
Chlamydia pneumoniae and Oxidative Stress in Cardiovascular Disease: State of the Art and Prevention Strategies




Abstract
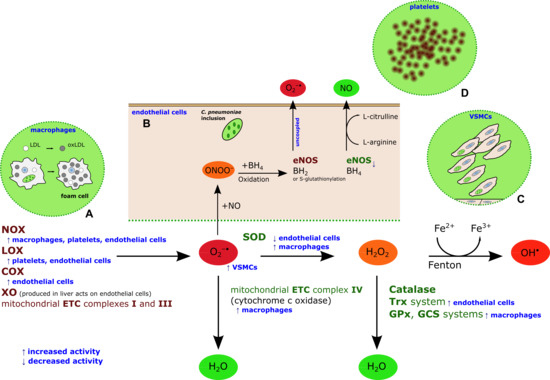
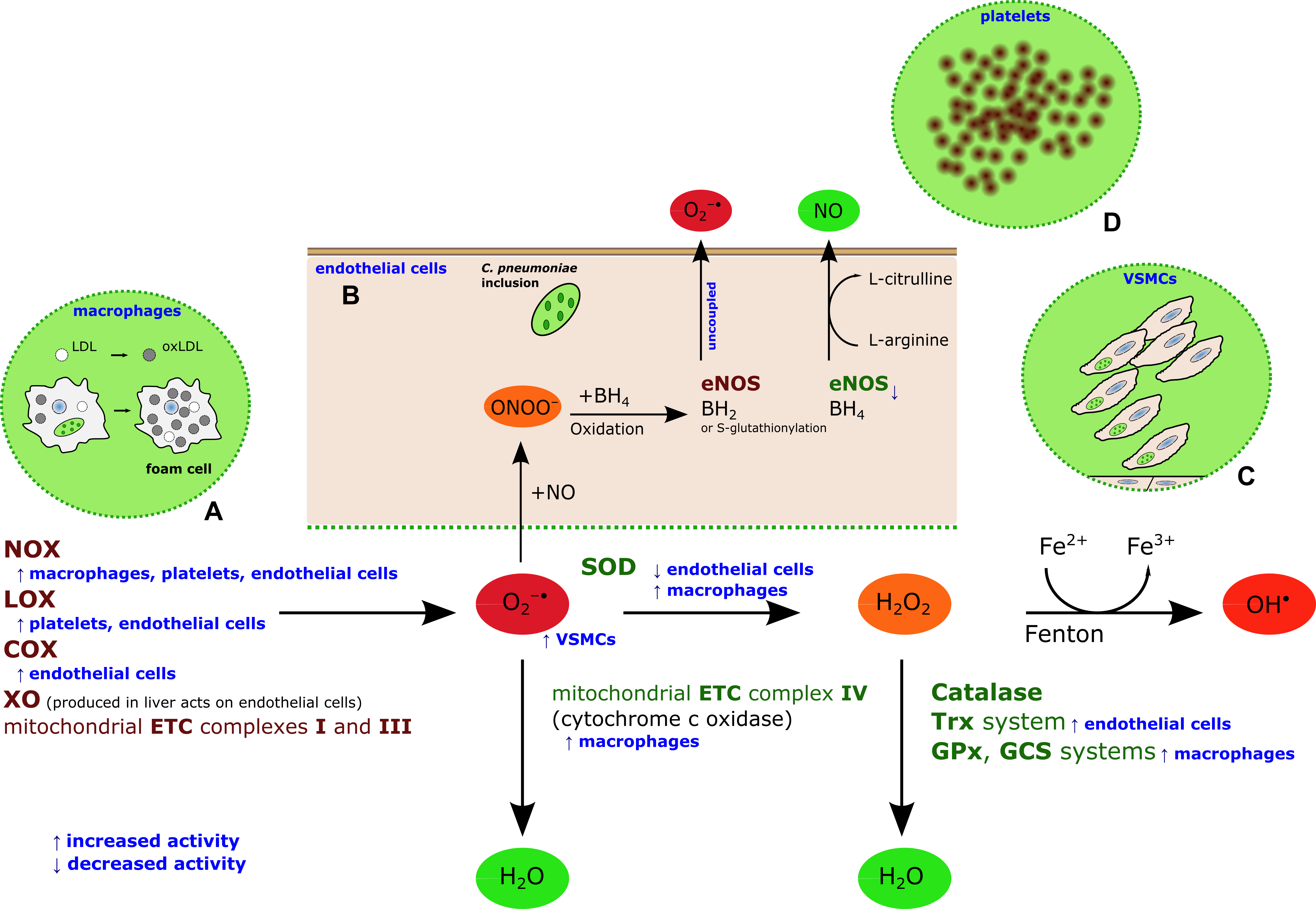
:
{kind=link}
{kind=link}
1. Introduction
2. Chlamydia pneumoniae-Induced Oxidative Stress
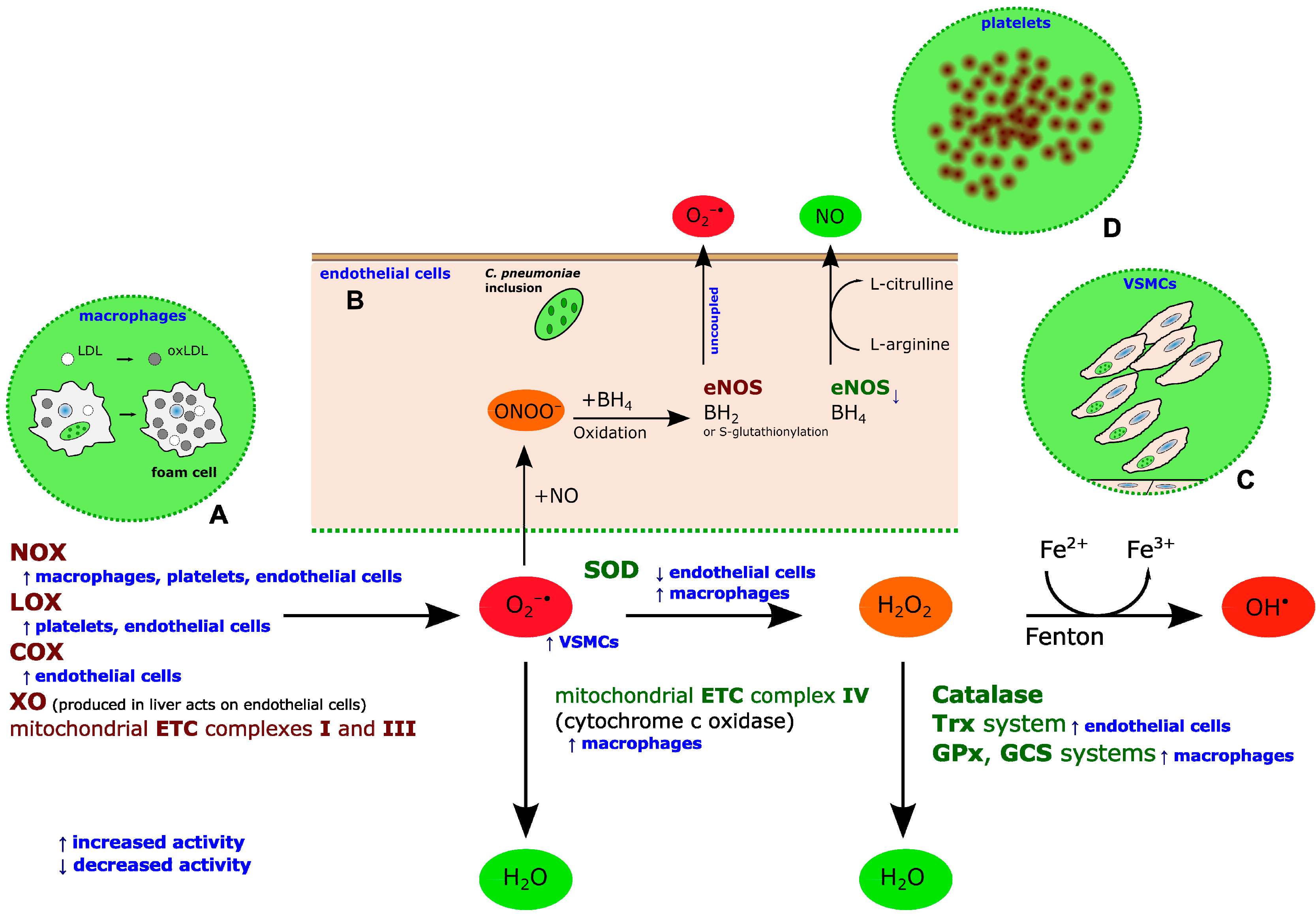
3. C. Pneumoniae-Induced Oxidative Stress as a Target for CVD Prevention
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mendis, S.; Puska, P.; Norrving, B.; World Health Organization; World Heart Federation; World Stroke Organization. Global Atlas on Cardiovascular Disease Prevention and Control. Mendis, S., Puska, P., Norrving, B., Eds.; Available online: http://whqlibdoc.who.int/publications/2011/9789241564373_eng.pdf (accessed on 2011).
