
The Effects of Endogenous Non-Peptide Molecule Isatin and Hydrogen Peroxide on Proteomic Profiling of Rat Brain Amyloid-β Binding Proteins: Relevance to Alzheimer’s Disease?

 ,
,
Abstract
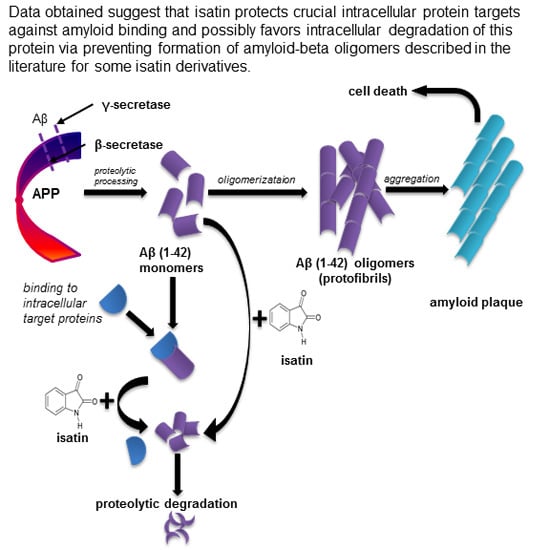
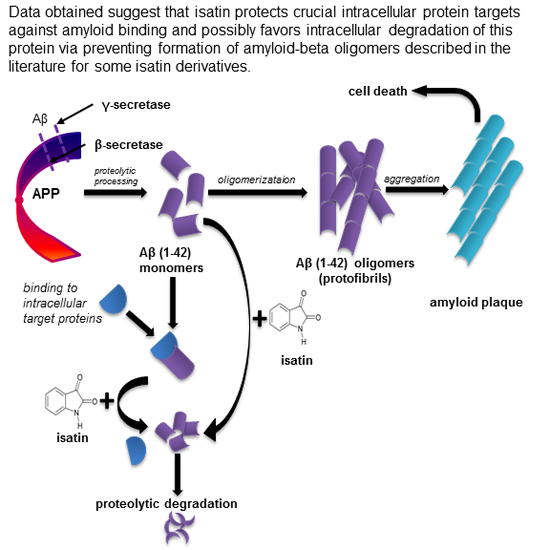
:
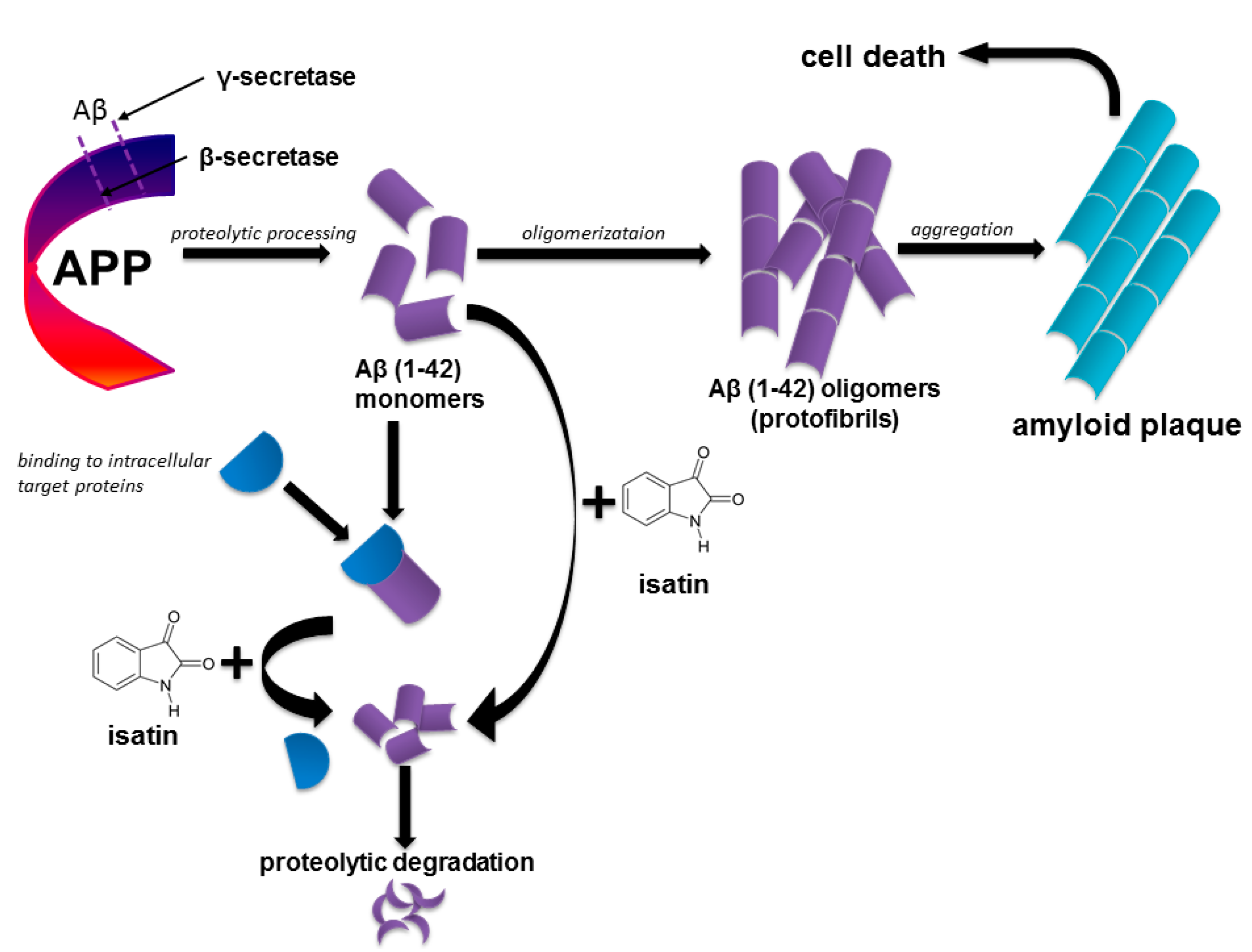
1. Introduction
2. Results
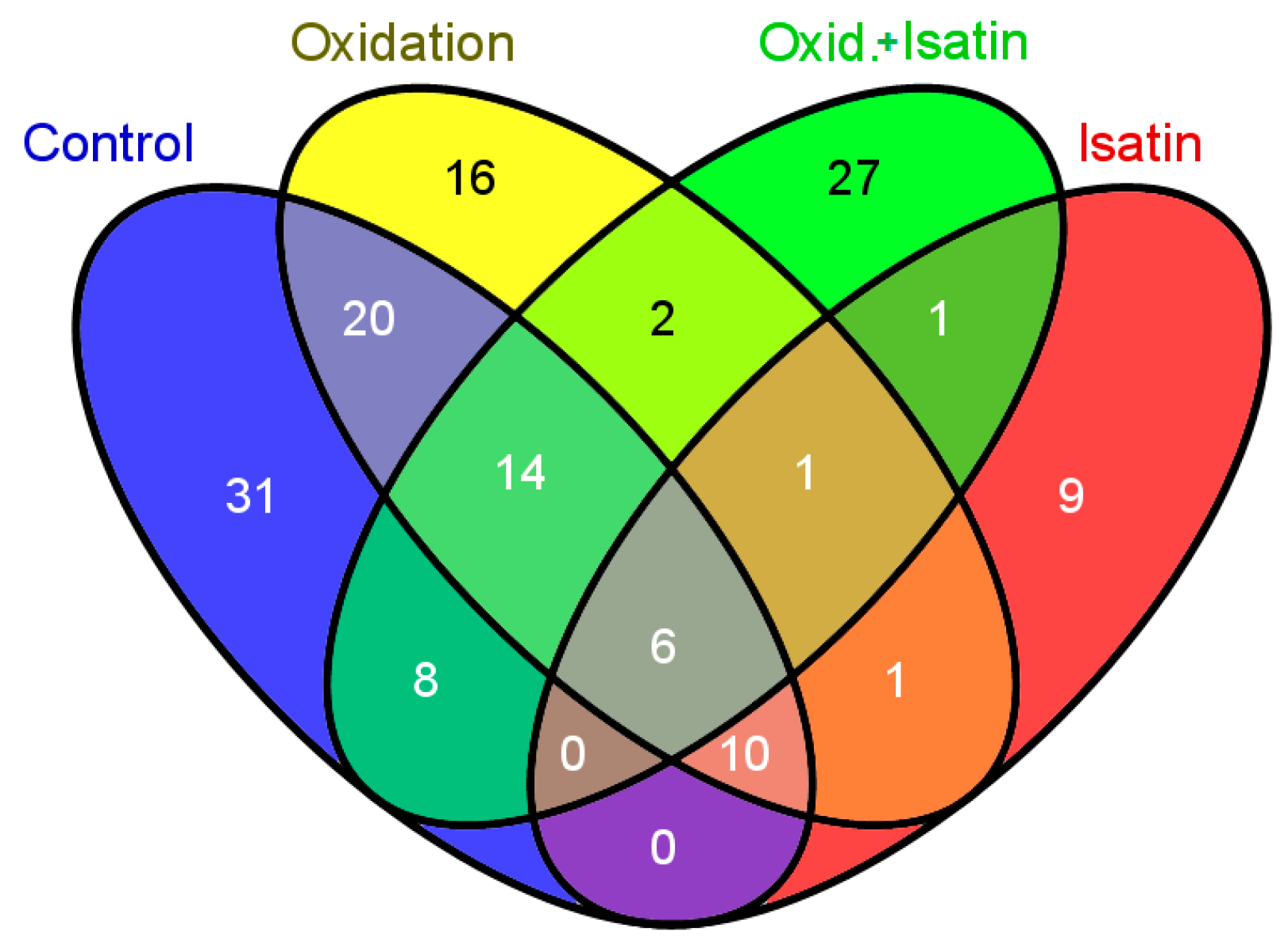
2.1. Proteomic Profiling of Amyloid-Binding Proteins of Intact Rat Brain Homogenate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Protein Name | Changes in Pathology or Experimental Model | Reference |
|---|---|---|---|
| 1 | Pyruvate kinase isozymes M1/M2 | OM, I | [11,33,35] |
| 2 | Actin, cytoplasmic 1 | OM, D | [36,37] |
| 3 | Heat shock cognate 71 kDa protein | OM | [38] |
| 4 | ATP synthase subunit β, mitochondrial | I | [39] |
| 5 | Alpha-enolase | OM, I, AALS | [33,35,40,41] |
| 6 | Myelin basic protein S | OM | [11] |
| 7 | Glutathione S-transferase α-2 | OM | [11] |
| 8 | Dihydropyrimidinase-related protein 2 | OM, I, D, AALS | [11,40,41,42] |
| 9 | 60 kDa Heat shock protein, mitochondrial | D | [39] |
| 10 | Glyceraldehyde-3-phosphate dehydrogenase | OM | [11,33] |
| 11 | Fructose-bisphosphate aldolase A | OM | [33] |
| 12 | ATP synthase subunit α, mitochondrial | D | [39] |
| 13 | Endoplasmin | OM | [33] |
| 14 | Peroxiredoxin-1 | I | [43] |
| 15 | Stress-70 protein, mitochondrial | I | [44,45,46] |
| 16 | Retinol-binding protein 4 OS | D | [47] |
| 17 | 78 kDa Glucose-regulated protein | OM, AALS | [33,41] |
| 18 | Peroxiredoxin-2 | OM, I | [33,43,48] |
| 19 | Glutamine synthetase | OM, I | [11,40] |
| 20 | Glutathione S-transferase P OS | OM | [11] |
| 21 | Glutamate dehydrogenase 1, mitochondrial | OM, AALS | [33,41] |
| 22 | Heat shock protein HSP 90-α | I | [49] |
| 23 | Spectrin α chain, non-erythrocytic 1 | OM | [11] |
| 24 | Phosphoglucomutase-1 | OM | [32] |
| 25 | Synaptotagmin-1 | D | [44,50] |
| 26 | Phosphatidylethanolamine-binding protein 1 | I | [51] |
| 27 | Calmodulin | D, AALS | [41,52] |
| 28 | Protein disulfide-isomerase | AALS | [41] |
| 29 | Superoxide dismutase [Mn] | OM | [53] |
| 30 | Tubulin α-1A chain | OM | [32] |
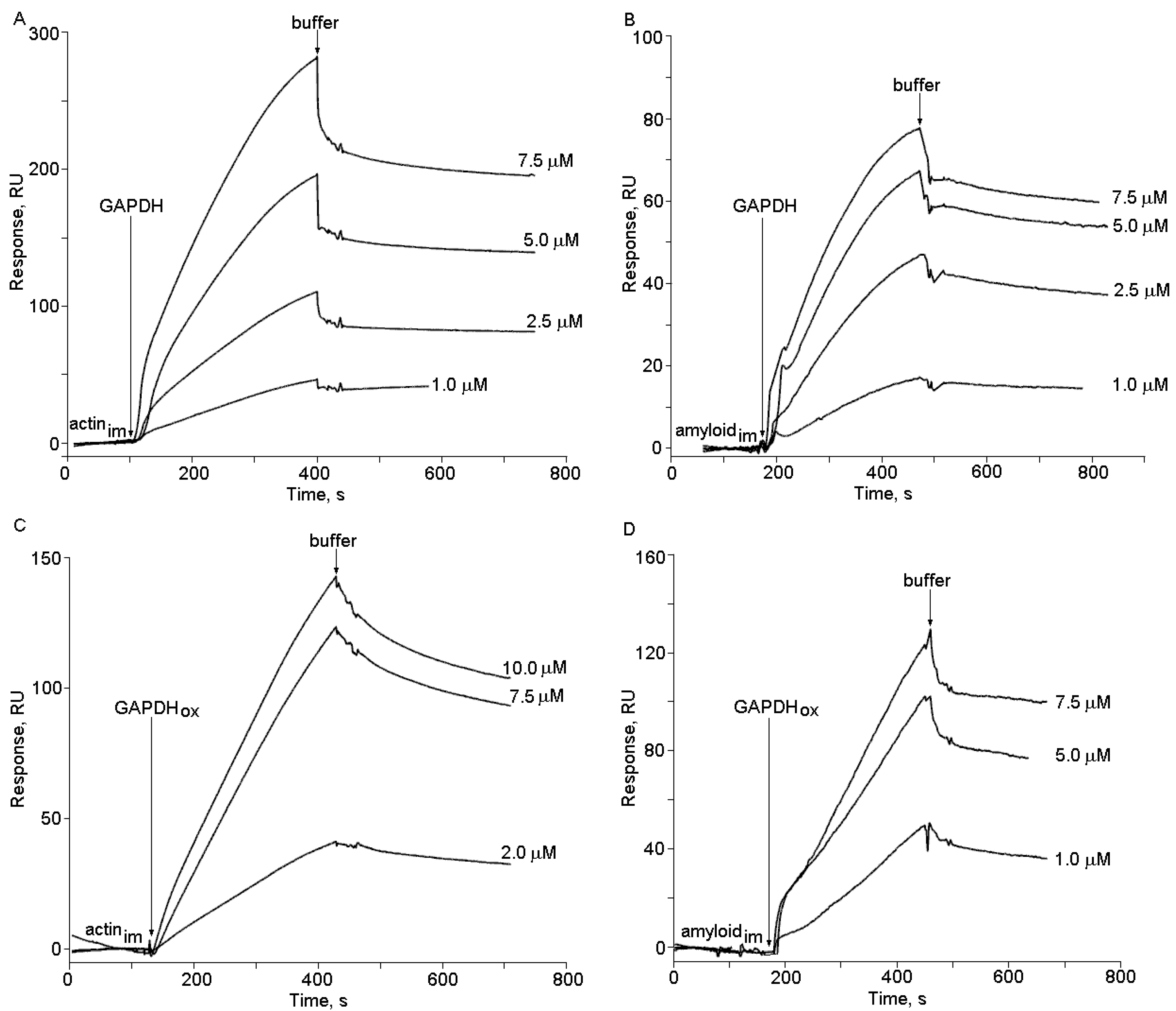
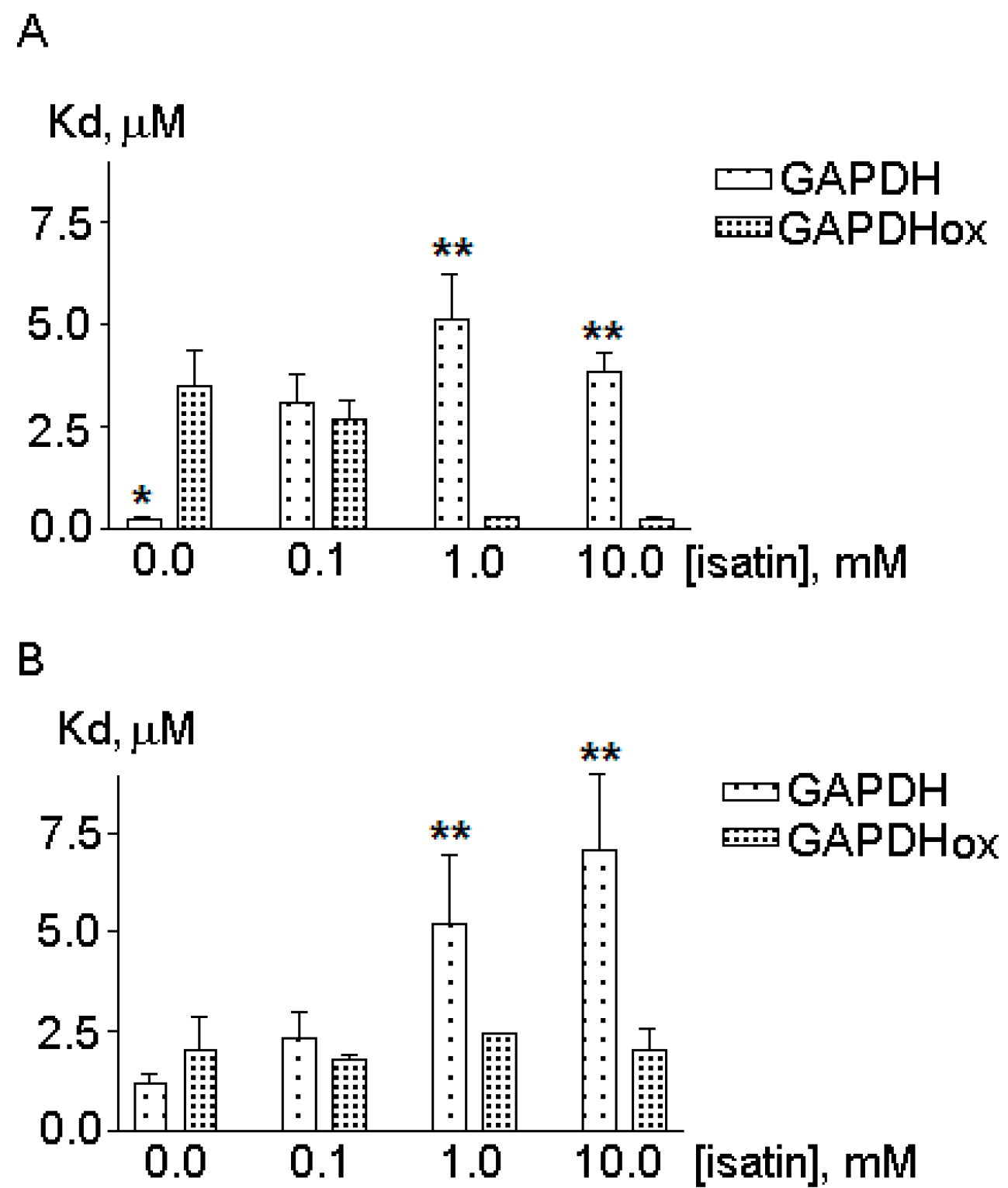
2.2. The Effects of Hydrogen Peroxide and Isatin on the Interaction of Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) with Immobilized Amyloid-β and Actin
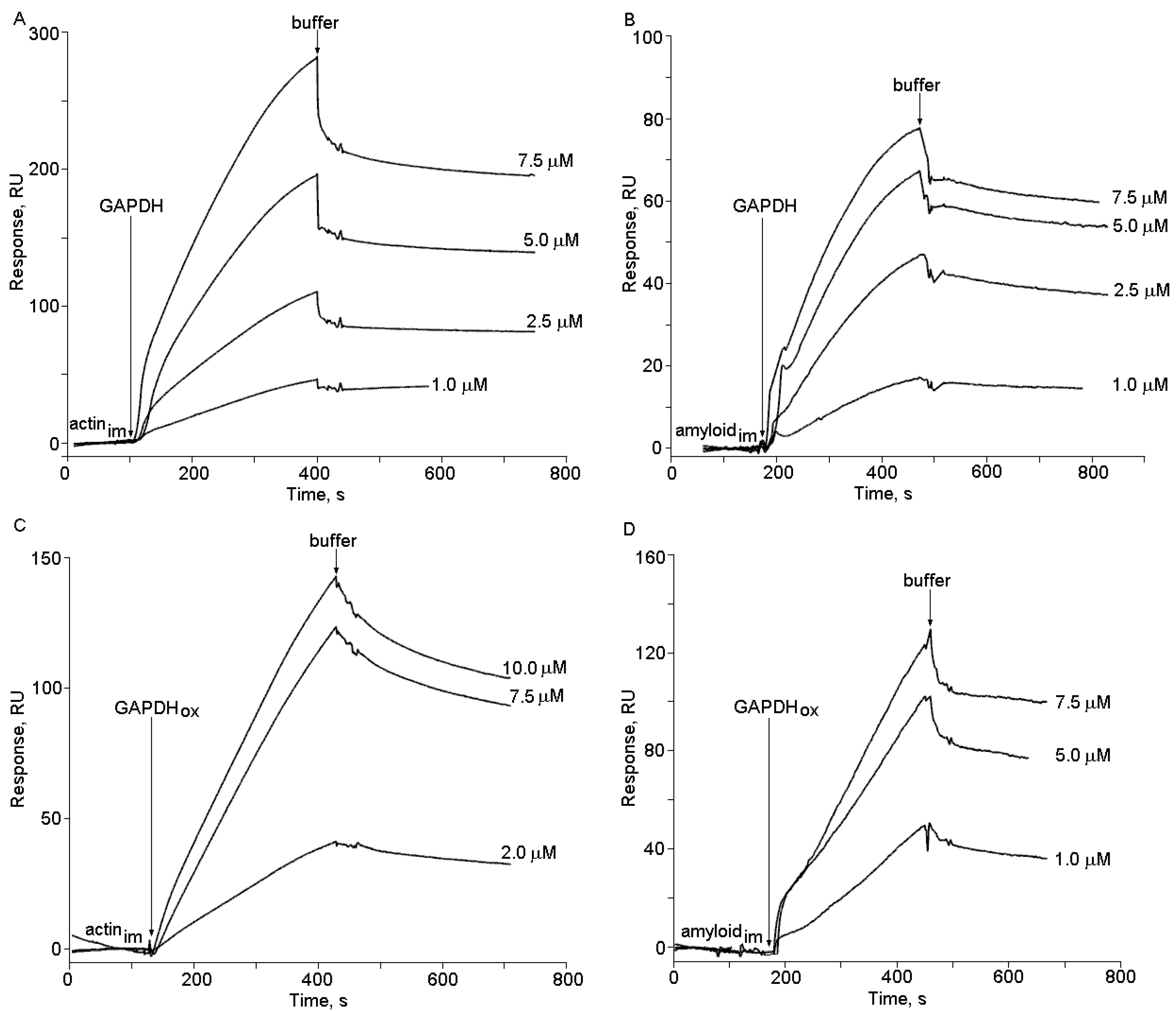
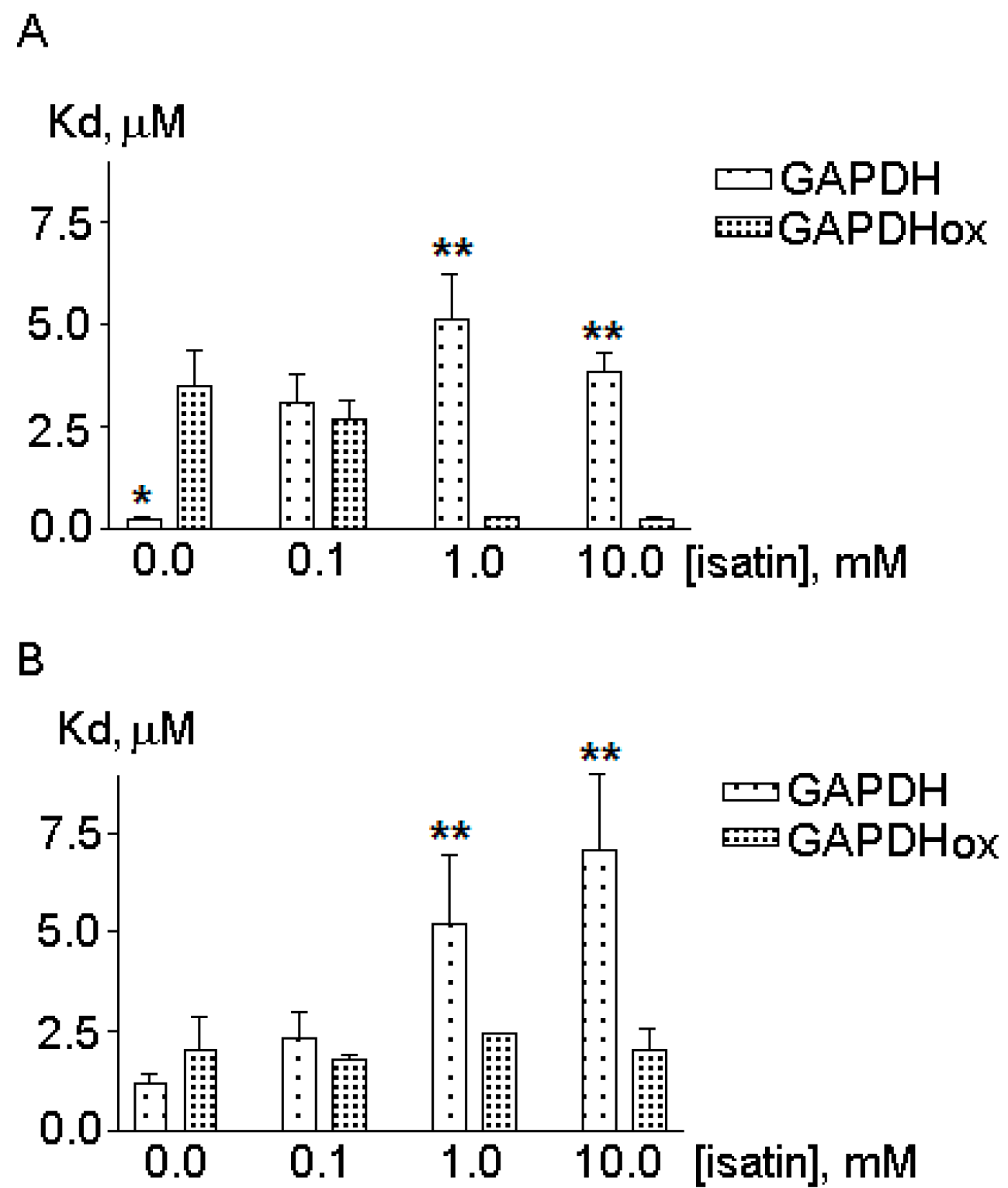
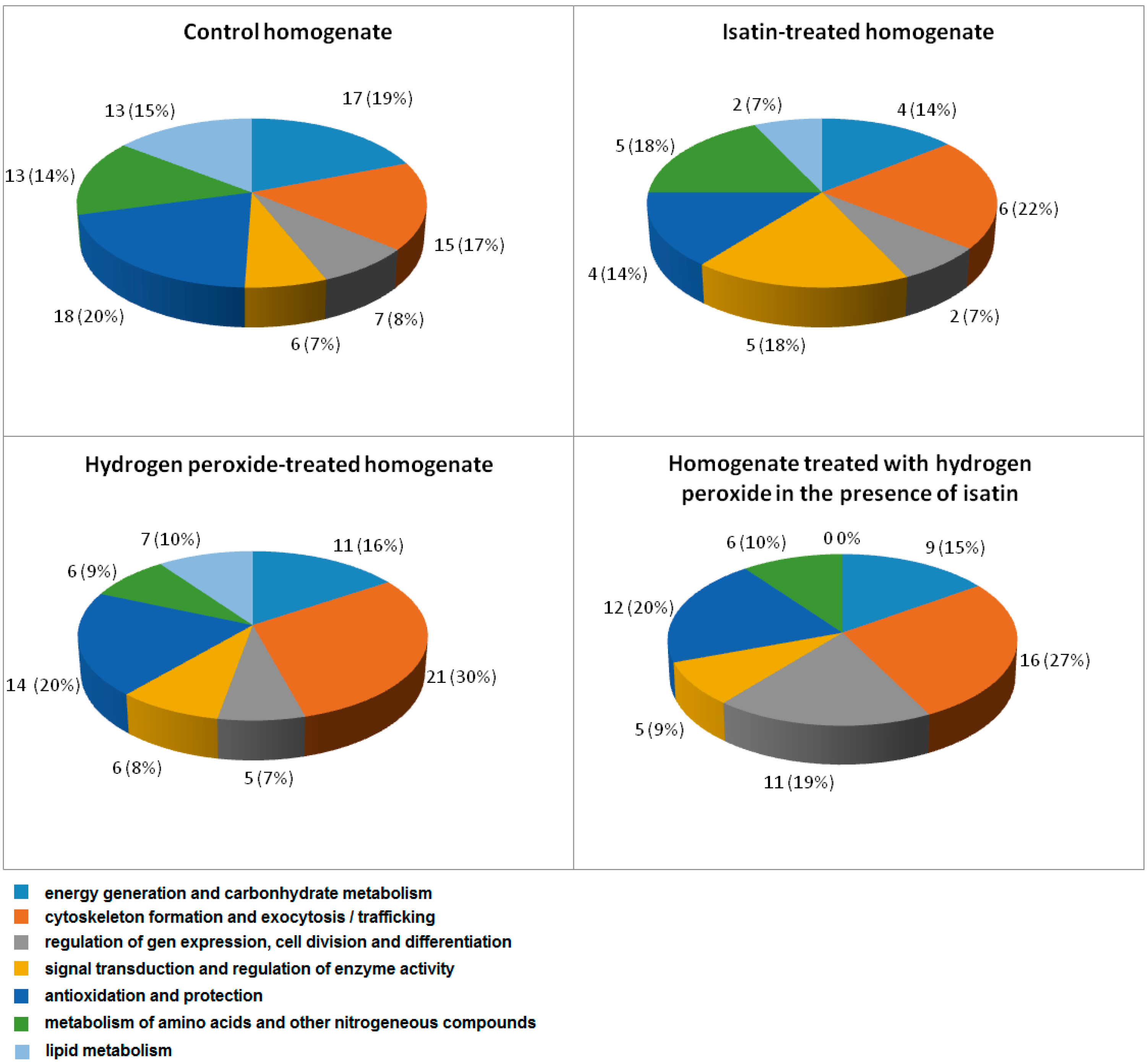
