
HCN Channels—Modulators of Cardiac and Neuronal Excitability

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Modulation, Expression and Functional Significance of Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels
2.1. Modulation by Cyclic Nucleotides, Phosphoinositides and Tyrosine Phosphorylation
2.2. Expression Profile
2.3. Overview of HCN Knockout Studies
3. Impact of HCN Channels on Cardiac Ventricular Excitability
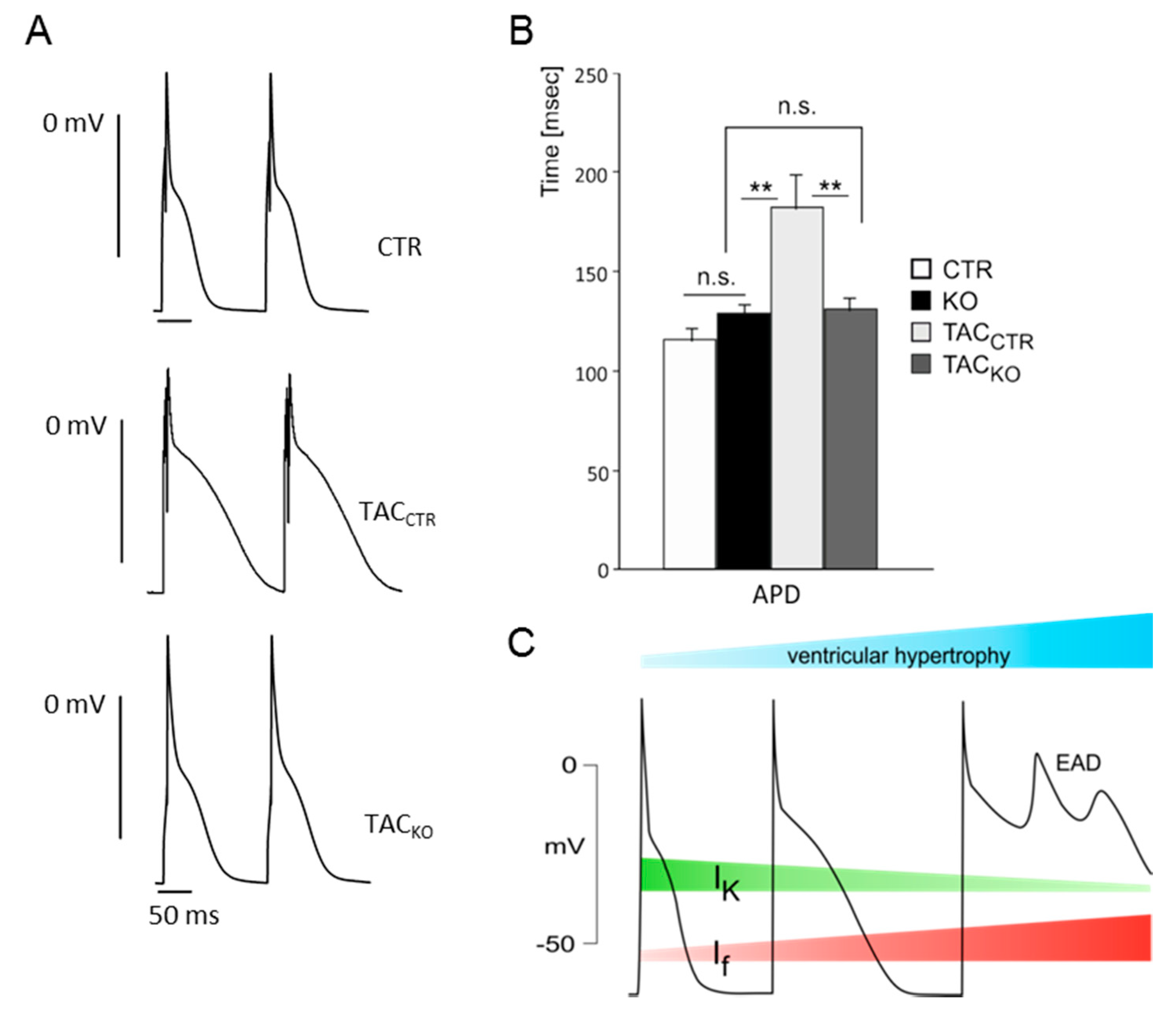
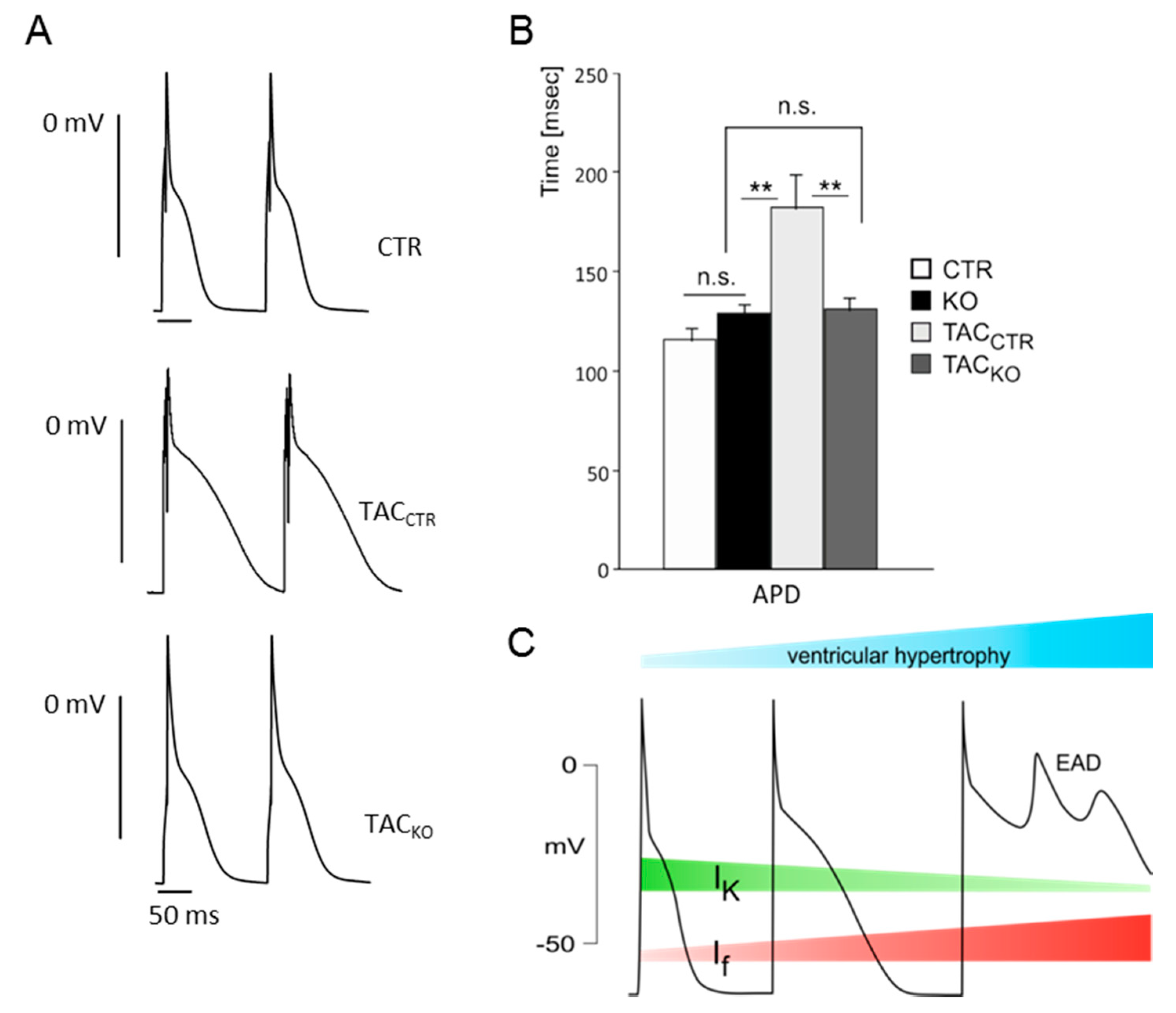
4. Contribution of HCN Channels to the Excitability of Peripheral Neurons
4.1. Participation of HCN Channels in Neuropathic Pain Behavior
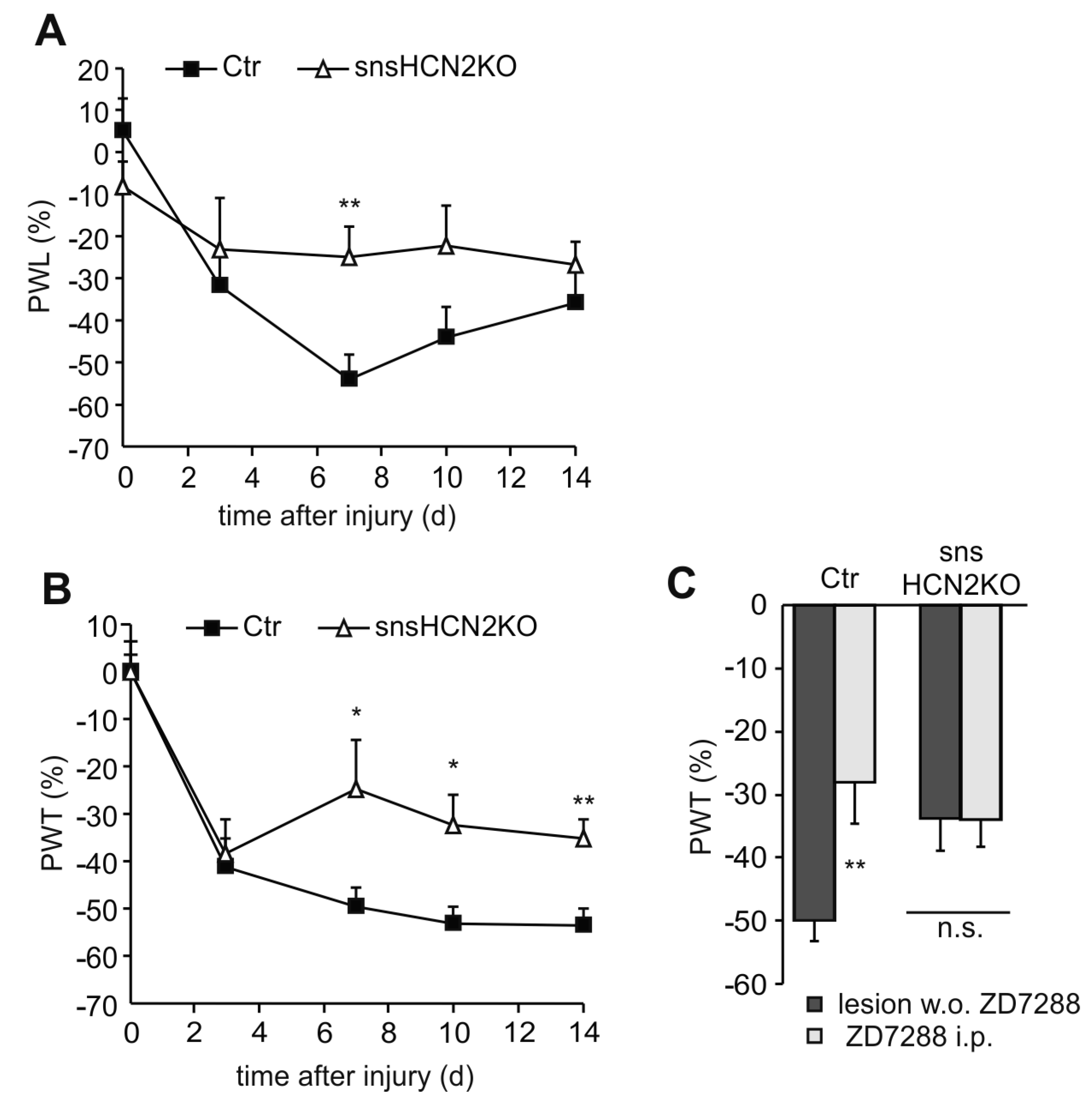
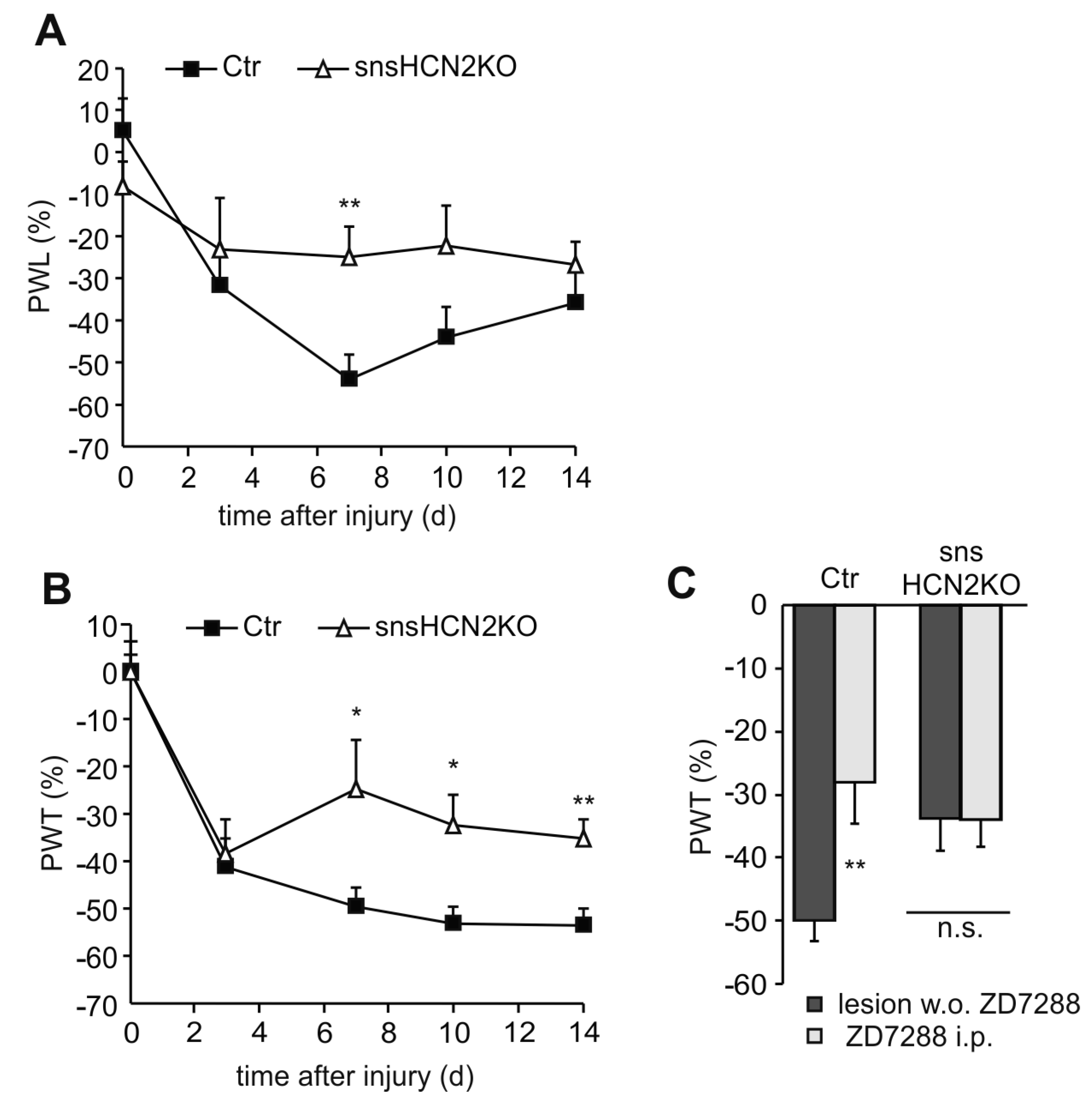
4.2. Participation of HCN Channels in Inflammatory Pain Behavior
5. HCN Channel Blockers—Current Therapeutic Use and Perspectives
5.1. Ivabradine in the Treatment of Coronary Artery Disease
5.2. Ivabradine in the Treatment of Heart Failure
5.3. Isoform-Selective HCN Channel Blockers
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ludwig, A.; Zong, X.; Jeglitsch, M.; Hofmann, F.; Biel, M. A family of hyperpolarization-activated mammalian cation channels. Nature 1998, 393, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Santoro, B.; Liu, D.T.; Yao, H.; Bartsch, D.; Kandel, E.R.; Siegelbaum, S.A.; Tibbs, G.R. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell 1998, 93, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Zong, X.; Stieber, J.; Hullin, R.; Hofmann, F.; Biel, M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999, 18, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Scholten, A.; Gauss, R.; Mincheva, A.; Lichter, P.; Kaupp, U.B. Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc. Natl. Acad. Sci. USA 1999, 96, 9391–9396. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; DeGennaro, M.; Santoro, B.; Siegelbaum, S.A.; Tibbs, G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 2001, 411, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Ulens, C.; Siegelbaum, S.A. Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron 2003, 40, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Pian, P.; Bucchi, A.; Robinson, R.B.; Siegelbaum, S.A. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 2006, 128, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Zolles, G.; Klocker, N.; Wenzel, D.; Weisser-Thomas, J.; Fleischmann, B.K.; Roeper, J.; Fakler, B. Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron 2006, 52, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.W.; Tibbs, G.R.; Picollo, A.; Abbas, S.Y.; Sanford, R.L.; Accardi, A.; Hofmann, F.; Ludwig, A.; Goldstein, P.A. PIP2-mediated HCN3 channel gating is crucial for rhythmic burst firing in thalamic intergeniculate leaflet neurons. J. Neurosci. 2011, 31, 10412–10423. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.E.; Zagotta, W.N. Molecular mechanism underlying phosphatidylinositol 4,5-bisphosphate-induced inhibition of SpIH channels. J. Biol. Chem. 2011, 286, 15535–15542. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.G.; Lu, Z.; Pan, Z.; Cohen, I.S. Tyrosine kinase inhibition differentially regulates heterologously expressed HCN channels. Pflugers Arch. 2004, 447, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Eckert, C.; Yuan, H.; Wahl-Schott, C.; Abicht, H.; Fang, L.; Li, R.; Mistrik, P.; Gerstner, A.; Much, B.; et al. A novel mechanism of modulation of hyperpolarization-activated cyclic nucleotide-gated channels by Src kinase. J. Biol. Chem. 2005, 280, 34224–34232. [Google Scholar] [CrossRef] [PubMed]
