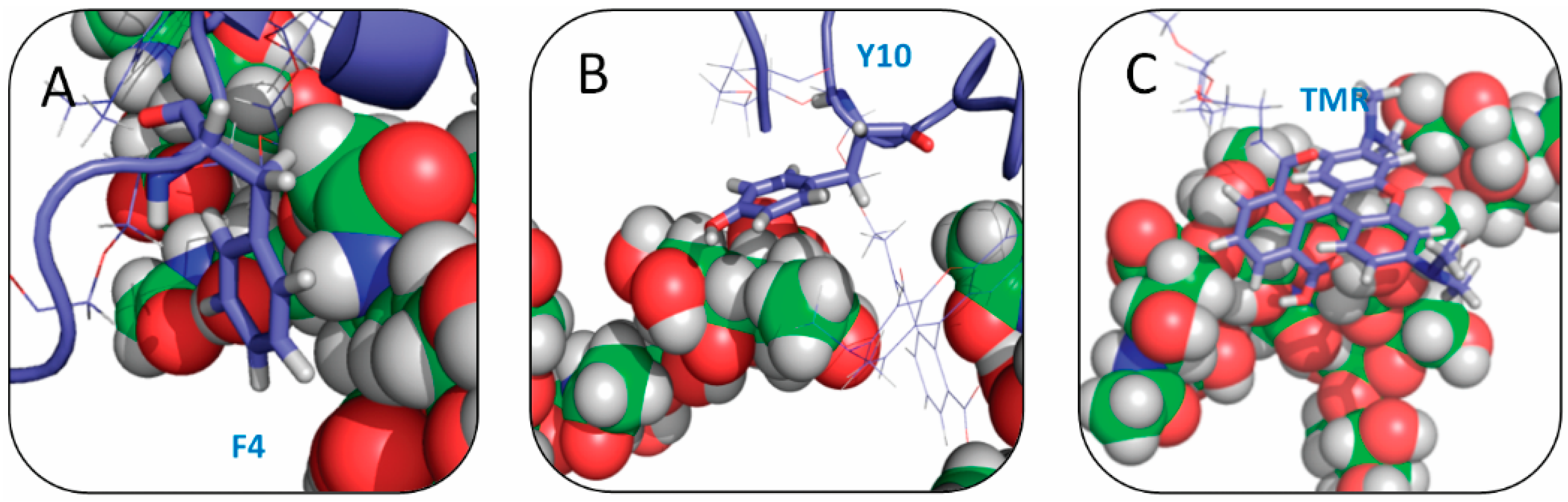
2.4. Residue Y10 and F4 Formed CH–π Interactions with GT1b Sugar Groups in E16 and K16 SBD Variants Respectively
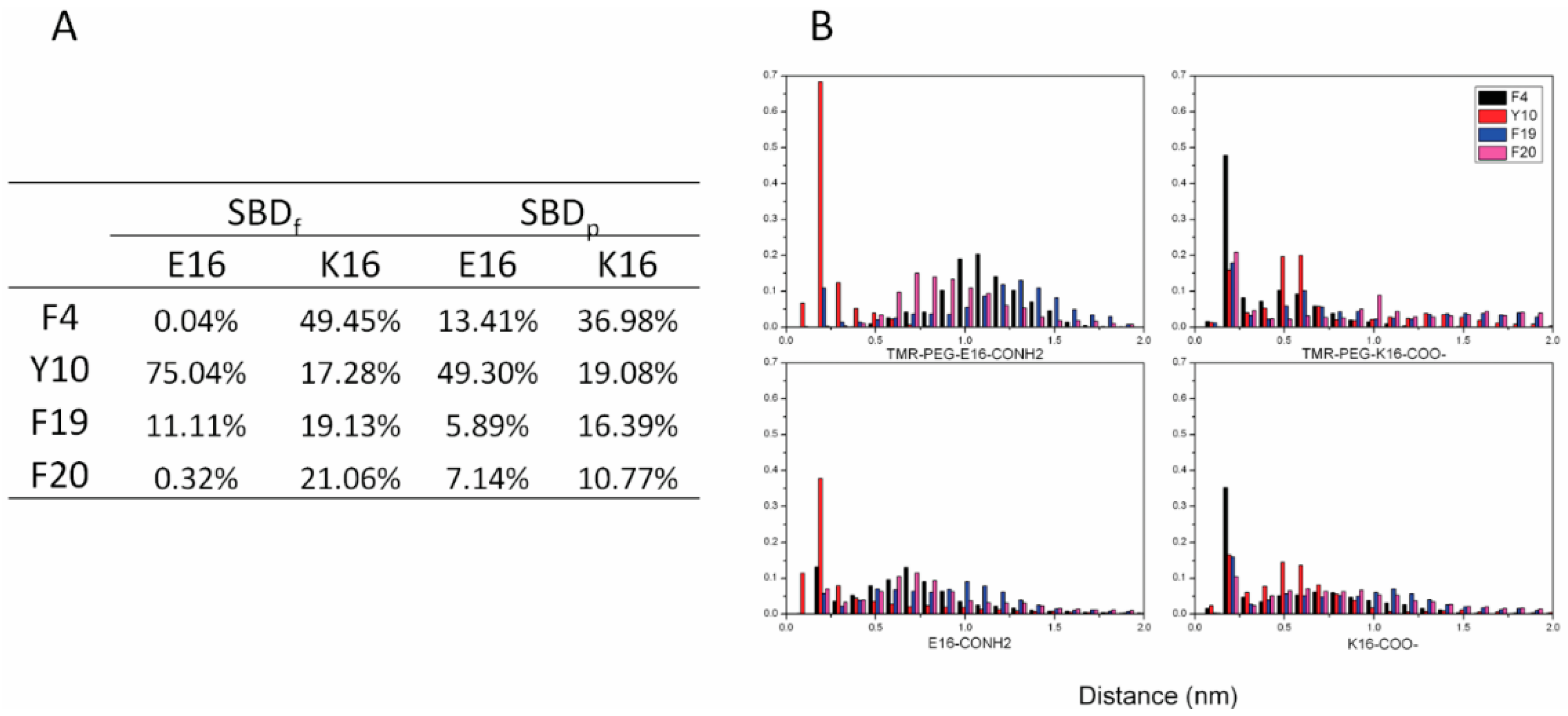
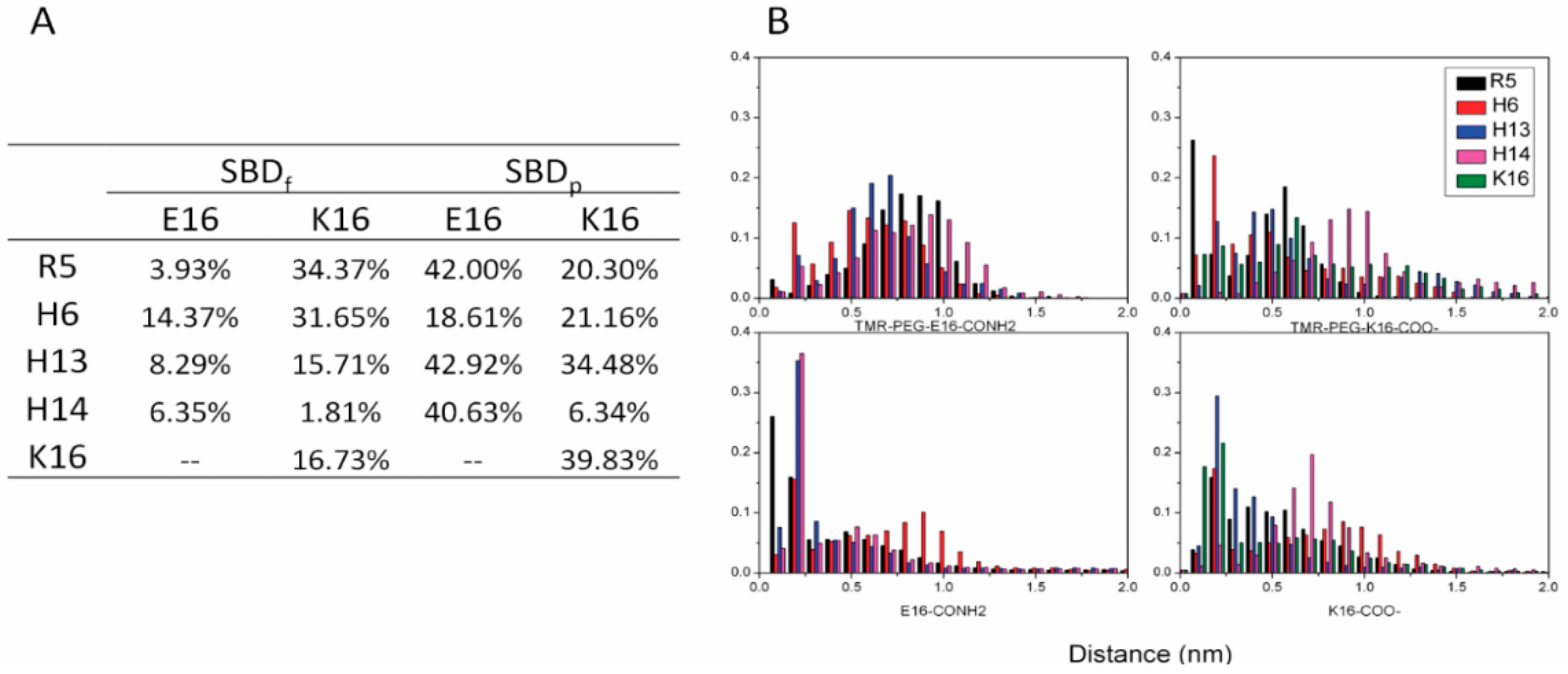
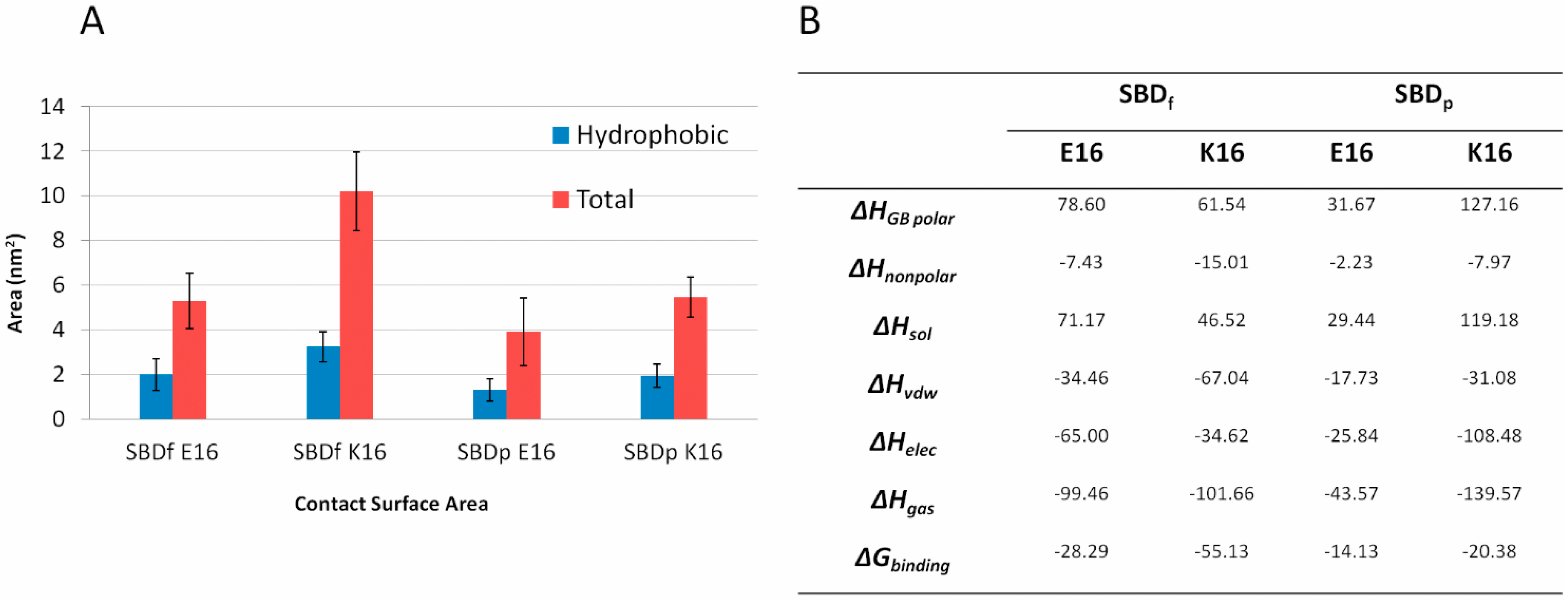
2.5. Binding Energy Calculations Showed Residues R5, Y10, H13 in E16 Variants and Residues R5, H13, K16 in K16 Variants Played Important Roles in Binding Modes to GT1b
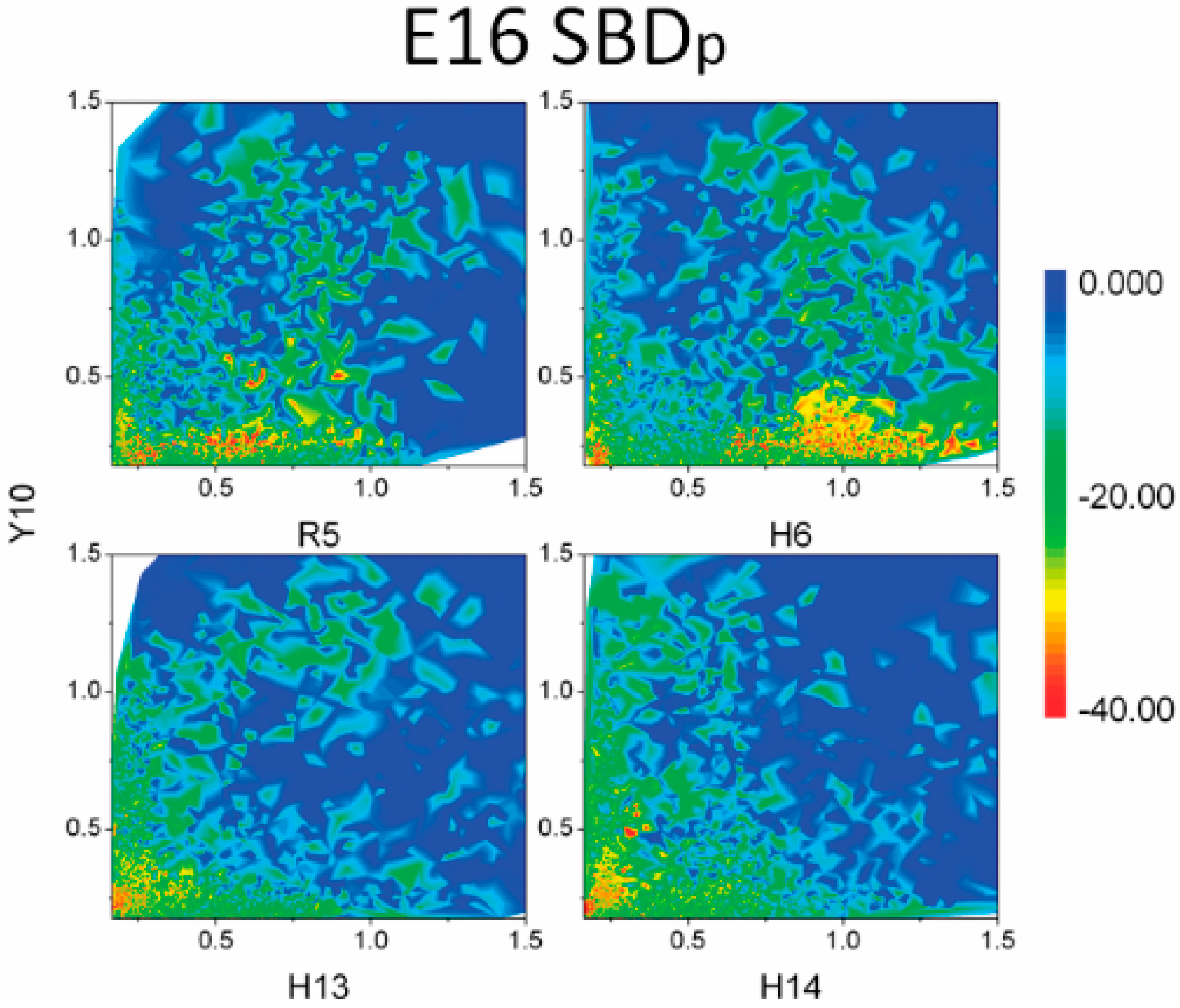
3. Discussion
4. Experimental Section
4.1. Modeling of the SBD Fluorescent-Tagged Probe and a GT1b Containing Plasma-Membrane-Like Bilayer
4.2. Measurement of Electrostatic and CH–π Interactions
4.3. MM-GBSA Binding Energy Calculations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the keystone symposium on lipid rafts and cell function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Mattner, J.; DeBord, K.L.; Ismail, N.; Goff, R.D.; Cantu, C.; Zhou, D.P.; Saint-Mezard, P.; Wang, V.; Gao, Y.; Yin, N.; et al. Exogenous and endogenous glycolipid antigens activate nkt cells during microbial infections. Nature 2005, 434, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A. Glycolipid receptors for verotoxin and helicobacter pylori: Role in pathology. Biochim. Biophys. Acta 1999, 1455, 375–386. [Google Scholar] [CrossRef]
- Lencer, W.I.; Saslowsky, D. Raft trafficking of ab5 subunit bacterial toxins. Biochim. Biophys. Acta 2005, 1746, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Gangliosides as influenza virus receptors. Variation of influenza viruses and their recognition of the receptor sialo-sugar chains. Prog. Lipid Res. 1994, 33, 429–457. [Google Scholar] [CrossRef]