- Laboz, C.; Mostaza, J.M. Atherosclerosis as a systemic disease. Rev. Esp. Cardiol. 2007, 60, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; di Pietro, M.; Filardo, S.; Turriziani, O. Infectious burden and atherosclerosis: A clinical issue. World J. Clin. Cases 2014, 2, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Hogan, R.J.; Mathews, S.A.; Mukhopadhyay, S.; Summersgill, J.T.; Timms, P. Chlamydial persistence: Beyond the biphasic paradigm. Infect. Immun. 2004, 72, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Schoborg, R.V. Chlamydia persistence—A tool to dissect chlamydia—Host interactions. Microbes Infect. 2011, 13, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Tramonti, A.; de Santis, F.; de Biase, D.; Schiavoni, G.; Filardo, S.; Zagaglia, C.; Sessa, R. Analysis of gene expression in penicillin G induced persistence of Chlamydia pneumoniae. J. Biol. Regul. Homeost. Agents 2012, 26, 277–284. [Google Scholar] [PubMed]
- Saikku, P.; Leinonen, M.; Mattila, K.; Ekman, M.R.; Nieminen, M.S.; Mäkelä, P.H.; Huttunen, J.K.; Valtonen, V. Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 1988, 2, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Khandelwal, B.; Joshi, D.; Gupta, O.P. Chlamydophila pneumoniae infection and cardiovascular disease. N. Am. J. Med. Sci. 2013, 5, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Sakurai-Komada, N.; Iso, H.; Koike, K.A.; Ikeda, A.; Umesawa, M.; Ikehara, S.; Inoue, M.; Tsugane, S. Association between Chlamydophila pneumoniae infection and risk of coronary heart disease for Japanese: The JPHC study. Atherosclerosis 2014, 233, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; di Pietro, M.; Schiavoni, G.; Petrucca, A.; Cipriani, P.; Zagaglia, C.; Nicoletti, M.; Santino, I.; del Piano, M. Measurement of Chlamydia pneumoniae bacterial load in peripheral blood mononuclear cells may be helpful to assess the state of chlamydial infection in patients with carotid atherosclerotic disease. Atherosclerosis 2007, 195, e224–e230. [Google Scholar] [CrossRef] [PubMed]
- Atik, B.; Johnston, S.C.; Dean, D. Association of carotid plaque Lp-PLA(2) with macrophages and Chlamydia pneumoniae infection among patients at risk for stroke. PLoS One 2010, 5, e11026. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Turu, M.M.; Rovira, N.; Juan-Babot, J.O.; Slevin, M.; Krupinski, J. Early atherosclerotic plaques show evidence of infection by Chlamydia pneumoniae. Front. Biosci. 2012, 4, 2423–2432. [Google Scholar] [CrossRef]
- Moazed, T.C.; Kuo, C.C.; Grayston, J.T.; Campbell, L.A. Evidence of systemic dissemination of Chlamydia pneumoniae via macrophages in the mouse. J. Infect. Dis. 1998, 177, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; di Pietro, M.; Schiavoni, G.; Santino, I.; Benedetti-Valentini, F.; Perna, R.; Romano, S.; del Piano, M. Chlamydia pneumoniae DNA in patients with symptomatic carotid atherosclerotic disease. J. Vasc. Surg. 2003, 37, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Filardo, S.; Cazzavillan, S.; Segala, C.; Bevilacqua, P.; Bonoldi, E.; D’Amore, E.S.; Rassu, M.; Sessa, R. Could past Chlamydial vascular infection promote the dissemination of Chlamydia pneumoniae to the brain? J. Biol. Regul. Homeost. Agents 2013, 27, 155–164. [Google Scholar] [PubMed]
