
Oxidative Stress in Obesity: A Critical Component in Human Diseases

 ,
,  ,
, 

{kind=link}
Abstract
:1. Oxidative Stress and Obesity
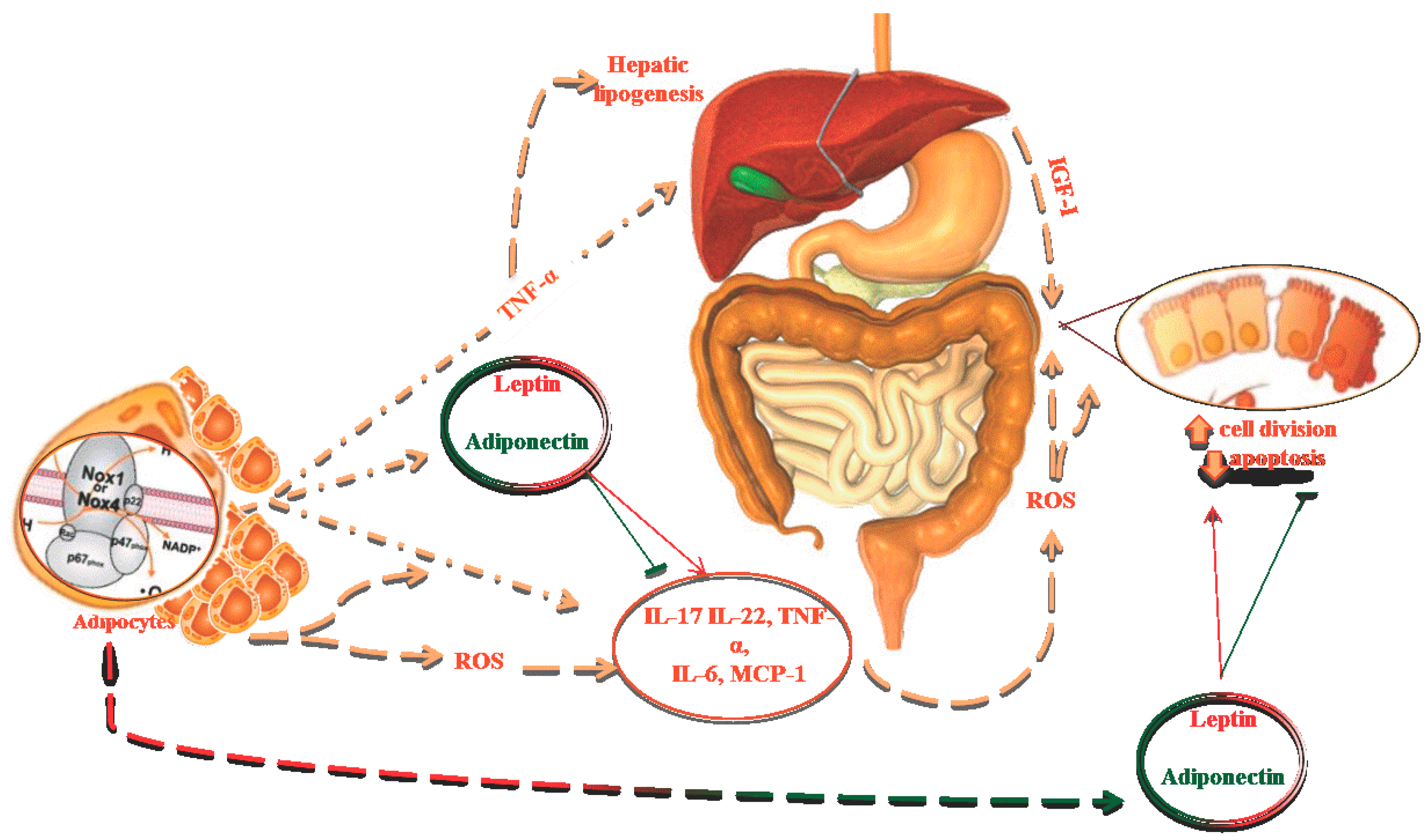
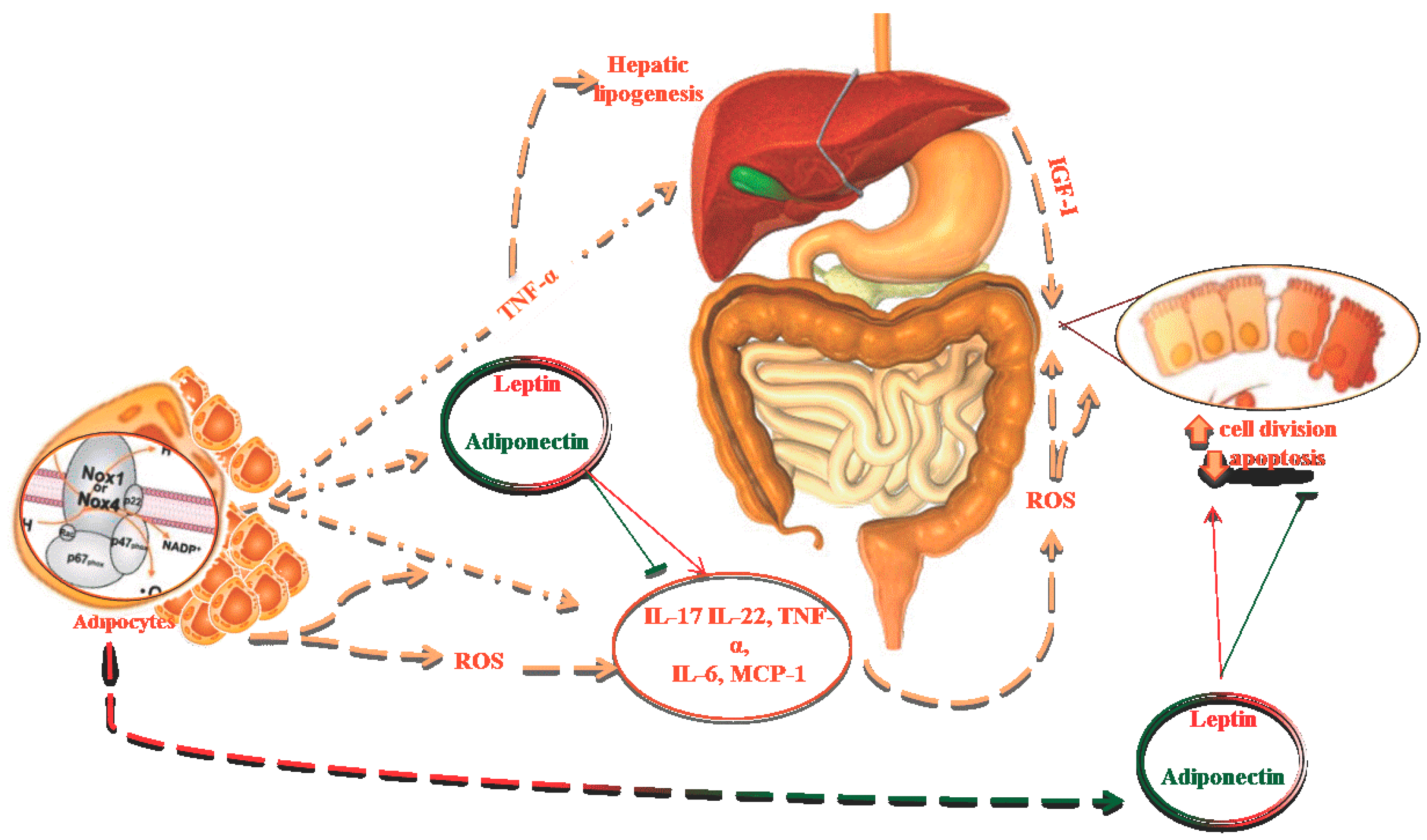
2. Oxidative Stress, Obesity and Metabolic Syndrome
3. Oxidative Stress, Obesity and Type 2 Diabetes Mellitus
4. Oxidative Stress, Obesity and Cardiovascular Diseases
4.1. Dyslipidemia
4.2. Hypertension
5. Oxidative Stress, Obesity and Liver Diseases
6. Oxidative Stress, Obesity and Cancer Susceptibility
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sikaris, K. The clinical biochemistry of obesity. Clin. Biochem. Rev. 2004, 25, 165–181. [Google Scholar] [PubMed]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.; Wang, J.; Thornton, J.C.; Mei, Z.; Sopher, A.B.; Pierson, R.N., Jr; Dietz, W.H.; Horlick, M. Classification of body fatness by body mass index-for-age categories among children. Arch. Pediatr. Adolesc. Med. 2009, 163, 801–811. [Google Scholar] [CrossRef]
- Office of the Surgeon General. The Surgeon General’s Vision for a Healthy and Fit Nation; External Web Site Icon.: Rockville, MD, USA, 2010. [Google Scholar]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Pre-eclampsia, the placenta and the maternal systemicinflammatory respons—A review. Placenta 2003, 24, 21–27. [Google Scholar] [CrossRef]
- Fonseca-Alaniz, M.H.; Takada, J.; Alonso-Vale, M.I.; Lima, F.B. Adipose tissue as an endocrine organ: From theory to practice. J. Pediatr. 2007, 83, 192–203. [Google Scholar] [CrossRef]
- Chandel, N.S.; Schumacker, P.T.; Arch, R.H. Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J. Biol. Chem. 2001, 276, 42728–42736. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Trayhurn, P. Acute and prolonged effects of TNF-α on the expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture. Pflüg. Arch. 2006, 452, 418–427. [Google Scholar] [CrossRef]
- Stienstra, R.; Tack, C.J.; Kanneganti, T.D.; Joosten, L.A.; Netea, M.G. The inflammasome puts obesity in the danger zone. Cell Metab. 2012, 15, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Curti, M.L.R.; Borges, P.; Rogero, M.C.; Ferreira, S.R. Studies of gene variants related to inflammation, oxidative stress, dyslipidemia, and obesity: Implications for a nutrigenetic approach. J. Obes. 2011, 2011, 1–30. [Google Scholar] [CrossRef]
- Stenlöf, K.; Wernstedt, I.; Fjällman, T.; Wallenius, V.; Wallenius, K.; Jansson, J.O. Interleukin-6 levels in the central nervous system are negatively correlated with fat mass in overweight/obese subjects. J. Clin. Endocrinol. Metab. 2003, 88, 4379–4383. [Google Scholar] [CrossRef] [PubMed]
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Herrero, L; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Frossi, B.; de Carli, M.; Daniel, K.C.; Rivera, J.; Pucillo, C. Oxidative stress stimulates IL-4 and IL-6 production in mast cells by an APE/Ref-1-dependent pathway. Eur. J. Immunol. 2003, 33, 2168–2177. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Umemoto, T.; Omer, M. NADPH oxidase derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 2012, 287, 10379–10393. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar]