2.3. Effects of Hydrogen Peroxide and Isatin on Proteomic Profiles of Rat Brain Amyloid-Binding Proteins
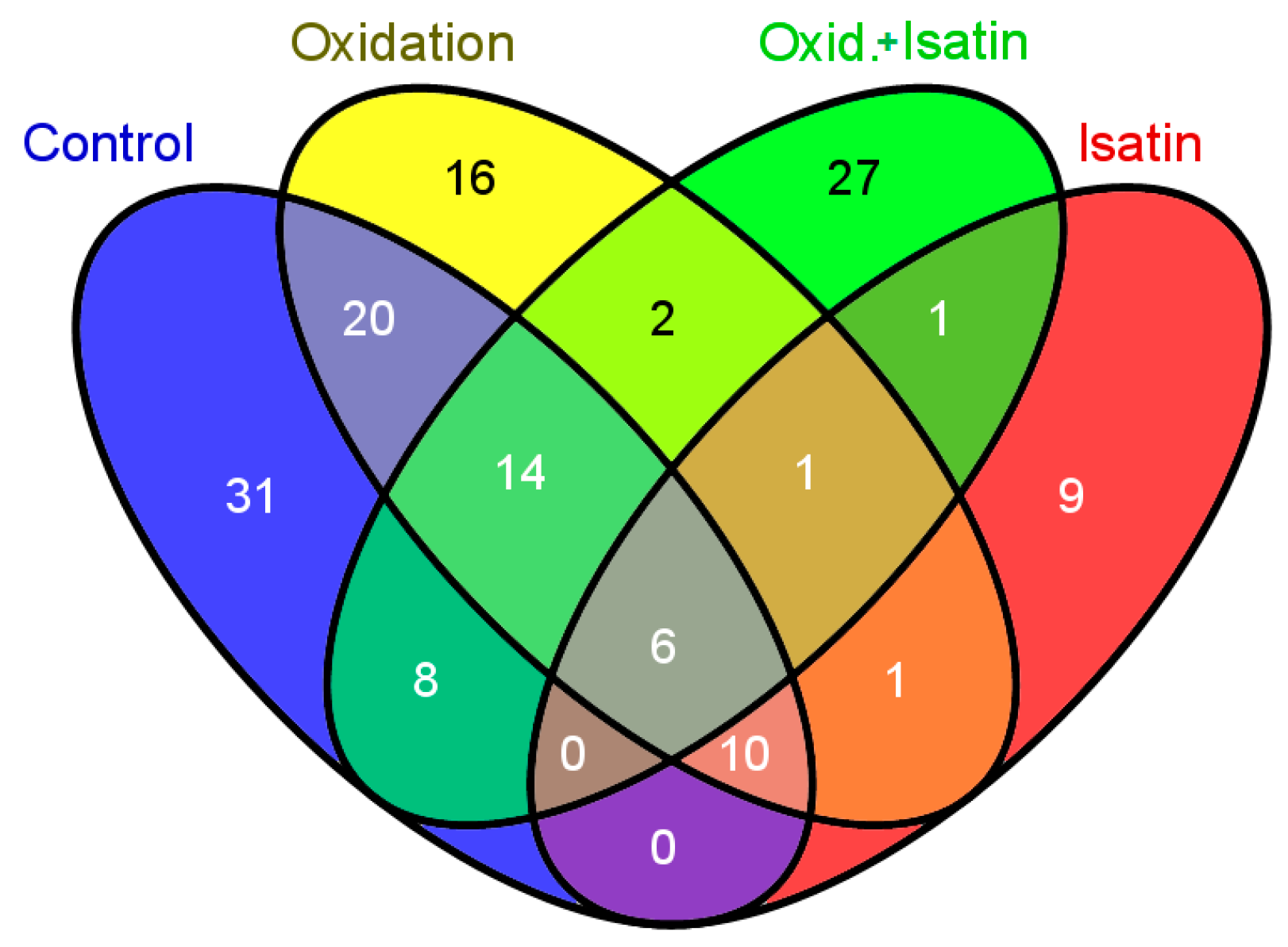
| No. | Protein Name | Intracellular Localization * | Oxidatively Modified/Altered in AD Brain and Different AD Models ** |
|---|---|---|---|
| 1 | Carbamoyl-phosphate synthase [ammonia], mitochondrial | M | No |
| 2 | Betaine-homocysteine S-methyltransferase 1 | C | No |
| 3 | Fructose-bisphosphate aldolase B | C, L, N, ER, CM | No |
| 4 | Endoplasmin ** | ER | Yes |
| 5 | Stress-70 protein, mitochondrial ** | M | Yes |
| 6 | Keratin, type II cytoskeletal 73 | C | No |
3. Discussion

4. Experimental Section
4.1. Materials
4.2. Animals, Brain Homogenate Preparation and Incubations
4.3. Immobilization of Amyloid-β Protein Fragment 1–42 on Affi-Gel 10
4.4. Affinity Interactions
4.5. SPR (Surface Plasmon Resonance) Measurements
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 6, a006262. [Google Scholar]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 2013, 4, e939. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally applied synthetic peptide isoAsp-Aβ(1–42) triggers cerebral β-amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Upadhaya, A.; Kosterin, I.; Kumar, S.; von Arnim, C.A.; Yamaguchi, H.; Fändrich, M.; Walter, J.; Thal, D.R. Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 2014, 137, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Maynard, C.; Cappai, R.; McLean, C.A.; Cherny, R.A.; Lynch, T.; Culvenor, J.G.; Trevaskis, J.; Tanner, J.E.; Bailey, K.A.; et al. Intracellular accumulation of detergent-soluble amyloidogenic Aβ fragment of Alzheimer’s disease precursor protein in the hippocampus of aged transgenic mice. J. Neurochem. 1999, 72, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Aβ accumulation precedes plaque formation in β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Sultana, R.; Poon, H.F. Redox proteomics: A new approach to investigate oxidative stress in Alzheimer’s disease. In Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases; Dalle-Donne, I., Scaloni, A., Butterfield, D.A., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2006; pp. 563–603. [Google Scholar]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Spuch, C.; Ortolano, S.; Navarro, C. New insights in the amyloid-beta interaction with mitochondria. J. Aging Res. 2012, 324968. [Google Scholar]
- Wu, Z.; Zhu, Y.; Cao, X.; Sun, S.; Zhao, B. Mitochondrial toxic effects of Aβ through mitofusins in the early pathogenesis of Alzheimer’s disease. Mol. Neurobiol. 2004, 50, 986–996. [Google Scholar] [CrossRef]