- Arinsburg, S.S.; Cohen, I.S.; Yu, H.G. Constitutively active Src tyrosine kinase changes gating of HCN4 channels through direct binding to the channel proteins. J. Cardiovasc. Pharmacol. 2006, 47, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Zhang, Q.; Teng, B.; Mustafa, S.J.; Huang, J.Y.; Yu, H.G. Src tyrosine kinase alters gating of hyperpolarization-activated HCN4 pacemaker channel through Tyr531. Am. J. Physiol. Cell Physiol. 2008, 294, C355–C362. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Layh, B.; Ludwig, A. Novel insights into the distribution of cardiac HCN channels: An expression study in the mouse heart. J. Mol. Cell. Cardiol. 2011, 51, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.; Krause, S.C.; Hassan, S.I.; Becirovic, E.; Auer, F.; Bernard, R.; Kupatt, C.; Lange, P.; Ziegler, T.; Wotjak, C.T.; et al. Sick sinus syndrome in HCN1-deficient mice. Circulation 2013, 128, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.; Mader, R.; Scharr, A.; Paparizos, C.; Cao-Ehlker, X.; Michalakis, S.; Shaltiel, L.; Weidinger, M.; Stieber, J.; Feil, S.; et al. HCN3 contributes to the ventricular action potential waveform in the murine heart. Circ. Res. 2011, 109, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Biel, M.; Hofmann, F.; Ludwig, A. Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol. Chem. 1999, 380, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Shigemoto, R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J. Comp. Neurol. 2004, 471, 241–276. [Google Scholar] [CrossRef] [PubMed]
- Much, B.; Wahl-Schott, C.; Zong, X.; Schneider, A.; Baumann, L.; Moosmang, S.; Ludwig, A.; Biel, M. Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 2003, 278, 43781–43786. [Google Scholar] [CrossRef] [PubMed]
- Brewster, A.L.; Bernard, J.A.; Gall, C.M.; Baram, T.Z. Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol. Dis. 2005, 19, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.Y.; Ying, S.W.; Goldstein, P.A. Compartmental distribution of hyperpolarization-activated cyclic-nucleotide-gated channel 2 and hyperpolarization-activated cyclic-nucleotide-gated channel 4 in thalamic reticular and thalamocortical relay neurons. Neuroscience 2006, 141, 1811–1825. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, G.M.; Angoli, D.; Nazzari, H.; Shigemoto, R.; Accili, E.A. HCN2 and HCN4 isoforms self-assemble and co-assemble with equal preference to form functional pacemaker channels. J. Biol. Chem. 2007, 282, 22900–22909. [Google Scholar] [CrossRef] [PubMed]
- Zolles, G.; Wenzel, D.; Bildl, W.; Schulte, U.; Hofmann, A.; Muller, C.S.; Thumfart, J.O.; Vlachos, A.; Deller, T.; Pfeifer, A.; et al. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron 2009, 62, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.S.; Vaidya, S.P.; Blaiss, C.A.; Liu, Z.; Stoub, T.R.; Brager, D.H.; Chen, X.; Bender, R.A.; Estep, C.M.; Popov, A.B.; et al. Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice. J. Neurosci. 2011, 31, 7424–7440. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.F.; Malleret, G.; Lee, K.H.; Gibbs, E.; Dudman, J.T.; Santoro, B.; Yin, D.; Thompson, R.F.; Siegelbaum, S.A.; Kandel, E.R.; et al. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar purkinje cells. Cell 2003, 115, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.F.; Malleret, G.; Dudman, J.T.; Buhl, D.L.; Santoro, B.; Gibbs, E.; Vronskaya, S.; Buzsaki, G.; Siegelbaum, S.A.; Kandel, E.R.; et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 2004, 119, 719–732. [Google Scholar] [PubMed]