- Amirkhizi, F.; Siassi, F.; Minaie, S.; Djalali, M.; Rahimi, A.; Chamari, M. Is obesity associated with increased plasma lipid peroxidación and oxidative stress in women. ARYA Atheroscler. J. 2007, 2, 189–192. [Google Scholar]
- Ozata, M.; Mergen, M.; Oktenli, C.; Aydin, A.; Sanisoglu, S.Y.; Bolu, E.; Yilmaz, M.I.; Sayal, A.; Isimer, A.; Ozdemir, I.C. Increased oxidative stress and hypozincemia in male obesity. Clin. Biochem. 2002, 35, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Rzheshevsky, A.V. Fatal “Triad”: Lipotoxicity, oxidative stress, and phenoptosis. Biochemistry 2013, 78, 991–1000. [Google Scholar] [PubMed]
- Tereshin, E.V. A role of fatty acids in the development of oxidative stress in aging. A hypothesis. Adv. Gerontol. 2007, 20, 59–65. [Google Scholar] [PubMed]
- Duvnjak, M.; Lerotic, I.; Barsic, N.; Tomasic, V.; Virovic Jukic, L.; Velagic, V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 4539–4550. [Google Scholar] [PubMed]
- Goossens, G.H. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol. Behav. 2008, 94, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Naz, L.; Yasmeen, G. Obesity: An independent risk factor systemic oxidative stress. Pak. J. Pharm. Sci. 2006, 19, 62–69. [Google Scholar] [PubMed]
- Hukshorn, C.J.; Lindeman, J.H.; Toet, K.H.; Saris, W.H.; Eilers, P.H.; Westerterp-Plantenga, M.S.; Kooistra, T. Leptin and the proinflammatory state associated with human obesity. J. Clin. Endocrinol. Metab. 2004, 89, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.; Desideri, G.; Valenti, M.; Bellini, C.; Pasin, M.; Santucci, A.; de Mattia, G. Early up-regulation of endothelial adhesion molecules in obese hypertensive men. Hypertension 1999, 34, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G.; Faggioni, R. Leptin in the regulation of immunity, inflammation, and haematopoiesis. J. Leukoc. Biol. 2000, 68, 437–446. [Google Scholar] [PubMed]
- Deng, Y.; Scherer, P.E. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. N. Y. Acad. Sci. 2010, 1212, E1–E19. [Google Scholar] [CrossRef] [PubMed]
- Ouedraogo, R.; Gong, Y.; Berzins, B.; Wu, X.; Mahadev, K.; Hough, K.; Chan, L.; Goldstein, B.J.; Scalia, R. Adiponectin deficiency increases leukocyte-endothelium interactions via up-regulation of endothelial cell adhesion molecules in vivo. J. Clin. Investig. 2007, 117, 1718–1761. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nishizawa, H.; Funahashi, T.; Shimomura, I.; Shimabukuro, M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circulation 2006, 70, 1437–1442. [Google Scholar] [CrossRef]
- Beltowski, J. Apelin and visfatin: Unique beneficial adipokines up-regulated in obesity? Med. Sci. Monit. 2006, 12, 112–119. [Google Scholar]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Cuppari, C.; Salpietro, V.; Filippelli, M.; Chirico, V.; Gitto, E.; Salpietro, C.; Arrigo, T. The role of visfatin in pregnancy, complications and procreation. J. Pediatr. Biochem. 2014, in press. [Google Scholar]
