FOXO1, TGF-β Regulation and Wound Healing

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammation
3. Matrix Metalloproteinases
4. Oxidative Stress and Wound Healing
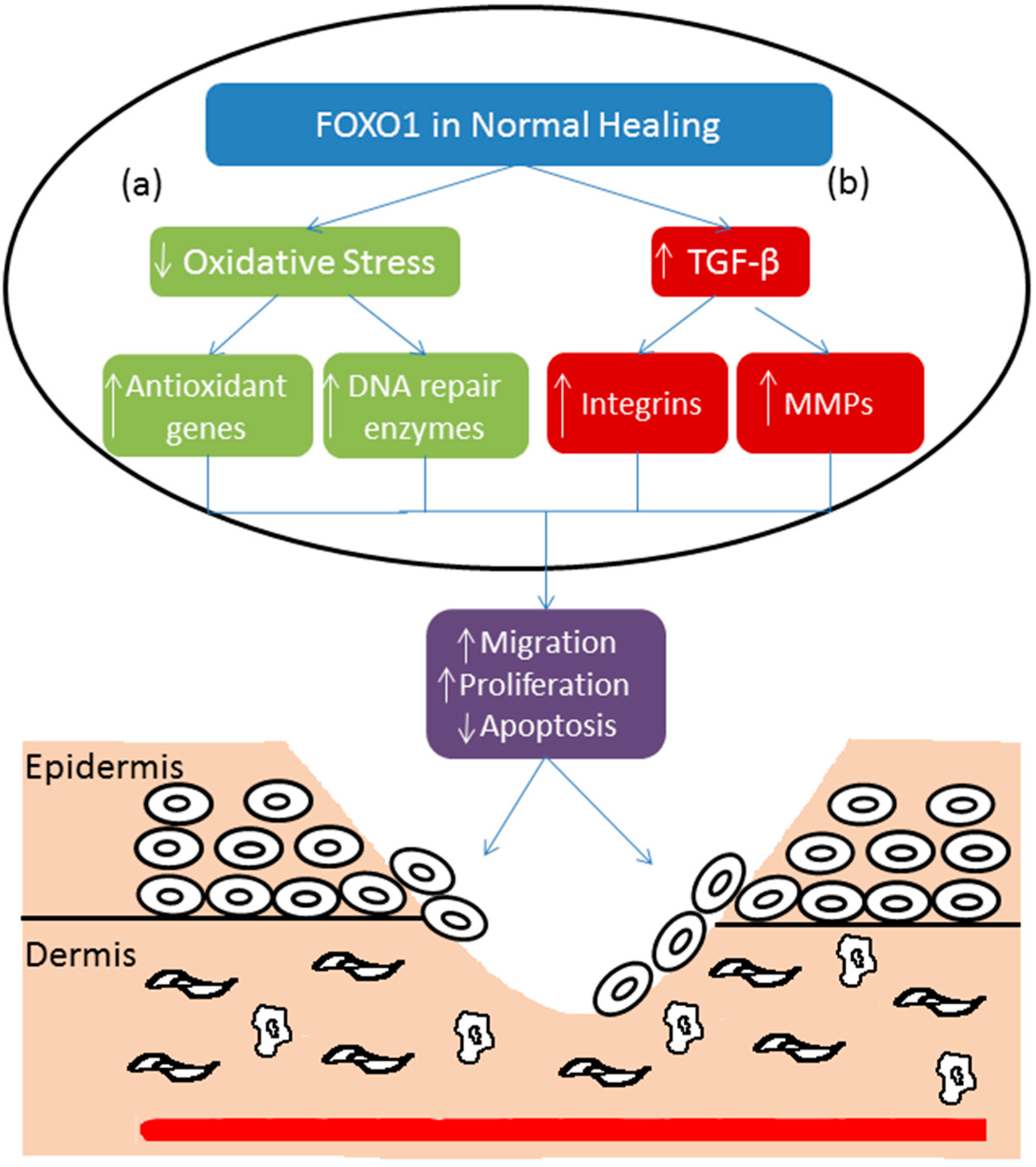
5. Forkhead BoxO-1 (FOXO1) and Re-Epithelialization
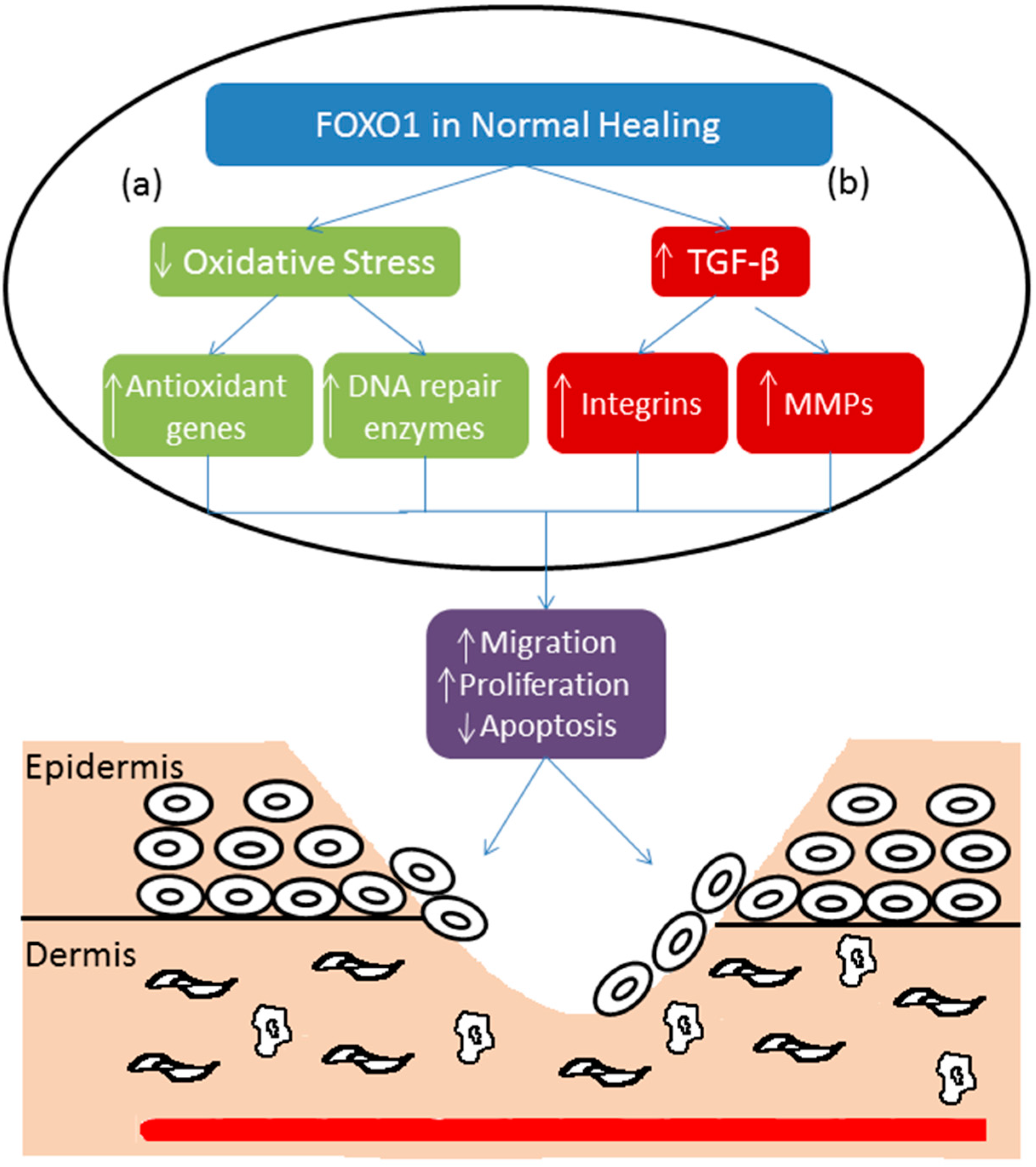