- Ludwig, A.; Budde, T.; Stieber, J.; Moosmang, S.; Wahl, C.; Holthoff, K.; Langebartels, A.; Wotjak, C.; Munsch, T.; Zong, X.; et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003, 22, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.K.; Shin, M.; Jaramillo, T.C.; Leibel, R.L.; LeDuc, C.A.; Fischer, S.G.; Tzilianos, E.; Gheith, A.A.; Lewis, A.S.; Chetkovich, D.M. Absence epilepsy in apathetic, a spontaneous mutant mouse lacking the h channel subunit, HCN2. Neurobiol. Dis. 2009, 33, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Institute of Pharmacology and Toxicology, Technische Universität München, München, Germany. Unpublished observations. 2000.
- Stieber, J.; Herrmann, S.; Feil, S.; Loster, J.; Feil, R.; Biel, M.; Hofmann, F.; Ludwig, A. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc. Natl. Acad. Sci. USA 2003, 100, 15235–15240. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Stieber, J.; Stockl, G.; Hofmann, F.; Ludwig, A. HCN4 provides a “depolarization reserve” and is not required for heart rate acceleration in mice. EMBO J. 2007, 26, 4423–4432. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, E.; Stieber, J.; Herrmann, S.; Feil, S.; Tybl, E.; Hofmann, F.; Feil, R.; Ludwig, A. Tamoxifen-inducible gene deletion in the cardiac conduction system. J. Mol. Cell. Cardiol. 2008, 45, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene HCN4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Hofmann, F.; Stieber, J.; Ludwig, A. HCN channels in the heart: Lessons from mouse mutants. Br. J. Pharmacol. 2012, 166, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Mesirca, P.; Alig, J.; Torrente, A.G.; Muller, J.C.; Marger, L.; Rollin, A.; Marquilly, C.; Vincent, A.; Dubel, S.; Bidaud, I.; et al. Cardiac arrhythmia induced by genetic silencing of “funny” (f) channels is rescued by GIRK4 inactivation. Nat. Commun. 2014, 5, 4664. [Google Scholar] [CrossRef]
- Shi, W.; Wymore, R.; Yu, H.; Wu, J.; Wymore, R.T.; Pan, Z.; Robinson, R.B.; Dixon, J.E.; McKinnon, D.; Cohen, I.S. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ. Res. 1999, 85, e1–e6. [Google Scholar] [CrossRef]
- Marionneau, C.; Couette, B.; Liu, J.; Li, H.; Mangoni, M.E.; Nargeot, J.; Lei, M.; Escande, D.; Demolombe, S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 2005, 562, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Stillitano, F.; Lonardo, G.; Zicha, S.; Varro, A.; Cerbai, E.; Mugelli, A.; Nattel, S. Molecular basis of funny current (If) in normal and failing human heart. J. Mol. Cell. Cardiol. 2008, 45, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Cerbai, E.; Barbieri, M.; Mugelli, A. Characterization of the hyperpolarization-activated current, If, in ventricular myocytes isolated from hypertensive rats. J. Physiol. 