- Martos-Moreno, G.A.; Kratzsch, J.; Korner, A.; Barrios, V.; Hawkins, F.; Kiess, W.; Argente, J. Serum visfatin and vaspin levels in prepubertal children: Effect of obesity and weight loss after behavior modifications on their secretion and relationship with glucose metabolism. Int. J. Obes. 2011, 35, 1355–1362. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-κB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Maemura, K.; Takeda, N.; Harada, T.; Nojiri, T.; Imai, Y.; Manabe, I.; Utsunomiya, K.; Nagai, R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine-endothelial cell interactions. Biochem. Biophys. Res. Commun. 2004, 314, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Jiang, J.; Lü, J.M.; Chai, H.; Wang, X.; Lin, P.H.; Yao, Q. Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 193–201. [Google Scholar] [CrossRef]
- Than, A.; Zhang, X.; Leow, M.K.; Poh, C.L.; Chong, S.K.; Chen, P. Apelin attenuates oxidative stress in human adipocytes. J. Biol. Chem. 2014, 289, 3763–3774. [Google Scholar] [CrossRef]
- Pisarenko, O.I.; Bespalova, ZhD.; Lankin, V.Z.; Timoshin, A.A.; Serebriakova, L.I.; Shulzhenko, V.S.; Pelogeikina, IuA.; Studneva, I.M.; Tskitishvili, O.V.; Azmuko, A.A.; et al. Antioxidant properties of apelin-12 and its structural analogue in experimental ischemia and reperfusion. Kardiologiia 2013, 53, 61–67. [Google Scholar] [PubMed]
- Gottschling-Zeller, H.; Birgel, M.; Rohrig, K.; Hauner, H. Effect of tumor necrosis factor α and transforming growth factor β 1 on plasminogen activator inhibitor-1 secretion from subcutaneous and omental human fat cells in suspension culture. Metabolism 2000, 49, 666–671. [Google Scholar] [CrossRef] [PubMed]
- To, M.; Takagi, D.; Akashi, K.; Kano, I.; Haruki, K.; Barnes, P.J.; Ito, K. Sputum plasminogen activator inhibitor-1 elevation by oxidative stress-dependent nuclear factor-κB activation in COPD. Chest 2013, 144, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Overstreet, J.M.; Higgins, S.P.; Higgins, P.J. TGF-β1→SMAD/p53/USF2→PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res. 2012, 347, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Vulin, A.I.; Stanley, F.M. Oxidative stress activates the plasminogen activator inhibitor type 1 (PAI-1) promoter through an AP-1 response element and cooperates with insulin for additive effects on PAI-1 transcription. J. Biol. Chem. 2004, 279, 25172–25178. [Google Scholar] [CrossRef] [PubMed]
- De Taeye, B.; Smith, L.H.; Vaughan, D.E. Plasminogen activator inhibitor-1: A common denominator in obesity, diabetes and cardiovascular disease. Curr. Opin. Pharmacol. 2005, 5, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lam, K.S.; Wang, Y.; Wu, D.; Lam, M.C.; Shen, J.; Wong, L.; Hoo, R.L.; Zhang, J.; Xu, A. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem. Biophys. Res. Commun. 2006, 341, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Pihl, E.; Zilmer, K.; Kullisaar, T.; Kairane, C.; Magi, A.; Zilmer, M. Atherogenic inflammatory and oxidative stress markers in relation to overweight values in male former athletes. Int. J. Obes. 2006, 30, 141–146. [Google Scholar] [CrossRef]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Skoumas, I.; Papademetriou, L.; Economou, M.; Stefanadis, C. The implication of obesity on total antioxidant capacity apparently healthy men and women: The ATTICA study. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Allison, D.B.; Heymsfield, S.B.; Kelley, D.E.; Leibel, R.L.; Nonas, C.; Kahn, R. Waist circumference and cardiometabolic risk: A consensus from shaping America’s health: Association for weight management and Obesity Prevention; NAASO, The Obesity Society; the American Society for Nutrition; and American Diabetes Association. Am. J. Clin. Nutr. 2007, 30, 1647–1652. [Google Scholar]