- Xu, G.; Stevens, S.M., Jr.; Moore, B.D.; McClung, S.; Borchelt, D.R. Cytosolic proteins lose solubility as amyloid deposits in a transgenic mouse model of Alzheimer-type amyloidosis. Hum. Mol. Genet. 2013, 22, 2765–2774. [Google Scholar] [CrossRef] [PubMed]
- Habib, L.; Lee, M.T.C.; Yang, J. Inhibitors of catalase-amyloid interactions protect cells from β-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 2010, 285, 38933–38943. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Zimbron, L.F.; Luna-Munoz, J.; Mena, R.; Vazquez-Ramirez, R.; Kubli-Garfias, C.; Cribbs, D.H.; Manoutcharian, K.; Gevorkian, G. Amyloid-β peptide binds to cytochrome C oxidase subunit 1. PLoS One 2012, 7, e42344. [Google Scholar] [CrossRef] [PubMed]
- Calero, M.; Rostagno, A.; Ghiso, J. Search for amyloid-binding proteins by affinity chromatography. Methods Mol. Biol. 2012, 849, 213–223. [Google Scholar] [PubMed]
- Schulze, H.; Schuler, A.; Stuber, D.; Dobeli, H.; Langen, H.; Huber, G. Rat brain glyceraldehyde-3-phosphate dehydrogenase interacts with the recombinant cytoplasmic domain of Alzheimer’s beta-amyloid precursor protein. J. Neurochem. 1993, 60, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Verdier, Y.; Földi, I.; Sergeant, N.; Fülöp, L.; Penke, Z.; Janáky, T.; Szücs, M.; Penke, B. Characterization of the interaction between Aβ 1–42 and glyceraldehyde phosphodehydrogenase. J. Pept. Sci. 2008, 14, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Naletova, I.; Schmalhausen, E.; Kharitonov, A.; Katrukha, A.; Saso, L.; Caprioli, A.; Muronetz, V. Non-native glyceraldehyde-3-phosphate dehydrogenase can be an intrinsic component of amyloid structures. Biochim. Biophys. Acta 2008, 1784, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Hardas, S.S.; Bader Lange, M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer disease: Many pathways to neurodegeneration. J. Alzheimers Dis. 2010, 20, 369–393. [Google Scholar] [PubMed]
- Medvedev, A.E.; Clow, A.; Sandler, M.; Glover, V. Isatin–A link between natriuretic peptides and monoamines? Biochem. Pharmacol. 1996, 52, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.; Igosheva, N.; Crumeyrolle-Arias, M.; Glover, V. Isatin: Role in stress and anxiety. Stress 2005, 8, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.; Buneeva, O.; Gnedenko, O.; Fedchenko, V.; Medvedeva, M.; Ivanov, Y.; Glover, V.; Sandler, M. Isatin interaction with glyceraldehyde-3-phosphate dehydrogenase, a putative target for neuroprotective drugs: Partial agonism with deprenyl. J. Neural. Transm. Suppl. 2006, 71, 195–203. [Google Scholar]