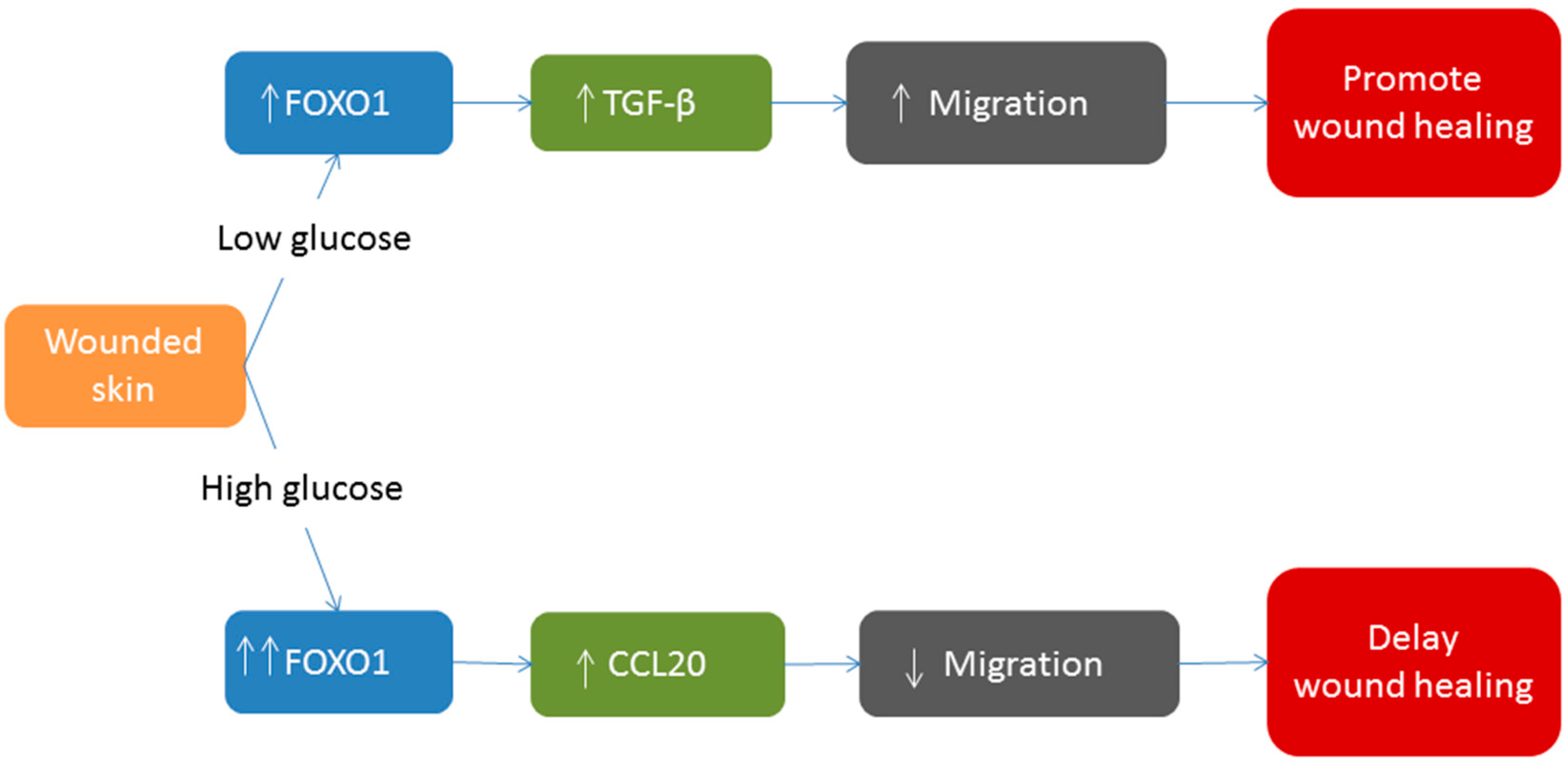
6. FOXO1 and Diabetic Wound Healing
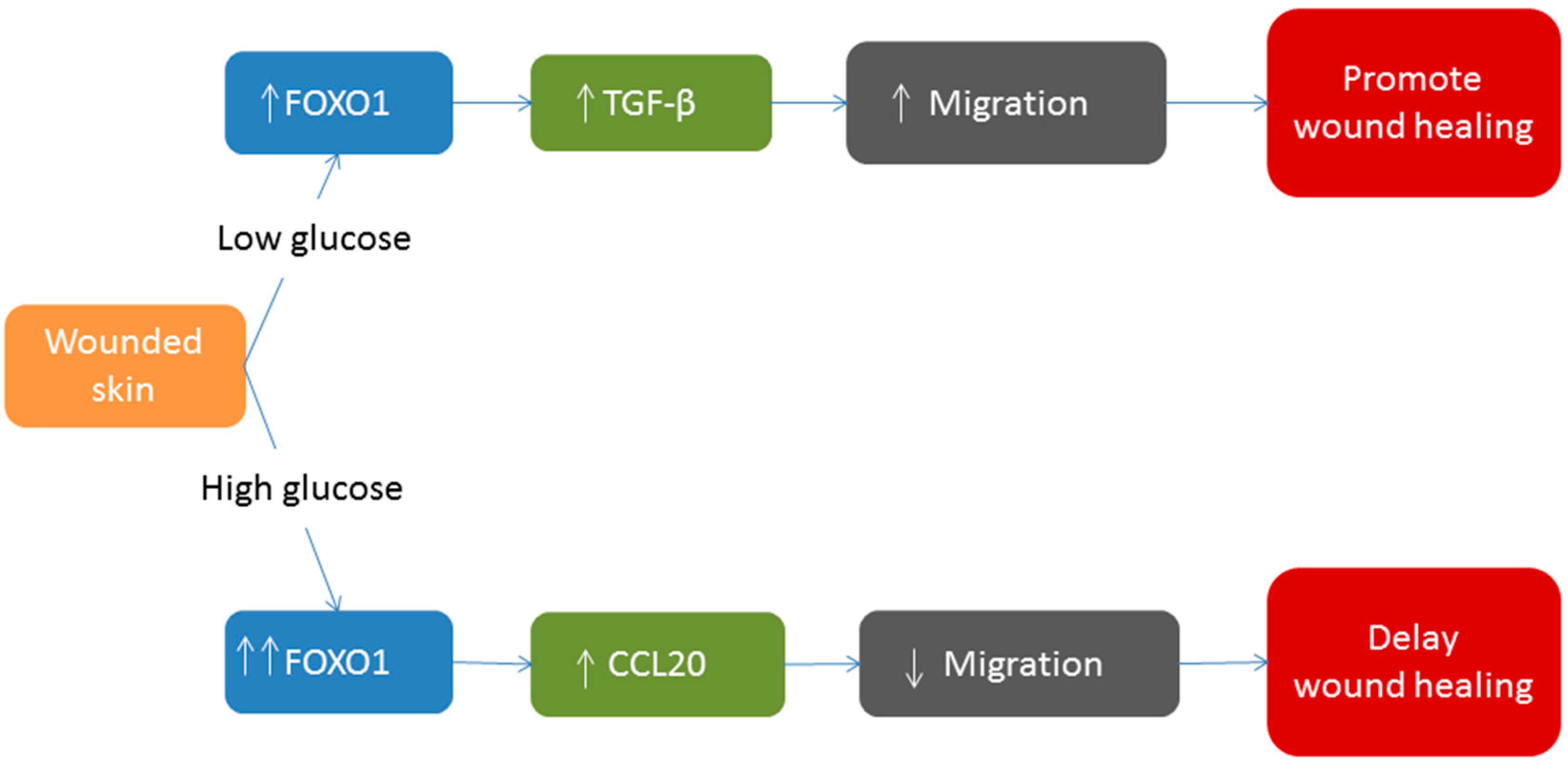
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reinke, J.M.; Sorg, H. Woud repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Nguyen, D.T.; Pietramaggiori, G.; Scherer, S.; Chen, B.; Zhan, Q.; Ogawa, R.; Yannas, I.V.; Wagers, A.J.; Orgill, D.P.; et al. Use of the parabiotic model in studies of cutaneous wound healing to define the participation of circulating cells. Int. J. Tissue Repair Regen. 2010, 18, 426–432. [Google Scholar]
- Fathke, C.; Wilson, L.; Hutter, J.; Kapoor, V.; Smith, A.; Hocking, A.; Isik, F. Contribution of bone marrow-derived cells to skin: Collagen deposition and wound repair. Stem Cells 2004, 22, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Mercandetti, M.; Cohen, A.J. Wound Healing and Repair. MedScape. Available online: http://emedicine.medscape.com/article/1298129-overview#aw2aab6b6 (accessed on 28 February 2014).