- Juge-Aubry, C.E.; Henrichot, E.; Meier, C.A. Adipose tissue: A regulator of inflammation. J. Clin. Endocrinol. Metab. 2005, 19, 547–566. [Google Scholar]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signalling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Sy, M.; Pavlovich, K.; Leibel, R.L; Hirsch, J. Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J. Clin. Investig. 2008, 118, 2583–2591. [Google Scholar] [PubMed]
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.I. Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases. Curr. Pharm. Des. 2009, 15, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Lago, F.; Dieguez, C.; G´omez-Reino, G.; Gualillo, O. Adipokines as emerging mediators of immune response and inflammation. Nat. Clin. Pract. Rheumatol. 2007, 3, 716–724. [Google Scholar] [CrossRef]
- Grundy, S.M. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2005, 109, 433–438. [Google Scholar] [CrossRef]
- Matsuoka, T. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J. Clin. Investig. 1997, 99, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Maddux, B.A. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by micromolar concentrations of α-lipoic acid. Diabetes 2001, 50, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Rudich, A. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Hopps, E.; Noto, D.; Caimi, G.; Averna, M.R. A novel comoponent of the metabolic syndrome: The oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Palmieri, V.O.; Portincasa, P.; Moschetta, A.; Palasciano, G. Oxidative stress-induced risk factors associated with the metabolic syndrome: A unifying hyopothesis. J. Nutr. Biochem. 2008, 19, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Mokdad, A.H.; Giles, W.H.; Brown, D.W. The metabolic syndrome and antioxidant concentrations: Findings from the Third National Health and Nutrition Examination Survey. Diabetes 2003, 52, 2346–2352. [Google Scholar] [CrossRef] [PubMed]
- Mullarkey, C.J.; Edelstein, D.; Brownee, M. Free radical generation by early glycation products: A mechanism for accelerated atherogenesis in diabetes. Biochem. Biophys. Res. Commun. 1990, 173, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and β-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, K.; Jiang, Z.Y.; Takahara, N.; Ha, S.W.; Igarashi, M.; Yamauchi, T.; Feener, E.P.; Herbert, T.P.; Rhodes, C.J.; King, G.L. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: A specific vascular action of insulin. Circulation 2000, 101, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.B.; Seftel, A. Glucose-induced changes in protein kinase Cand nitric oxide are prevented by vitamin E. Am. J. Physiol. Endocrinol. Metab. 2000, 278, 146–152. [Google Scholar]