1994, 481, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Cerbai, E.; Barbieri, M.; Mugelli, A. Occurrence and properties of the hyperpolarization-activated current If in ventricular myocytes from normotensive and hypertensive rats during aging. Circulation 1996, 94, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Cerbai, E.; Pino, R.; Porciatti, F.; Sani, G.; Toscano, M.; Maccherini, M.; Giunti, G.; Mugelli, A. Characterization of the hyperpolarization-activated current, If, in ventricular myocytes from human failing heart. Circulation 1997, 95, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, U.C.; Jansen, E.; Sudkamp, M.; Beuckelmann, D.J. Hyperpolarization-activated inward current in ventricular myocytes from normal and failing human hearts. Circulation 1998, 97, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Stilli, D.; Sgoifo, A.; Macchi, E.; Zaniboni, M.; De Iasio, S.; Cerbai, E.; Mugelli, A.; Lagrasta, C.; Olivetti, G.; Musso, E. Myocardial remodeling and arrhythmogenesis in moderate cardiac hypertrophy in rats. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H142–H150. [Google Scholar] [PubMed]
- Fernandez-Velasco, M.; Goren, N.; Benito, G.; Blanco-Rivero, J.; Bosca, L.; Delgado, C. Regional distribution of hyperpolarization-activated current (If) and hyperpolarization-activated cyclic nucleotide-gated channel mRNA expression in ventricular cells from control and hypertrophied rat hearts. J. Physiol. 2003, 553, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Fabritz, L.; Stieber, J.; Schmitt, J.; Kirchhof, P.; Ludwig, A.; Herrmann, S. Ventricular HCN channels decrease the repolarization reserve in the hypertrophic heart. Cardiovasc. Res. 2012, 95, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Giles, W.R. Supraventricular pacemaker activity in the canine heart: contributions from HCN channels in control conditions and in a model of heart failure. Cardiovasc. Res. 2005, 66, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; van Ginneken, A.C.; Wilders, R. Pacemaker activity of the human sinoatrial node: role of the hyperpolarization-activated current, If. Int. J. Cardiol. 2009, 132, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.; Krause, S.; Biel, M.; Wahl-Schott, C. The role of HCN channels in ventricular repolarization. Trends Cardiovasc. Med. 2011, 21, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Michael, G.; Xiao, L.; Qi, X.Y.; Dobrev, D.; Nattel, S. Remodelling of cardiac repolarization: How homeostatic responses can lead to arrhythmogenesis. Cardiovasc. Res. 2009, 81, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Brunet, S.; Flagg, T.P.; Pilgram, T.K.; Demolombe, S.; Nerbonne, J.M. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ. Res. 2008, 102, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Kuwahara, K.; Takano, M.; Kinoshita, H.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Igata, S.; Usami, S.; Minami, T.; et al. Increased expression of HCN channels in the ventricular myocardium contributes to enhanced arrhythmicity in mouse failing hearts. J. Am. Heart Assoc. 2013, 2, e000150. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Saito, Y.; Takano, M.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Takahashi, N.; Adachi, Y.; Takemura, G.; Horie, M.; et al. NRSF regulates fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003, 22, 6310–6321. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.; Barbier, S.; Chagraoui, A.; Richard, V.; Henry, J.P.; Lallemand, F.; Renet, S.; Lerebours, G.; Mahlberg-Gaudin, F.; Thuillez, C. Long-term heart rate reduction induced by the selective If current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation 2004, 109, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Maczewski, M.; Mackiewicz, U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc. Res. 2008, 79, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Doesch, A.O.; Ammon, K.; Konstandin, M.; Celik, S.; Kristen, A.; Frankenstein, L.; Buss, S.; Hardt, S.; Sack, F.U.; Katus, H.A.; et al. Heart rate reduction for 12 months with ivabradine reduces left ventricular mass in cardiac allograft recipients. Transplantation 2009, 88, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Milliez, P.; Messaoudi, S.; Nehme, J.; Rodriguez, C.; Samuel, J.L.; Delcayre, C. Beneficial effects of delayed ivabradine treatment on cardiac anatomical and electrical remodeling in rat severe chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H435–H441. [Google Scholar] [CrossRef] [PubMed]
- Ceconi, C.; Comini, L.; Suffredini, S.; Stillitano, F.; Bouly, M.; Cerbai, E.; Mugelli, A.; Ferrari, R. Heart rate reduction with ivabradine prevents the global phenotype of left ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H366–H373. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; O’Meara, E.; Komajda, M.; Bohm, M.; Borer, J.S.; Ford, I.; Tavazzi, L.; Swedberg, K.; Investigators, S. Effects of selective heart rate reduction with ivabradine on left ventricular remodelling and function: Results from the SHIFT echocardiography substudy. Eur. Heart J. 2011, 32, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Sartiani, L.; Stillitano, F.; Cerbai, E.; Mugelli, A. Electrophysiologic changes in heart failure: Focus on pacemaker channels. Can. J. Physiol. Pharmacol. 2009, 87, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Westbrook, G.L. A voltage-clamp analysis of inward (anomalous) rectification in mouse spinal sensory ganglion neurones. J. Physiol. 1983, 340, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.; Difrancesco, D. Voltage-clamp investigations of membrane currents underlying pace-maker activity in rabbit sino-atrial node. J. Physiol. 1980, 308, 331–351. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, K.; Irisawa, H. Inward current activated during hyperpolarization in the rabbit sinoatrial node cell. Pflugers Arch. 1980, 385, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Scroggs, R.S.; Todorovic, S.M.; Anderson, E.G.; Fox, A.P. Variation in IH, IIR, and ILEAK between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Neurophysiol. 1994, 71, 271–279. [Google Scholar] [PubMed]
- Luo, L.; Chang, L.; Brown, S.M.; Ao, H.; Lee, D.H.; Higuera, E.S.; Dubin, A.E.; Chaplan, S.R. Role of peripheral hyperpolarization-activated cyclic nucleotide-modulated channel pacemaker channels in acute and chronic pain models in the rat. Neuroscience 2007, 144, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Kouranova, E.V.; Strassle, B.W.; Ring, R.H.; Bowlby, M.R.; Vasilyev, D.V. Hyperpolarization-activated cyclic nucleotide-gated channel mRNA and protein expression in large versus small diameter dorsal root ganglion neurons: Correlation with hyperpolarization-activated current gating. Neuroscience 2008, 153, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Smith, T.; Sathish, J.; Djouhri, L. Chronic inflammatory pain is associated with increased excitability and hyperpolarization-activated current (Ih) in C- but not Aδ-nociceptors. Pain 2012, 153, 900–914. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; McMullan, S.; Djouhri, L.; Gao, L.; Watkins, R.; Berry, C.; Dempsey, K.; Lawson, S.N. HCN1 and HCN2 in Rat DRG neurons: Levels in nociceptors and non-nociceptors, NT3-dependence and influence of CFA-induced skin inflammation on HCN2 and NT3 expression. PLoS One 2012, 7, e50442. [Google Scholar] [CrossRef] [PubMed]
- Chaplan, S.R.; Guo, H.Q.; Lee, D.H.; Luo, L.; Liu, C.; Kuei, C.; Velumian, A.A.; Butler, M.P.; Brown, S.M.; Dubin, A.E. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J. Neurosci. 2003, 23, 1169–1178. [Google Scholar] [PubMed]
- Tu, H.; Deng, L.; Sun, Q.; Yao, L.; Han, J.S.; Wan, Y. Hyperpolarization-activated, cyclic nucleotide-gated cation channels: Roles in the differential electrophysiological properties of rat primary afferent neurons. J. Neurosci. Res. 2004, 76, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Antal, M.; Papp, I.; Bahaerguli, N.; Veress, G.; Vereb, G. Expression of hyperpolarization-activated and cyclic nucleotide-gated cation channel subunit 2 in axon terminals of peptidergic nociceptive primary sensory neurons in the superficial spinal dorsal horn of rats. Eur. J. Neurosci. 2004, 19, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Q.; Xing, G.G.; Wang, S.L.; Tu, H.Y.; Chi, Y.N.; Li, J.; Liu, F.Y.; Han, J.S.; Wan, Y. Axonal accumulation of hyperpolarization-activated cyclic nucleotide-gated cation channels contributes to mechanical allodynia after peripheral nerve injury in rat. Pain 2008, 137, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Schnorr, S.; Eberhardt, M.; Kistner, K.; Rajab, H.; Kasser, J.; Hess, A.; Reeh, P.; Ludwig, A.; Herrmann, S. HCN2 channels account for mechanical (but not heat) hyperalgesia during long-standing inflammation. Pain 2014, 155, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Momin, A.; Cadiou, H.; Mason, A.; McNaughton, P.A. Role of the hyperpolarization-activated current Ih in somatosensory neurons. J. Physiol. 2008, 586, 5911–5929. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.L.; McMullan, S.; Djouhri, L.; Acosta, C.; Harper, A.A.; Lawson, S.N. Expression and properties of hyperpolarization-activated current in rat dorsal root ganglion neurons with known sensory function. J. Physiol. 2012, 590, 4691–705. [Google Scholar] [CrossRef] [PubMed]
- Emery, E.C.; Young, G.T.; Berrocoso, E.M.; Chen, L.; McNaughton, P.A. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 2011, 333, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Donnelly, D.F.; Ma, C.; LaMotte, R.H. Upregulation of the hyperpolarization-activated cation current after chronic compression of the dorsal root ganglion. J. Neurosci. 2003, 23, 2069–2074. [Google Scholar] [PubMed]