- Medvedev, A.; Buneeva, O.; Glover, V. Biological targets for isatin and its analogues: Implications for therapy. Biologics 2007, 1, 151–162. [Google Scholar] [PubMed]
- Crumeyrolle-Arias, M.; Buneeva, O.; Zgoda, V.; Kopylov, A.; Cardona, A.; Tournaire, M.-C.; Pozdnev, V.; Glover, V.; Medvedev, A. Isatin binding proteins in rat brain: In situ imaging, quantitative characterization of specific [3H]isatin binding, and proteomic profiling. J. Neurosci. Res. 2009, 87, 2763–2772. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Gnedenko, O.; Zgoda, V.; Kopylov, A.; Glover, V.; Ivanov, A.; Medvedev, A.; Archakov, A. Isatin binding proteins of rat and mouse brain: Proteomic identification and optical biosensor validation. Proteomics 2010, 10, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.A.; Kopylov, A.T.; Tikhonova, O.V.; Zgoda, V.G.; Medvedev, A.E.; Archakov, A.I. Effect of affinity sorbent on proteomic profiling of isatin binding proteins of mouse brain. Biochemistry (Moscow) 2012, 77, 1326–1328. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Poon, H.F.; St Clair, D.; Keller, J.N.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol. Dis. 2006, 22, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Liu, L.; Ahmed, E.K.; Friguet, B. Protein oxidative damage at the crossroads of cellular senescence, aging, and age-related diseases. Oxid. Med. Cell. Longev. 2012, 2012, 919832. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Witzmann, F.A.; Arnold, R.J.; Bai, F.; Hrncirova, P.; Kimpel, M.W.; Mechref, Y.S.; McBride, W.J.; Novotny, M.V.; Pedrick, N.M.; Ringham, H.N.; et al. A proteomic survey of rat cerebral cortical synaptosomes. Proteomics 2005, 5, 2177–2201. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Sultana, R.; Butterfield, D.A. Redox proteomics of oxidatively modified brain proteins in mild cognitive impairment. In Neuroproteomics; Alzate, O., Ed.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Butterfield, D.A. Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. Proteomics: A new approach to investigate oxidative stress in Alzheimer’s disease brain. Brain Res. 2004, 1000, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Aksenov, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, α-enolase andheat shock cognate 71. J. Neurochem. 