- Ha, H.; Yu, M.R.; Choi, Y.J.; Kitamura, M.; Lee, H.B. Role of high glucose-induced nuclear factor-κB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J. Am. Soc. Nephrol. 2002, 13, 894–902. [Google Scholar] [PubMed]
- Yamagishi, S.; Maeda, T.; Matsui, S.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta 2012, 1820, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Kristina, I.R. Diabetes treatment—Bridging the divide. N. Engl. J. Med. 2007, 356, 1499–1501. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Cosentino, F. Insulin resistance, diabetes, and cardiovascular risk. Curr. Atheroscler. Rep. 2014, 16, 419. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Trumpower, B.L. The protonmotive Q cycle: Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 1990, 265, 11409–11412. [Google Scholar] [PubMed]
- Pitocco, D.; Tesauro, M.; Alessandro, R.; Ghirlanda, G.; Cardillo, C. Oxidative stress in diabetes: Implications for vascular and other complications. Int. J. Mol. Sci. 2013, 14, 21525–21550. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Huang, H.; Wei, Y. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochem. Biophys. Res. Commun. 2003, 300, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; Collins, S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. J. Am. Med. Assoc. 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Peraldi, P.; Spiegelman, B. TNF-α and insulin resistance: Summary and future prospects. Mol. Cell. Biochem. 1998, 182, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 2004, 27, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor-α and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Dinarello, C.A.; Mier, J.W. IL-6 and APPs: Anti-inflammatory and immunosuppressive mediators. Immunol. Today 1997, 18, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kang, J.; Guan, Y. The mechanisms linking adiposopathy to type 2 diabetes. Front. Med. 2013, 7, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Bolkent, S.; Yanardag, R.; Bolkent, S.; Doger, M.M. Beneficial effects of combined treatment with niacin and chromium on the liver of hyperlipemic rats. Biol. Trace Elem. Res. 2004, 101, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.L.; Shi, Y.H.; Hao, G. Increasing oxidative stress with progressive hyperlipidemia in human: Relation between malondialdehyde and atherogenic index. J. Clin. Biochem. Nutr. 2008, 43, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Taboga, C.; Tonutti, L.; Quagliaro, L.; Piconi, L.; Bais, D.; da Ros, R.; Motz, E. Evidence for an independent and cumulativeeffect of postprandial hypertriglyceridemia and hyper-glycemia on endothelial dysfunction and oxidative stressgeneration: Effects of short- and long-term simvastatin treatment. Circulation 2002, 106, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, M. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [PubMed]