- Di Pietro, M.; Schiavoni, G.; Sessa, V.; Pallotta, F.; Costanzo, G.; Sessa, R. Chlamydia pneumoniae and osteoporosis-associated bone loss: A new risk factor? Osteoporos. Int. 2013, 24, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.A. Isolation of Chlamydia pneumoniae from the coronary artery of a patient with coronary atherosclerosis. The Chlamydia pneumoniae/Atherosclerosis Study Group. Ann. Intern. Med. 1996, 125, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Maass, M.; Bartels, C.; Engel, P.M.; Mamat, U.; Sievers, H.H. Endovascular presence of viable Chlamydia pneumoniae is a common phenomenon in coronary artery disease. J. Am. Coll. Cardiol. 1998, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Apfalter, P.; Loidl, M.; Nadrchal, R.; Makristathis, A.; Rotter, M.; Bergmann, M.; Polterauer, P.; Hirschl, A.M. Isolation and continuous growth of Chlamydia pneumoniae from arterectomy specimens. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Blessing, E.; Campbell, L.A.; Rosenfeld, M.E.; Chough, N.; Kuo, C.C. Chlamydia pneumoniae infection accelerates hyperlipidemia induced atherosclerotic lesion development in C57BL/6J mice. Atherosclerosis 2001, 158, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Nicoletti, M.; di Pietro, M.; Schiavoni, G.; Santino, I.; Zagaglia, C.; del Piano, M.; Cipriani, P. Chlamydia pneumoniae and atherosclerosis: Current state and future prospectives. Int. J. Immunopathol. Pharmacol. 2009, 22, 9–14. [Google Scholar] [PubMed]
- Chen, S.; Shimada, K.; Zhang, W.; Huang, G.; Crother, T.R.; Arditi, M. IL-17A is proatherogenic in high-fat diet-induced and Chlamydia pneumoniae infection-accelerated atherosclerosis in mice. J. Immunol. 2010, 185, 5619–5627. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Shih, C.M.; Tsao, N.W.; Chen, Y.H.; Li, C.Y.; Chang, Y.J.; Chang, N.C.; Ou, K.L.; Lin, C.Y.; Lin, Y.W.; et al. GroEL1, from Chlamydia pneumoniae, induces vascular adhesion molecule 1 expression by p37(AUF1) in endothelial cells and hypercholesterolemic rabbit. PLoS One 2012, 7, e42808. [Google Scholar] [CrossRef] [PubMed]
- Gaydos, C.A. Growth in vascular cells and cytokine production by Chlamydia pneumoniae. J. Infect. Dis. 2000, 181, S473–S478. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Kullberg, B.J.; Galama, J.M.; Stalenhoef, A.F.; Dinarello, C.A.; van der Meer, J.W. Non-LPS components of Chlamydia pneumoniae stimulate cytokine production through Toll-like receptor 2-dependent pathways. Eur. J. Immunol. 2002, 32, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Coriolan, D.; Schultz, K.; Golenbock, D.T.; Beasley, D. Toll-like receptor 2 mediates persistent chemokine release by Chlamydia pneumoniae-infected vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2308–2314. [Google Scholar] [CrossRef] [PubMed]
- Högdahl, M.; Söderlund, G.; Kihlström, E. Expression of chemokines and adhesion molecules in human coronary artery endothelial cells infected with Chlamydia (Chlamydophila) pneumoniae. APMIS 2008, 116, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Al-Bannawi, A.; Al-Wesebai, K.; Taha, S.; Bakhiet, M. Chlamydia pneumoniae induces chemokine expression by platelets in patients with atherosclerosis. Med. Princ. Pract. 2011, 20, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Al-Kuhlani, M.; Johnston, S.C.; Ojcius, D.M.; Chou, J.; Dean, D. Transcription factor complex AP-1 mediates inflammation initiated by Chlamydia pneumoniae infection. Cell Microbiol. 2013, 15, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Roivainen, M.; Viik-Kajander, M.; Palosuo, T.; Toivanen, P.; Leinonen, M.; Saikku, P.; Tenkanen, L.; Manninen, V.; Hovi, T.; Mänttäri, M. Infections, inflammation, and the risk of coronary heart disease. Circulation 2000, 101, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; di Pietro, M.; Ronco, C.; de Cal, M.; Cazzavillan, S.; Rassu, M.; Nicoletti, M.; del Piano, M.; Sessa, R. Chlamydia pneumoniae infection as a risk factor for accelerated atherosclerosis in hemodialysis patients. J. Biol. Regul. Homeost. Agents 2010, 24, 367–375. [Google Scholar] [PubMed]