2002, 82, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Shiozaki, A.; Kohno, R.; Yoshizato, K.; Shimohama, S. Proteomic profiling and neurodegeneration in Alzheimer’s disease. Neurochem. Res. 2002, 27, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxyterminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Owen, J.B. Lectin-affinity chromatography brain glycoproteomics and Alzheimer’s disease: Insights into protein alterations consistent with the pathology and progression of this dementing disorder. Proteomics Clin. Appl. 2011, 5, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lubec, G.; Nonaka, M.; Krapfenbauer, K.; Gratzer, M.; Cairns, N.; Fountoulakis, M. Expression of the dihydropyrimidinase related protein 2 (DRP-2) in Down syndrome and Alzheimer’s disease brain is downregulated at the mRNA and dysregulated at the protein level. J. Neural. Transm. Suppl. 1999, 57, 161–177. [Google Scholar] [PubMed]
- Krapfenbauer, K.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003, 967, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Cairns, N.; Fountoulakis, M.; Lubec, G. Synaptosomal proteins, beta-soluble N-ethylmaleimidesensitive factor attachment protein (β-SNAP), γ-SNAP and synaptotagmin I in brain of patients with Down syndrome and Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2001, 12, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Vlkolinsky, R.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Differential expression of molecular chaperones in brain of patients with Down syndrome. Electrophoresis 2001, 22, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Puchades, M.; Hansson, S.F.; Nilsson, C.L.; Andreasen, N.; Blennow, K.; Davidsson, P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Mol. Brain Res. 2003, 118, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J. Neural. Transm. Suppl. 2001, 61, 223–235. [Google Scholar] [PubMed]
- Liao, L.; Cheng, D.; Wang, J.; Duong, D.M.; Losik, T.G.; Gearing, M.; Rees, H.D.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 2004, 279, 37061–37068. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.K.; Etheridge, N.; Dodd, P.R.; Nouwens, A.S. Targeted quantitative analysis of synaptic proteins in Alzheimer’s disease brain. Neurochem. Int. 2014, 75, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Andreev, V.P.; Petyuk, V.A.; Brewer, H.M.; Karpievitch, Y.V.; Xie, F.; Clarke, J.; Camp, D.; Smith, R.D.; Lieberman, A.P.; Albin, R.L.; et al. Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lu, F.F.; Seeman, P.; Liu, F. Quantitative proteomic analysis of human substantia nigra in Alzheimer’s disease, Huntington’s disease and multiple sclerosis. Neurochem. Res. 2012, 37, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2013, 256C, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Schmalhausen, E.V.; Nagradova, N.K.; Boschi-Muller, S.; Branlant, G.; Muronetz, V.I. Mildly oxidized GAPDH: The coupling of the dehydrogenase and acyl phosphatase activities. FEBS Lett. 1999, 452, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, K.; Takahashi, H.; Chuang, D.M.; Ishitani, R. Glyceraldehyde-3-phosphate dehydrogenase is over-expressed during apoptotic death of neuronal cultures and is recognized by a monoclonal antibody against amyloid plaques from Alzheimer’s brain. Neurosci. Lett. 1995, 200, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Tamaoka, A.; Endoh, R.; Shoji, S.; Takahashi, H.; Hirokawa, K.; Teplow, D.B.; Selkoe, D.J.; Mori, H. Antibodies to amyloid beta protein (Aβ) crossreact with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Neurobiol. Aging 1996, 17, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.M.; Hough, C.; Senatorov, V.V. Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Cumming, R.C.; Schubert, D. Amyloid-beta induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. FASEB J. 2005, 19, 2060–2062. [Google Scholar] [PubMed]
- Ishitani, R.; Tajima, H.; Takata, H.; Tsuchiya, K.; Kuwae, T.; Yamada, M.; Takahashi, H.; Tatton, N.A.; Katsube, N. Proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase: A possible site of action of antiapoptotic drugs. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Tatton, W.; Chalmers-Redman, R.; Tatton, N. Neuroprotection by deprenyl and other propargylamines: Glyceraldehyde-3-phosphate dehydrogenase rather than monoamine oxidase B. J. Neural Transm. 2003, 110, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.D.; Bereiter-Hahn, J. Glyceraldehyde-3-phosphate dehydrogenase associates with actin filaments in serum deprived NIH 3T3 cells only. Cell Biol. Int. 2002, 26, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Waingeh, V.F.; Gustafson, C.D.; Kozliak, E.I.; Lowe, S.L.; Knull, H.R.; Thomasson, K.A. Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys. J. 2006, 90, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Oyama, R.; Yamamoto, H.; Titani, K. Glutamine synthetase, hemoglobin alphachain, and macrophage migration inhibitory factor binding to amyloid β-protein: Their identification in rat brain by a novel affinity chromatography and in Alzheimer’s disease brain by