- Nishimura, I.; Manabe, M. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 2007, 56, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Merkel, M.; Heeren, J.; Dudeck, W.; Rinninger, F.; Radner, H.; Breslow, J.L.; Goldberg, I.J.; Zechner, R.; Greten, H. Inactive lipoprotein lipase (LPL) alone increases selective cholesterol ester uptake in vivo, whereas in the presence of active LPL it also increases triglyceride hydrolysis and whole particle lipoprotein uptake. J. Biol. Chem. 2002, 277, 7405–7411. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Stehouwer, C.D.; Schalkwijk, C.G. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr. Opin. Lipidol. 2013, 24, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Patryn, E.; Hotowy, K.; Czapinska, E.; Majda, J.; Kustrzeba-Wojcicka, I.; Noczynska, A.; Gamian, A. Paraoxonase (PON)-1 activity in overweight and obese children and adolescents: Association with obesity-related inflammation and oxidative stress. Adv. Clin. Exp. Med. 2013, 22, 229–236. [Google Scholar] [PubMed]
- Manea, A.; Simionescu, M. Nox enzymes and oxidative stress in atherosclerosis. Front. Biosci. 2012, 4, 651–670. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef]
- Redon, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Saez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Pedro-Botet, J.; Covas, M.I.; Martin, S.; Rubies-Prat, J. Decreased endogenous antioxidant enzymatic status in essential hypertension. J. Hum. Hypertens. 2000, 14, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Parmer, R.J.; Lacy, F.; Kailasam, M.T.; O’Connor, D.T.; Schmid-Schonbein, G.W.; Parmer, R.J. Plasma hydrogen peroxide production in human essential hypertension: Role of heredity, gender, and ethnicity. Hypertension 2000, 36, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Edmunds, E.; Nuttall, S.L.; Landray, M.J.; Blann, A.D.; Beevers, D.G. Oxidative stress in malignant and non-malignant phase hypertension. J. Hum. Hypertens. 2002, 16, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, L.; Aronne, L.J.; Beilin, L.J.; Burke, V.; Igel, L.I.; LIoyd-Jones, D.; Sowers, J. Obesity-related hypertension: Pathogenesis, cardiovascular risk, and treatment: A position paper of The Obesity Society and the American Society of Hypertension. J. Clin. Hypertens. 2013, 15, 14–33. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, F.; Ren, J.; Driscoll, M.L.; Culver, B. High dietary fat induces NADPH oxidase-associated oxidativestress and inflammation in rat cerebral cortex. Exp. Neurol. 2005, 191, 318–225. [Google Scholar] [CrossRef] [PubMed]
- Nagae, A.; Fujita, M.; Kawarazaki, H.; Matsui, H.; Ando, K.; Fujita, T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation 2009, 119, 978–986. [Google Scholar] [CrossRef]
- Ferrante, AW. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Kumagi, T.; Hirooka, M.; Furukawa, S.; Kawasaki, K.; Koizumi, M.; Todo, Y.; Yamamoto, S.; Nunoi, H.; Tokumoto, Y.; et al. Significance of exercise in nonalcoholic fatty liver disease in men: A community-based large cross-sectional study. J. Gastroenterol. 2014, in press. [Google Scholar]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef] [PubMed]