- Papp, I.; Hollo, K.; Antal, M. Plasticity of hyperpolarization-activated and cyclic nucleotid-gated cation channel subunit 2 expression in the spinal dorsal horn in inflammatory pain. Eur. J. Neurosci. 2010, 32, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Pian, P.; Bucchi, A.; Decostanzo, A.; Robinson, R.B.; Siegelbaum, S.A. Modulation of cyclic nucleotide-regulated HCN channels by PIP2 and receptors coupled to phospholipase C. Pflugers Arch. 2007, 455, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Zhou, Y. Novel role of KT5720 on regulating hyperpolarization-activated cyclic nucleotide-gated channel activity and dorsal root ganglion neuron excitability. DNA Cell Biol. 2013, 32, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Ingram, S.L.; Williams, J.T. Modulation of the hyperpolarization-activated current (Ih) by cyclic nucleotides in guinea-pig primary afferent neurons. J. Physiol. 1996, 492, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.G.; Mar, L.P.; Vysokanov, A.V.; Arnold, P.B.; Cardenas, L.M.; Surmeier, D.J.; Scroggs, R.S. Serotonergic modulation of hyperpolarization-activated current in acutely isolated rat dorsal root ganglion neurons. J. Physiol. 1999, 518, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Weinreich, D. Substance P regulates Ih via a NK-1 receptor in vagal sensory neurons of the ferret. J. Neurophysiol. 1998, 79, 769–777. [Google Scholar] [PubMed]
- Dalle, C.; Eisenach, J.C. Peripheral block of the hyperpolarization-activated cation current (Ih) reduces mechanical allodynia in animal models of postoperative and neuropathic pain. Reg. Anesth. Pain Med. 2005, 30, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Takasu, K.; Ono, H.; Tanabe, M. Spinal hyperpolarization-activated cyclic nucleotide-gated cation channels at primary afferent terminals contribute to chronic pain. Pain 2010, 151, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Papp, I.; Szucs, P.; Hollo, K.; Erdelyi, F.; Szabo, G.; Antal, M. Hyperpolarization-activated and cyclic nucleotide-gated cation channel subunit 2 ion channels modulate synaptic transmission from nociceptive primary afferents containing substance P to secondary sensory neurons in laminae I-IIo of the rodent spinal dorsal horn. Eur. J. Neurosci. 2006, 24, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Aradi, I.; Santhakumar, V.; Soltesz, I. H-channels in epilepsy: New targets for seizure control? Trends Pharmacol. Sci. 2002, 23, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Chevaleyre, V.; Castillo, P.E. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc. Natl. Acad. Sci. USA 2002, 99, 9538–9543. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Vasilyev, D.; Lu, P.; Cummons, T.; Bowlby, M.R. Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and pain. Curr. Pharm. Des. 2009, 15, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Young, G.T.; Emery, E.C.; Mooney, E.R.; Tsantoulas, C.; McNaughton, P.A. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain 2014, 155, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Reinold, H.; Ahmadi, S.; Depner, U.B.; Layh, B.; Heindl, C.; Hamza, M.; Pahl, A.; Brune, K.; Narumiya, S.; Muller, U.; et al. Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. J. Clin. Investig. 2005, 115, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Wurmsee, S.; Herrmann, S.; Institut für Experimentelle und Klinische Pharmakologie und Toxikologie, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91054 Erlangen. Unpublished work. 2013.