- Swierszcz, J.; Jacek, D.S.; Milewicz, T.; Krzysiek, J.; Sztefko, K.; Galicka-Latała, D. One-year observation of inflammatory markers in patients with aortic valve stenosis who expressed high or low Chlamydia pneumoniae antibody titers. J. Heart Valve Dis. 2012, 21, 599–607. [Google Scholar] [PubMed]
- Di Pietro, M.; Filardo, S.; de Santis, F.; Sessa, R. Chlamydia pneumoniae infection in atherosclerotic lesion development through oxidative stress: A brief overview. Int. J. Mol. Sci. 2013, 14, 15105–15120. [Google Scholar] [CrossRef] [PubMed]
- Yaraei, K.; Campbell, L.A.; Zhu, X.; Liles, W.C.; Kuo, C.C.; Rosenfeld, M.E. Chlamydia pneumoniae augments the oxidized low-density lipoprotein-induced death of mouse macrophages by a caspase-independent pathway. Infect. Immun. 2005, 73, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Nazzal, D.; Cantero, A.V.; Therville, N.; Segui, B.; Negre-Salvayre, A.; Thomsen, M.; Benoist, H. Chlamydia pneumoniae alters mildly oxidized low-density lipoprotein-induced cell death in human endothelial cells, leading to necrosis rather than apoptosis. J. Infect. Dis. 2006, 193, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. New insights on oxidative stress in the arterial wall. J. Thromb. Haemost. 2005, 3, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kälvegren, H.; Bylin, H.; Leanderson, P.; Richter, A.; Grenegård, M.; Bengtsson, T. Chlamydia pneumoniae induces nitric oxide synthase and lipoxygenase-dependent production of reactive oxygen species in platelets. Effects on oxidation of low density lipoproteins. Thromb. Haemost. 2005, 94, 327–335. [Google Scholar] [PubMed]
- Azenabor, A.A.; Yang, S.; Job, G.; Adedokun, O.O. Elicitation of reactive oxygen species in Chlamydia pneumoniae-stimulated macrophages: A Ca2+-dependent process involving simultaneous activation of NADPH oxidase and cytochrome oxidase genes. Med. Microbiol. Immunol. 2005, 194, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Azenabor, A.A.; Muili, K.; Akoachere, J.F.; Chaudhry, A. Macrophage antioxidant enzymes regulate Chlamydia pneumoniae chronicity: Evidence of the effect of redox balance on host-pathogen relationship. Immunobiology 2006, 211, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Kreutmayer, S.; Csordas, A.; Kern, J.; Maass, V.; Almanzar, G.; Offterdinger, M.; Öllinger, R.; Maass, M.; Wick, G. Chlamydia pneumoniae infection acts as an endothelial stressor with the potential to initiate the earliest heat shock protein 60-dependent inflammatory stage of atherosclerosis. Cell Stress Chaperones 2013, 18, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chai, H.; Wang, X.; Lin, P.H.; Yao, Q. Chlamydia heat shock protein 60 decreases expression of endothelial nitric oxide synthase in human and porcine coronary artery endothelial cells. Cardiovasc. Res. 2009, 83, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.E.; Wolf, K. C. pneumoniae disrupts eNOS trafficking and impairs NO production in human aortic endothelial cells. Cell Microbiol. 2014. [Google Scholar] [CrossRef]