- Wu, Y.S.; Chen, S.N. Apoptotic cell: Linkage of inflammation and wound healing. Front. Pharmacol. 2014, 5, 1–6. [Google Scholar] [PubMed]
- Martin, P.; Leibovich, S.J. Inflammatory cells during wound repair: The good, the bad and the ugly. Trends Cell Biol. 2005, 154, 599–607. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, C.; Graves, D.T. Abnormal cell responses and role of TNF-α in impaired diabetic wound healing. BioMed Res. Int. 2013, 2013, 1–9. [Google Scholar]
- Plikus, M.V.; Gay, D.L.; Treffeisen, E.; Wang, A.; Supapannachart, R.J.; Cotsarelis, G. Epithelial stem cells and implications for wound repair. Semin. Cell Dev. Biol. 2012, 23, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Liu, Y.; Yang, Z.; Nguyen, J.; Liang, F.; Morris, R.J.; Cotsarelis, G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 2005, 11, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.L.; Caley, M.; O’Toole, E.A. Matrix metalloproteinases and epidermal wound repair. Cell Tissue Res. 2013, 351, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Salonurmi, T.; Parikka, M.; Kontusaari, S.; Pirila, E.; Munaut, C.; Salo, T.; Tryggvason, K. Over-expression of TIMP-1 under the MMP-9 promoter interferes with wound healing in transgenic mice. Cell Tissue Res. 2004, 315, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kyriakides, T.R.; Wulsin, D.; Skokos, E.A.; Fleckman, P.; Pirrone, A.; Shipley, J.M.; Senior, R.M.; Bornstein, P. Mice that lack matrix metalloproteinase-9 display delayed wound healing associated with delayed reepithelization and disordered collagen fibrillogenesis. Matrix Biol. 2009, 28, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Koppel, A.C.; Kiss, A.; Hindes, A.; Burns, C.J.; Marmer, B.L.; Goldberg, G.; Blumenberg, M.; Efimova, T. Delayed skin wound repair in proline-rich protein tyrosine kinase 2 (Pyk2) knockout mice. Am. J. Physiol. Cell Physiol. 2014, 306, C899–C909. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Wu, Y.; Badadani, M. Pyk2 contributes to reepithelialization by promoting MMP expression. Focus on “Delayed skin wound repair in proline-rich protein tyrosine kinase 2 knockout mice”. Am. J. Physiol. Cell Physiol. 2014, 306, C887–C888. [Google Scholar]
- Ponugoti, B.; Dong, G.; Graves, D.T. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp. Diabetes Res. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Moseley, R.; Hilton, J.R.; Waddington, R.J.; Harding, K.G.; Stephens, P.; Thomas, D.W. Comparison of oxidative stress biomarker profiles between acute and chronic wound environments. Wound Repair Regen. 2004, 12, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Scharffetter-Kochanek, K. Oxidative stress in chronic venous leg ulcers. Wound Repair Regen. 2005, 13, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar]
- Soneja, A.; Drews, M.; Malinski, T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol. Rep. 2005, 57, 108–119. [Google Scholar] [PubMed]
- Deveci, M.; Gilmont, R.R.; Dunham, W.R.; Mudge, B.P.; Smith, D.J.; Marcelo, C.L. Glutathione enhances fibroblast collagen contraction and protects keratinocytes from apoptosis in hyperglycaemic culture. Br. J. Dermatol. 2005, 152, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.V.; Raffetto, J.D.; Phillips, T.; Menzoian, J.O.; Park, H.Y. The proliferative capacity of neonatal skin fibroblasts is reduced after exposure to venous ulcer wound fluid: A potential mechanism for senescence in venous ulcers. J. Vasc. Surg. 1999, 30, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A. Oxidative stress and “senescent” fibroblasts in non-healing wounds as potential therapeutic targets.v. J. Investig. Dermatol. 2008, 128, 2361–2364. [Google Scholar]