- Antes, P.; Schwarzmann, G.; Sandhoff, K. Detection of protein mediated glycosphingolipid clustering by the use of resonance energy transfer between fluorescent labelled lipids. A method established by applying the system ganglioside gm1 and cholera toxin b subunit. Chem. Phys. Lipids 1992, 62, 269–280. [Google Scholar] [CrossRef]
- Tettamanti, G.; Bonali, F.; Marchesini, S.; Zambotti, V. A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta 1973, 296, 160–170. [Google Scholar] [CrossRef]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Vajn, K.; Viljetic, B.; Degmecic, I.V.; Schnaar, R.L.; Heffer, M. Differential distribution of major brain gangliosides in the adult mouse central nervous system. PLoS ONE 2013, 8, e75720. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, H.; Aoyagi, M.; Kasama, T.; Handa, S.; Hirakawa, K.; Taki, T. Gt1b in human metastatic brain tumors: Gt1b as a brain metastasis-associated ganglioside. Biochim. Biophys. Acta 1999, 1437, 93–99. [Google Scholar] [CrossRef]
- Rogers, T.B.; Snyder, S.H. High affinity binding of tetanus toxin to mammalian brain membranes. J. Biol. Chem. 1981, 256, 2402–2407. [Google Scholar] [PubMed]
- Schnaar, R.L. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010, 584, 1741–1747. [Google Scholar] [CrossRef]
- Kozaki, S.; Kamata, Y.; Watarai, S.; Nishiki, T.; Mochida, S. Ganglioside GT1b as a complementary receptor component for clostridium botulinum neurotoxins. Microb. Pathog. 1998, 25, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Onishi, A.; Sato, T. Carbohydrate recognition by pentadecapeptide ligands for a series of sialylated oligosaccharides. Bioorg. Med. Chem. 2012, 20, 6452–6458. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.L.; Lee, K.Y. Number of sialic acid residues in ganglioside headgroup affects interactions with neighboring lipids. Biophys. J. 2013, 105, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Venkateshwari, S.; Veluraja, K. Conformational analysis of GT1B ganglioside and its interaction with botulinum neurotoxin type B: A study by molecular modeling and molecular dynamics. J. Biomol. Struct. Dyn. 2012, 30, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J. How sphingolipids bind and shape proteins: Molecular basis of lipid-protein interactions in lipid shells, rafts and related biomembrane domains. Cell. Mol. Life Sci. 2003, 60, 1027–1032. [Google Scholar] [PubMed]
- Lauterbach, T.; Manna, M.; Ruhnow, M.; Wisantoso, Y.; Wang, Y.; Matysik, A.; Oglęcka, K.; Mu, Y.; Geifman-Shochat, S.; Wohland, T.; et al. Weak glycolipid binding of a microdomain-tracer peptide correlates with aggregation and slow diffusion on cell membranes. PLoS ONE 2012, 7, e51222. [Google Scholar] [CrossRef] [PubMed]
- Yahi, N.; Aulas, A.; Fantini, J. How cholesterol constrains glycolipid conformation for optimal recognition of alzheimer’s β amyloid peptide (a β (1–40)). PLoS ONE 2010, 5, e9079. [Google Scholar] [CrossRef] [PubMed]
- Delezay, O.; Hammache, D.; Fantini, J.; Yahi, N. SPC3, a V3 loop-derived synthetic peptide inhibitor of HIV-1 infection, binds to cell surface glycosphingolipids. Biochemistry 1996, 35, 15663–15671. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, R.; Garmy, N.; Maresca, M.; Yahi, N.; Puigserver, A.; Fantini, J. Identification of a common sphingolipid-binding domain in alzheimer, prion, and HIV-1 proteins. J. Biol. Chem. 2002, 277, 11292–11296. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of the shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb(3). Biochemistry 1998, 37, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. Molecular basis for the glycosphingolipid-binding specificity of α-synuclein: Key role of tyrosine 39 in membrane insertion. J. Mol. Biol. 2011, 408, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Verdier, Y.; Zarandi, M.; Penke, B. Amyloid β-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for alzheimer’s disease. J. Pept. Sci. 2004, 10, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Odaka, A.; Suzuki, N.; Ihara, Y. Gm1 ganglioside-bound amyloid β-protein (a β): A possible form of preamyloid in alzheimer’s disease. Nat. Med. 1995, 1, 1062–1066. [Google Scholar] [CrossRef]
- Kakio, A.; Nishimoto, S.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: Importance of GM1 ganglioside-bound form as an endogenous seed for alzheimer amyloid. Biochemistry 2002, 41, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Igbabvoa, U.; Shimada, Y.; Ohno-Iwashita, Y.; Kobayashi, M.; Wood, W.G.; Fujita, S.C.; Yanagisawa, K. Accelerated aβ aggregation in the presence of GM1-ganglioside-accumulated synaptosomes of aged apoe4-knock-in mouse brain. FEBS Lett. 2004, 569, 135–139. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Franklin, T.; Fraser, P.E.; Chakrabartty, A. Structural transitions associated with the interaction of alzheimer β-amyloid peptides with gangliosides. J. Biol. Chem. 1998, 273, 4506–4515. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Yang, D.; Yip, C.M.; Fraser, P.E. Review: Modulating factors in amyloid-β fibril formation. J. Struct. Biol. 2000, 130, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.M.; McLaurin, J. Amyloid-β peptide assembly: A critical step in fibrillogenesis and membrane disruption. Biophys. J. 2001, 80, 1359–1371. [Google Scholar] [CrossRef]
- Yip, C.M.; Darabie, A.A.; McLaurin, J. Aβ42-peptide assembly on lipid bilayers. J. Mol. Biol. 2002, 318, 97–107. [Google Scholar] [CrossRef]