- Abrahamsen, B.; Zhao, J.; Asante, C.O.; Cendan, C.M.; Marsh, S.; Martinez-Barbera, J.P.; Nassar, M.A.; Dickenson, A.H.; Wood, J.N. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 2008, 321, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.J.; McIlwrath, S.L.; Woodbury, C.J.; Davis, B.M.; Koerber, H.R. TRPV1 unlike TRPV2 is restricted to a subset of mechanically insensitive cutaneous nociceptors responding to heat. J. Pain 2008, 9, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, D.J.; Lee, H.; Lo, L.; Shields, S.D.; Zylka, M.J.; Basbaum, A.I.; Anderson, D.J. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc. Natl. Acad. Sci. USA 2009, 106, 9075–9080. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Ford, I.; Tendera, M.; Bourassa, M.G.; Fox, K.; Investigators, I. Efficacy of ivabradine, a new selective If inhibitor, compared with atenolol in patients with chronic stable angina. Eur. Heart J. 2005, 26, 2529–2536. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Ponikowski, P.; Kahan, T.; Investigators, A.S. Efficacy of the If current inhibitor ivabradine in patients with chronic stable angina receiving β-blocker therapy: A 4-month, randomized, placebo-controlled trial. Eur. Heart J. 2009, 30, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Werdan, K.; Ebelt, H.; Nuding, S.; Hopfner, F.; Hack, G.; Muller-Werdan, U. Ivabradine in combination with beta-blocker improves symptoms and quality of life in patients with stable angina pectoris: results from the ADDITIONS study. Clin. Res. Cardiol. 2012, 101, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Ferrari, R.; Investigators, B. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Robertson, M.; Ferrari, R.; investigators, B. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A subgroup analysis of a randomised controlled trial. Lancet 2008, 372, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.; Ford, I.; Steg, P.G.; Tardif, J.C.; Tendera, M.; Ferrari, R.; Investigators, S. Ivabradine in stable coronary artery disease without clinical heart failure. N. Engl. J. Med. 2014, 371, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, K.; Komajda, M.; Boehm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R. Ivabradine in heart failure—No paradigm SHIFT yet. Lancet 2010, 376, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, L.; Demontis, G.C.; Gargini, C. Cellular mechanisms underlying the pharmacological induction of phosphenes. Br. J. Pharmacol. 2007, 150, 383–390. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, S.; Schnorr, S.; Ludwig, A. HCN Channels—Modulators of Cardiac and Neuronal Excitability. Int. J. Mol. Sci. 2015, 16, 1429-1447. https://doi.org/10.3390/ijms16011429
Herrmann S, Schnorr S, Ludwig A. HCN Channels—Modulators of Cardiac and Neuronal Excitability. International Journal of Molecular Sciences. 2015; 16(1):1429-1447. https://doi.org/10.3390/ijms16011429
Chicago/Turabian StyleHerrmann, Stefan, Sabine Schnorr, and Andreas Ludwig. 2015. "HCN Channels—Modulators of Cardiac and Neuronal Excitability" International Journal of Molecular Sciences 16, no. 1: 1429-1447. https://doi.org/10.3390/ijms16011429
APA StyleHerrmann, S., Schnorr, S., & Ludwig, A. (2015). HCN Channels—Modulators of Cardiac and Neuronal Excitability. International Journal of Molecular Sciences, 16(1), 1429-1447. https://doi.org/10.3390/ijms16011429