- Lin, Y.W.; Huang, C.Y.; Chen, Y.H.; Shih, C.M.; Tsao, N.W.; Lin, C.Y.; Chang, N.C.; Tsai, C.S.; Tsai, H.Y.; Tsai, J.C.; et al. GroEL1, a heat shock protein 60 of Chlamydia pneumoniae, impairs neovascularization by decreasing endothelial progenitor cell function. PLoS One 2013, 8, e84731. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.Y.; Lin, Y.W.; Huang, C.Y.; Chang, Y.J.; Tsao, N.W.; Chang, N.C.; Ou, K.L.; Chen, T.L.; Shih, C.M.; Chen, Y.H. GroEL1, a heat shock protein 60 of Chlamydia pneumoniae, induces lectin-like oxidized low-density lipoprotein receptor 1 expression in endothelial cells and enhances atherogenesis in hypercholesterolemic rabbits. J. Immunol. 2011, 186, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Walduck, A.K.; Strugnell, R.A.; Sobey, C.G.; Drummond, G.R. Chlamydia pneumoniae induces a pro-inflammatory phenotype in murine vascular smooth muscle cells independently of elevating reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2012, 39, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Deniset, J.F.; Pierce, G.N. Possibilities for therapeutic interventions in disrupting Chlamydophila pneumoniae involvement in atherosclerosis. Fundam. Clin. Pharmacol. 2010, 24, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.A.; Rosenfeld, M.E. Persistent, C. pneumoniae infection in atherosclerotic lesions: Rethinking the clinical trials. Front. Cell. Infect. Microbiol. 2014, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Deby-Dupont, G.; Mouithys-Mickalad, A.; Serteyn, D.; Lamy, M.; Deby, C. Resveratrol and curcumin reduce the respiratory burst of Chlamydia-primed THP-1 cells. Biochem. Biophys. Res. Commun. 2005, 333, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; de Santis, F.; Schiavoni, G.; Filardo, S.; Sessa, R. Resveratrol in Chlamydia pneumoniae induced foam cell formation and interleukin-17A sythesis. J. Biol. Regul. Homeost. Agents 2013, 27, 509–518. [Google Scholar] [PubMed]
- Mouithys-Mickalad, A.; Deby-Dupont, G.; Dogne, J.M.; de Leval, X.; Kohnen, S.; Navet, R.; Sluse, F.; Hoebeke, M.; Pirotte, B.; Lamy, M. Effects of COX-2 inhibitors on ROS produced by Chlamydia pneumoniae-primed human promonocytic cells (THP-1). Biochem. Biophys. Res. Commun. 2004, 325, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Margaritis, M.; Shirodaria, C.; Antoniades, C. Translating the effects of statins: from redox regulation to suppression of vascular wall inflammation. Thromb. Haemost. 2012, 108, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Kothe, H.; Dalhoff, K.; Rupp, J.; Müller, A.; Kreuzer, J.; Maass, M.; Katus, H.A. Hydroxymethylglutaryl coenzyme A reductase inhibitors modify the inflammatory response of human macrophages and endothelial cells infected with Chlamydia pneumoniae. Circulation 2000, 101, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Dechend, R.; Gieffers, J.; Dietz, R.; Joerres, A.; Rupp, J.; Luft, F.C.; Maass, M. Hydroxymethylglutaryl coenzyme A reductase inhibition reduces Chlamydia pneumoniae-induced cell interaction and activation. Circulation 2003, 108, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Prochnau, D.; Rödel, J.; Prager, K.; Kuersten, D.; Heller, R.; Straube, E.; Figulla, H.R. Induced expression of lectin-like oxidized LDL receptor-1 in vascular smooth muscle cells following Chlamydia pneumoniae infection and its down-regulation by fluvastatin. Acta Microbiol. Immunol. Hung. 2010, 57, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Kang, S.W. Targeting cellular antioxidant enzymes for treating atherosclerotic vascular disease. Biomol. Ther. 2013, 21, 89–96. [Google Scholar] [CrossRef]