- Li, G.; Gustafson-Brown, C.; Hanks, S.K.; Nason, K.; Arbeit, J.M.; Pogliano, K.; Wisdom, R.M.; Johnson, R.S. C-Jun is essential for organization of the epidermal leading edge. Dev. Cell 2003, 4, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Feige, J.N.; Gelman, L.; Pedrazzini, T.; Keller, H.; Desvergne, B.; Wahli, W. Selective expression of a dominant-negative form of peroxisome proliferator-activated receptor in keratinocytes leads to impaired epidermal healing. Mol. Endocrinol. 2005, 19, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Schmuth, M.; Jiang, Y.J.; Dubrac, S.; Elias, P.M.; Feingold, K.R. Thematic review series: Skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J. Lipid Res. 2008, 49, 499–509. [Google Scholar]
- Michalik, L.; Desvergne, B.; Tan, N.S.; Basu-Modak, S.; Escher, P.; Rieusset, J.; Peters, J.M.; Kaya, G.; Gonzalez, F.J.; Zakany, J.; et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. J. Cell Biol. 2001, 154, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Coffer, P.J. The extending network of FOXO transcriptional target genes. Antioxid. Redox Signal. 2011, 14, 579–592. [Google Scholar]
- Szydłowski, M.; Jabłońska, E.; Juszczyński, P. FOXO1 transcription factor: A critical effector of the PI3K-AKT axis in B-Cell development. Int. Rev. Immunol. 2014, 33, 146–157. [Google Scholar]
- Liu, P.; Kao, T.P.; Huang, H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 2008, 27, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Roupé, K.M.; Alberius, P.; Schmidtchen, A.; Sørensen, O.E. Gene expression demonstrates increased resilience toward harmful inflammatory stimuli in the proliferating epidermis of human skin wounds. Exp. Dermatol. 2010, 19, 329–332. [Google Scholar]
- Ponugoti, B.; Xu, F.; Zhang, C.; Tian, C.; Pacios, S.; Graves, D.T. FOXO1 promotes wound healing through the up-regulation of TGF-β1 and prevention of oxidative stress. J. Cell Biol. 2013, 203, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Alexandrow, M.G.; Moses, H.L. Transforming growth factor beta and cell cycle regulation. Cancer Res. 1995, 55, 1452–1457. [Google Scholar] [PubMed]
- Wu, Z.; Sofronic-Milosavljevic, L.J.; Nagano, I.; Takahashi, Y. Trichinella spiralis: Nurse cell formation with emphasis on analogy to muscle cell repair. Parasit Vectors 2008, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Peplow, P.V.; Chatterjee, M.P. A review of the influence of growth factors and cytokines in in vitro human keratinocyte migration. Cytokine 2013, 62, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ferrer, M.; Afshar-Sherif, A.R.; Uwamariya, C.; de Crombrugghe, B.; Davidson, J.M.; Bhowmick, N.A. Dermal transforming growth factor-beta responsiveness mediates wound contraction and epithelial closure. Am. J. Pathol. 2010, 176, 98–107. [Google Scholar]
- Roberts, A.B. Transforming growth factor-beta: Activity and efficacy in animal models of wound healing. Wound Repair Regen. 1995, 3, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Gaudino, G. Cellular and molecular facets of keratinocyte reepithelization during wound healing. Exp. Cell Res. 2005, 304, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Gailit, J.; Welch, M.P.; Clark, R.A. TGF-β1 stimulates expression of keratinocyte integrins during re-epithelialization of cutaneous wounds. J. Investig. Dermatol. 1994, 103, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Roupe, K.M.; Veerla, S.; Olson, J.; Stone, E.L.; Sorensen, O.E.; Hedrick, S.M.; Nizet, V. Transcription factor binding site analysis identifies FOXO transcription factors as regulators of the cutaneous wound healing process. PLoS One 2014, 9, e89274. [Google Scholar] [CrossRef] [PubMed]