- Kakio, A.; Nishimoto, S.I.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Cholesterol-dependent formation of gm1 ganglioside-bound amyloid β-protein, an endogenous seed for alzheimer amyloid. J. Biol. Chem. 2001, 276, 24985–24990. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Plaque busters: Strategies to inhibit amyloid formation in alzheimer’s disease. Mol. Med. Today 1999, 5, 343–350. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2010, 12, e27. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, S.; Lee, E.; Manna, M.; Steinert, S.; Kumar, G.S.; Wenk, M.; Wohland, T.; Kraut, R. A fluorescent sphingolipid binding domain peptide probe interacts with sphingolipids and cholesterol-dependent raft domains. J. Lipid Res. 2008, 49, 1077–1089. [Google Scholar] [CrossRef]
- Steinert, S.; Lee, E.; Tresset, G.; Zhang, D.; Hortsch, R.; Wetzel, R.; Hebbar, S.; Sundram, J.R.; Kesavapany, S.; Boschke, E.; et al. A fluorescent glycolipid-binding peptide probe traces cholesterol dependent microdomain-derived trafficking pathways. PLoS ONE 2008, 3, e2933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Manna, M.; Wohland, T.; Kraut, R. Alternate raft pathways cooperate to mediate slow diffusion and efficient uptake of a sphingolipid tracer to degradative and recycling compartments. J. Cell Sci. 2009, 122, 3715–3728. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, J.; Manna, M.; Guo, L.; Kraut, R.; Wohland, T. Diffusion, transport, and cell membrane organization investigated by imaging fluorescence cross-correlation spectroscopy. Biophys. J. 2009, 97, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Ariga, T.; Kobayashi, K.; Hasegawa, A.; Kiso, M.; Ishida, H.; Miyatake, T. Characterization of high-affinity binding between gangliosides and amyloid β-protein. Arch. Biochem. Biophys. 