immunoprecipitation. Biochim. Biophys. Acta 2000, 1479, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Mawatari, K.; Segawa, M.; Masatsuka, R.; Hanawa, Y.; Iinuma, F.; Watanabe, M. Fluorimetric determination of isatin in human urine and serum by liquid chromatography postcolumn photoirradiation. Analyst 2001, 126, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Igosheva, N.; Matta, S.; Glover, V. Effect of acute stress and gender on isatin in rat tissues and serum. Physiol. Behav. 2004, 80, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Panova, N.G.; Zemskova, M.A.; Axenova, L.N.; Medvedev, A.E. Does isatin interact with rat brain monoamine oxidases in vivo? Neurosci. Lett. 1997, 233, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Pandeya, S.N.; Smitha, S.; Jyoti, M.; Sridhar, S.K. Biological activities of isatin and its derivatives. Acta Pharm. 2005, 55, 27–46. [Google Scholar] [PubMed]
- Zhou, Y.; Zhao, Z.Q.; Xie, J.X. Effects of isatin on rotational behavior and DA levels in caudate putamen in Parkinsonian rats. Brain Res. 2001, 917, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Hamaue, N.; Minami, M.; Terado, M.; Hirafuji, M.; Endo, T.; Machida, M.; Hiroshige, T.; Ogata, A.; Tashiro, K.; Saito, H.; et al. Comparative study of the effects of isatin, an endogenous MAO-inhibitor, and selegiline on bradykinesia and dopamine levels in a rat model of Parkinson’s disease induced by the Japanese encephalitis virus. Neurotoxicology 2004, 25, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Crumeyrolle-Arias, M.; Medvedev, A.; Cardona, A.; Barritault, D.; Glover, V. In situ imaging of specific binding of [3H]isatin in rat brain. J. Neurochem. 2003, 84, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.; Crumeyrolle-Arias, M.; Cardona, A.; Sandler, M.; Glover, V. Natriuretic peptide interaction with [3H]isatin binding sites in rat brain. Brain Res. 2005, 1042, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Goodwin, D.L.; Sandler, M.; Glover, V. Efficacy of isatin analogues as antagonists of rat brain and heart atrial natriuretic peptide receptors coupled to particulate guanylate cyclase. Biochem. Pharmacol. 1999, 57, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Campagna, F.; Catto, M.; Purgatorio, R.; Altomare, C.D.; Carotti, A.; de Stradis, A.; Palazzo, G. Synthesis and biophysical evaluation of arylhydrazono-1H-2-indolinones as β-amyloid aggregation inhibitors. Eur. J. Med. Chem. 2011, 46, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Quirante, J.; Nieto, J.; Almeida, M.R.; Saraiva, M.J.; Planas, A.; Arsequell, G.; Valencia, G. Isatin derivatives, a novel class of transthyretin fibrillogenesis inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5270–5273. [Google Scholar] [CrossRef] [PubMed]