- Fukuoka, K.; Sawabe, A.; Sugimoto, T.; Koga, M.; Okuda, H.; Kitayama, T.; Shirai, M.; Komai, K.; Komemushi, S.; Matsuda, K. Inhibitory actions of several natural products on proliferation of rat vascular smooth muscle cells induced by Hsp60 from Chlamydia pneumoniae J138. J. Agric. Food Chem. 2004, 52, 6326–6329. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant supplements and mortality. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 40–44. [Google Scholar] [PubMed]
- Berger, R.G.; Lunkenbein, S.; Ströhle, A.; Hahn, A. Antioxidants in food: mere myth or magic medicine? Crit. Rev. Food Sci. Nutr. 2012, 52, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Brackman, G.; Cos, P.; Maes, L.; Nelis, H.J.; Coenye, T. Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob. Agents Chemother. 2011, 55, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.C.; Linington, R.G.; Auerbuch, V. Chemical inhibitors of the type three secretion system: disarming bacterial pathogens. Antimicrob. Agents Chemother. 2012, 56, 5433–5441. [Google Scholar] [CrossRef]
- Beeckman, D.S.; de Puysseleyr, L.; de Puysseleyr, K.; Vanrompay, D. Chlamydial biology and its associated virulence blockers. Crit. Rev. Microbiol 2014, 40, 313–328. [Google Scholar]
- Byrne, G.I.; Kalayoglu, M.V. Chlamydia pneumoniae and atherosclerosis: links to the disease process. Am. Heart J. 1999, 138, S488–S490. [Google Scholar] [CrossRef] [PubMed]
- Sasu, S.; LaVerda, D.; Qureshi, N.; Golenbock, D.T.; Beasley, D. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ. Res. 2001, 89, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, C.U.; Wantia, N.; Kirschning, C.J.; Busch, D.H.; Rodriguez, N.; Wagner, H.; Miethke, T. Heat shock protein 60 from Chlamydia pneumoniae elicits an unusual set of inflammatory responses via Toll-like receptor 2 and 4 in vivo. Eur. J. Immunol. 2004, 34, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Srivastava, P.; Prasad, J.; Mittal, A. Chlamydia pneumoniae heat shock protein 60 enhances expression of ERK, TLR-4 and IL-8 in atheromatous plaques of coronary artery disease patients. Immunol. Investig. 2011, 40, 206–222. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, M.; Filardo, S.; De Santis, F.; Mastromarino, P.; Sessa, R. Chlamydia pneumoniae and Oxidative Stress in Cardiovascular Disease: State of the Art and Prevention Strategies. Int. J. Mol. Sci. 2015, 16, 724-735. https://doi.org/10.3390/ijms16010724
Di Pietro M, Filardo S, De Santis F, Mastromarino P, Sessa R. Chlamydia pneumoniae and Oxidative Stress in Cardiovascular Disease: State of the Art and Prevention Strategies. International Journal of Molecular Sciences. 2015; 16(1):724-735. https://doi.org/10.3390/ijms16010724
Chicago/Turabian StyleDi Pietro, Marisa, Simone Filardo, Fiorenzo De Santis, Paola Mastromarino, and Rosa Sessa. 2015. "Chlamydia pneumoniae and Oxidative Stress in Cardiovascular Disease: State of the Art and Prevention Strategies" International Journal of Molecular Sciences 16, no. 1: 724-735. https://doi.org/10.3390/ijms16010724
APA StyleDi Pietro, M., Filardo, S., De Santis, F., Mastromarino, P., & Sessa, R. (2015). Chlamydia pneumoniae and Oxidative Stress in Cardiovascular Disease: State of the Art and Prevention Strategies. International Journal of Molecular Sciences, 16(1), 724-735. https://doi.org/10.3390/ijms16010724