2001, 388, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Choo-Smith, L.P.; Surewicz, W.K. The interaction between alzheimer amyloid β(1–40) peptide and ganglioside GM1-containing membranes. FEBS Lett. 1997, 402, 95–98. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of alzheimer’s β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, Y.; Nishio, M. CH/π interactions in the crystal structure of class I MHC antigens and their complexes with peptides. Bioorg. Med. Chem. 1998, 6, 2507–2515. [Google Scholar] [CrossRef]
- Umezawa, Y.; Tsuboyama, S.; Takahashi, H.; Uzawa, J.; Nishio, M. CH/π interaction in the conformation of peptides. A database study. Bioorg. Med. Chem. 1999, 7, 2021–2026. [Google Scholar] [CrossRef]
- Perutz, M.F. The role of aromatic rings as hydrogen-bond acceptors in molecular recognition. Philos. Trans. R. Soc. Lond. A 1993, 345, 105–112. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Albertí, M.; Aguilar, A.; Lucas, J.M.; Pirani, F. Competitive role of CH4–CH4 and CH–π interactions in C6H6–(CH4)n aggregates: The transition from dimer to cluster features. J. Phys. Chem. A 2012, 116, 5480–5490. [Google Scholar] [CrossRef] [PubMed]
- Plevin, M.J.; Bryce, D.L.; Boisbouvier, J. Direct detection of CH/π interactions in proteins. Nat. Chem. 2010, 2, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Mahmood, M.I.; Neya, S.; Matsuzaki, K.; Hoshino, T. Formation of GM1 ganglioside clusters on the lipid membrane containing sphingomyeline and cholesterol. J. Phys. Chem. B 2012, 116, 5111–5121. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Mahmood, M.I.; Mori, K.; Matsuzaki, K. Binding and aggregation mechanism of amyloid β-peptides onto the gm1 ganglioside-containing lipid membrane. J. Phys. Chem. B 2013, 117, 8085–8094. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, R.; Mylvaganam, M.; Lingwood, C.A.; Fantini, J. A novel soluble analog of the HIV-1 fusion cofactor, globotriaosylceramide (Gb(3)), eliminates the cholesterol requirement for high affinity gp120/Gb(3) interaction. J. Lipid Res. 2002, 43, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Iijima, K.; Yamamoto, N.; Yanagisawa, K.; Sato, T. Density of GM1 in nanoclusters is a critical factor in the formation of a spherical assembly of amyloid β-protein on synaptic plasma membranes. Langmuir 2013, 29, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A.; Paila, Y.D.; Shrivastava, S.; Tiwari, S.; Singh, P.; Fantini, J. Sphingolipid-binding domain in the serotonin(1A) receptor. Adv. Exp. Med. Biol. 2012, 749, 279–293. [Google Scholar] [PubMed]
- Fantini, J.; Garmy, N.; Yahi, N. Prediction of glycolipid-binding domains from the amino acid sequence of lipid raft-associated proteins: Application to HpaA, a protein involved in the adhesion of helicobacter pylori to gastrointestinal cells. Biochemistry 2006, 45, 10957–10962. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P.; Suzuki, Y.; Bourne, N.T.; Asakura, T. Binding of amyloid β-peptide to ganglioside micelles is dependent on histidine-13. Biochem. J. 2006, 397, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The REd. Tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, W.; Bogdanov, M.; Mileykovskaya, E. Functional Roles of Lipids in Membranes, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. Gromacs 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; Gonzalez-Outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. Glycam06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Zhou, R.H.; Harder, E.; Xu, H.F.; Berne, B.J. Efficient multiple time step method for use with ewald and particle mesh ewald for large biomolecular systems. J. Chem. Phys. 2001, 115, 2348–2358. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-Ralgds complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Gohlke, H.; Kuhn, L.A.; Case, D.A. Change in protein flexibility upon complex formation: Analysis of Ras-Raf using molecular dynamics and a molecular framework approach. Proteins 2004, 56, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Garmy, N.; Mahfoud, R.; Yahi, N. Lipid rafts: Structure, function and role in HIV, Alzheimers and prion diseases. Expert Rev. Mol. Med. 2002, 2002, 1–22. [Google Scholar] [CrossRef]
- Hall, A.; Rog, T.; Vattulainen, I. Effect of galactosylceramide on the dynamics of cholesterol-rich lipid membranes. J. Phys. Chem. B 2011, 115, 14424–14434. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Kraut, R.; Mu, Y. Aβ1-25-Derived Sphingolipid-Domain Tracer Peptide SBD Interacts with Membrane Ganglioside Clusters via a Coil-Helix-Coil Motif. Int. J. Mol. Sci. 2015, 16, 26318-26332. https://doi.org/10.3390/ijms161125955
Wang Y, Kraut R, Mu Y. Aβ1-25-Derived Sphingolipid-Domain Tracer Peptide SBD Interacts with Membrane Ganglioside Clusters via a Coil-Helix-Coil Motif. International Journal of Molecular Sciences. 2015; 16(11):26318-26332. https://doi.org/10.3390/ijms161125955
Chicago/Turabian StyleWang, Yaofeng, Rachel Kraut, and Yuguang Mu. 2015. "Aβ1-25-Derived Sphingolipid-Domain Tracer Peptide SBD Interacts with Membrane Ganglioside Clusters via a Coil-Helix-Coil Motif" International Journal of Molecular Sciences 16, no. 11: 26318-26332. https://doi.org/10.3390/ijms161125955
APA StyleWang, Y., Kraut, R., & Mu, Y. (2015). Aβ1-25-Derived Sphingolipid-Domain Tracer Peptide SBD Interacts with Membrane Ganglioside Clusters via a Coil-Helix-Coil Motif. International Journal of Molecular Sciences, 16(11), 26318-26332. https://doi.org/10.3390/ijms161125955