- Zala, D.; Hinckelmann, M.V.; Yu, H.; Lyra da Cunha, M.M.; Liot, G.; Cordelieres, F.P.; Marco, S.; Saudou, F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 2013, 153, 479–491. [Google Scholar] [CrossRef]
- Scopes, R.K.; Stoter, A. Purification of all glycolytic enzymes from one muscle extract. Methods Enzymol. 1982, 90, 479–490. [Google Scholar] [PubMed]
- Wisniewski, J.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- SwissProt database. Available online: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz (accessed on 24 December 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medvedev, A.E.; Buneeva, O.A.; Kopylov, A.T.; Gnedenko, O.V.; Medvedeva, M.V.; Kozin, S.A.; Ivanov, A.S.; Zgoda, V.G.; Makarov, A.A. The Effects of Endogenous Non-Peptide Molecule Isatin and Hydrogen Peroxide on Proteomic Profiling of Rat Brain Amyloid-β Binding Proteins: Relevance to Alzheimer’s Disease? Int. J. Mol. Sci. 2015, 16, 476-495. https://doi.org/10.3390/ijms16010476
Medvedev AE, Buneeva OA, Kopylov AT, Gnedenko OV, Medvedeva MV, Kozin SA, Ivanov AS, Zgoda VG, Makarov AA. The Effects of Endogenous Non-Peptide Molecule Isatin and Hydrogen Peroxide on Proteomic Profiling of Rat Brain Amyloid-β Binding Proteins: Relevance to Alzheimer’s Disease? International Journal of Molecular Sciences. 2015; 16(1):476-495. https://doi.org/10.3390/ijms16010476
Chicago/Turabian StyleMedvedev, Alexei E., Olga A. Buneeva, Arthur T. Kopylov, Oksana V. Gnedenko, Marina V. Medvedeva, Sergey A. Kozin, Alexis S. Ivanov, Victor G. Zgoda, and Alexander A. Makarov. 2015. "The Effects of Endogenous Non-Peptide Molecule Isatin and Hydrogen Peroxide on Proteomic Profiling of Rat Brain Amyloid-β Binding Proteins: Relevance to Alzheimer’s Disease?" International Journal of Molecular Sciences 16, no. 1: 476-495. https://doi.org/10.3390/ijms16010476
APA StyleMedvedev, A. E., Buneeva, O. A., Kopylov, A. T., Gnedenko, O. V., Medvedeva, M. V., Kozin, S. A., Ivanov, A. S., Zgoda, V. G., & Makarov, A. A. (2015). The Effects of Endogenous Non-Peptide Molecule Isatin and Hydrogen Peroxide on Proteomic Profiling of Rat Brain Amyloid-β Binding Proteins: Relevance to Alzheimer’s Disease? International Journal of Molecular Sciences, 16(1), 476-495. https://doi.org/10.3390/ijms16010476