- Başaranoğlu, M.; Örmeci, N. Nonalcoholic fatty liver disease: Diagnosis, pathogenesis, and management. Turk. J. Gastroenterol. 2014, 25, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Klintworth, H.; Kowdley, K.V. Iron metabolism in nonalcoholic fatty liver disease. Curr. Gastroenterol. Rep. 2012, 14, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Nikonorov, A.A.; Skalnaya, M.G.; Tinkov, A.A.; Skalny, A.V. Mutual interaction between iron homeostasis and obesity pathogenesis. J. Trace Elem. Med. Biol. 2014, in press. [Google Scholar]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; MAgrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Seki, S. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2014, 15, 7352–7379. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennet, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Manti, S.; Marseglia, L.; D’Angelo, G.; Filippelli, M.; Cuppari, C.; Gitto, E.; Romano, C.; Arrigo, T.; Salpietro, C. Portal hypertension as immune mediate disease. Hepat. Mon. 2014, 14, e18625. [Google Scholar]
- Basaranoglu, M.; Kayacetin, S.; Yilmaz, N.; Kayacetin, E.; Tarcin, O.; Sonsuz, A. Understanding mechanisms of the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 2223–2226. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Laiyemo, A.O. The risk of colonic adenomas and colonic cancer in obesity. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Tremblay, A. Genetic influences on the response of body fat and fat distribution to positive and negative energy balances in human identical twins. J. Nutr. 1997, 127, 943–947. [Google Scholar]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Deignan, J.L.; Qi, H.; Zhu, J.; Qian, S.; Zhong, J.; Torosyan, G.; Majid, S.; Falkard, B.; Kleinhanz, R.R.; et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat. Genet. 2009, 41, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Gulati, P.; Cheung, M.K.; Antrobus, R.; Church, C.D.; Harding, H.P.; Tung, Y.C.; Rimmington, D.; Ma, M.; Ron, D.; Lehner, P.J.; et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc. Natl. Acad. Sci. USA 2013, 110, 2557–2562. [Google Scholar] [CrossRef] [PubMed]
- Vongsuvanh, R.; George, J.; Qiao, L.; van der Poorten, D. Visceral adiposity in gastrointestinal and hepatic carcinogenesis. Cancer Lett. 2013, 330, 1–10. [Google Scholar] [CrossRef] [PubMed]
- El Yafi, F.; Winkler, R.; Delvenne, P.; Boussif, N.; Belaiche, J.; Louis, E. Altered expression of type I insulin-like growth factor receptor in Crohn’s disease. Clin. Exp. Immunol. 2005, 139, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Van Goudoever, J.B.; Corpeleijn, W.; Riedijk, M.; Schaart, M.; Renes, I.; van der Schoor, S. The impact of enteral insulin-like growth factor 1 and nutrition on gut permeability and amino acid utilization. J. Nutr. 2008, 138, 1829–1833. [Google Scholar]
- Demark-Wahnefried, W.; Platz, E.A.; Ligibel, J.A.; Blair, C.K.; Courneya, K.S.; Meyerhardt, J.A.; Ganz, P.A.; Rock, C.L.; Schmitz, K.H.; Wadden, T.; et al. The role of obesity in cancer survival and recurrence. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1244–1259. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, C.; Sánchez, C.; Climent, B.; Vázquez, A.; Iradi, A.; el Amrani, F.; Bediaga, A.; Sáez, G.T. Oxidative stress and DNA damage in obesity-related tumorigenesis. Adv. Exp. Med. Biol. 2014, 824, 5–17. [Google Scholar] [PubMed]
- Dubois, V.; Jarde, T.; Delort, L.; Billard, H.; Bernard-Gallon, D.; Berger, E.; Geloen, A.; Vasson, M.P.; Caldefie-Chezet, F. Leptin induces a proliferative response in breast cancer cells but not in normal breast cells. Nutr. Cancer 2014, 66, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Allott, E.H.; Masko, E.M.; Freedland, S.J. Obesity and prostate cancer: Weighing the evidence. Eur. Urol. 2013, 63, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.P.; Yin, P.H.; Chang, Y.C.; Lee, C.H.; Huang, S.Y.; Chi, C.W. Differential roles of leptin in regulating cell migration in thyroid cancer cells. Oncol. Rep. 2010, 123, 1721–1727. [Google Scholar]
- MacDougald, O.A.; Burant, C.F. The rapidly expanding family of adipokines. Cell Metab. 2007, 6, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Walsh, K. Obesity, adiponectin and vascular inflammatory disease. Curr. Opin. Lipidol. 2003, 14, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lam, K.S.; Xu, A. Adiponectin as a negative regulator in obesity-related mammary carcinogenesis. Cell Res. 2007, 17, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Ouchi, N.; Kihara, S.; Walsh, K. Adiponectin stimulates angiogenesis in response to tissue ischemia through stimulation of amp-activated protein kinase signaling. J. Biol. Chem. 2004, 279, 28670–28674. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Chi, M.M.; Scull, B.P.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 2010, 5, e12191. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Barzilai, N. Role of visceral adipose tissue in aging. Biochim. Biophys. Acta 2009, 1790, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Olefsky, J.M. Inflamed fat: What starts the fire? J. Clin. Investig. 2006, 116, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Sengenes, C.; Miranville, A.; Lolmede, K.; Curat, C.A.; Bouloumie, A. The role of endothelial cells in inflamed adipose tissue. J. Intern. Med. 2007, 262, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Arrigo, T.; Barberi, I.; Reiter, R.J.; Gitto, E. Oxidative stress-mediated aging during the fetal and perinatal periods. Oxid. Med. Cell Longev. 2014, 2014, 358375. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative Stress in Obesity: A Critical Component in Human Diseases. Int. J. Mol. Sci. 2015, 16, 378-400. https://doi.org/10.3390/ijms16010378
Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, Di Rosa G, Gitto E, Arrigo T. Oxidative Stress in Obesity: A Critical Component in Human Diseases. International Journal of Molecular Sciences. 2015; 16(1):378-400. https://doi.org/10.3390/ijms16010378
Chicago/Turabian StyleMarseglia, Lucia, Sara Manti, Gabriella D’Angelo, Antonio Nicotera, Eleonora Parisi, Gabriella Di Rosa, Eloisa Gitto, and Teresa Arrigo. 2015. "Oxidative Stress in Obesity: A Critical Component in Human Diseases" International Journal of Molecular Sciences 16, no. 1: 378-400. https://doi.org/10.3390/ijms16010378
APA StyleMarseglia, L., Manti, S., D’Angelo, G., Nicotera, A., Parisi, E., Di Rosa, G., Gitto, E., & Arrigo, T. (2015). Oxidative Stress in Obesity: A Critical Component in Human Diseases. International Journal of Molecular Sciences, 16(1), 378-400. https://doi.org/10.3390/ijms16010378