- Mori, R.; Tanaka, K.; de Kerckhove, M.; Okamoto, M.; Kashiyama, K.; Kim, S.; Kawata, T.; Komatsu, T.; Park, S.; Ikematsu, K.; et al. Reduced FOXO1 accelerates skin wound healing and attenuates scarring. Am. J. Pathol. 2014. [Google Scholar] [CrossRef]
- Xu, F.; Othman, B.; Batres, A.; Ponugoti, B.; Zhang, C.; Lim, J.; Yi, L.; Liu, J.; Tian, C.; Hameedaldeen, A.; et al. FOXO1 inhibits diabetic mucosal wound healing but enhances healing of normoglycemic wounds. Diabetes 2014, in press. [Google Scholar]
- Walker, K.S.; Deak, M.; Paterson, A.; Hudson, K.; Cohen, P.; Alessi, D.R. Activation of protein kinase B β and γ isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: Comparison with protein kinase B α. Biochem. J. 1998, 331, 299–308. [Google Scholar] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.S.; Orena, S.J.; Rafidi, K.; Torchia, A.J.; Stock, J.L.; Hildebrandt, A.L.; Coskran, T.; Black, S.C.; Brees, D.J.; Wicks, J.R.; et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB β. J. Clin. Investig. 2003, 112, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; Depinho, R.A.; Accili, D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor FOXO1 in liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Siegel-Axel, D.I.; Ullrich, S.; Stefan, N.; Rittig, K.; Gerst, F.; Klingler, C.; Schmidt, U.; Schreiner, B.; Randrianarisoa, E.; Schaller, H.E.; et al. Fetuin-A influences vascular cell growth and production of proinflammatory and angiogenic proteins by human perivascular fat cells. Diabetologia 2014, 57, 1057–1066. [Google Scholar]
- Alikhani, M.; Roy, S.; Graves, D.T. FOXO1 plays an essential role in apoptosis of retinal pericytes. Mol. Vis. 2010, 16, 408–415. [Google Scholar] [PubMed]
- Behl, Y.; Krothapalli, P.; Desta, T.; Roy, S.; Graves, D.T. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 2009, 58, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Trudeau, K.; Behl, Y.; Dhar, S.; Chronopoulos, A. New insights into hyperglycemia-induced molecular changes in microvascular cells. J. Dent. Res. 2010, 89, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Coudriet, G.M.; Hyun Kim, D.; Lu, Y.; Perdomo, G.; Qu, S.; Slusher, S.; Tse, H.M.; Piganelli, J.; Giannoukakis, N.; et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1β production in macrophages. Diabetes 2009, 58, 2624–2633. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.C.; Wu, C.S.; Kuo, H.Y.; Huang, S.M.; Chen, G.S. Hyperglycaemic conditions hamper keratinocyte locomotion via sequential inhibition of distinct pathways: New insights on poor wound closure in patients with diabetes. Br. J. Dermatol. 2009, 160, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Desta, T.; Li, J.; Chino, T.; Graves, D.T. Altered fibroblast proliferation and apoptosis in diabetic gingival wounds. J. Dent. Res. 2010, 89, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.L.; Almeida, M.E.; Vicente-Manzanares, M.; Horwitz, A.F.; Santos, M.F. High glucose-mediated oxidative stress impairs cell migration. PLoS One 2011, 6, e22865. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, M.F.; Li, J.; Chehab, L.; Desta, T.; Chino, T.; Krothpali, N.; Behl, Y.; Alikhani, M.; Yang, J.; Braasch, C.; et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-α dysregulation and associated with enhanced activation of forkhead boxO1 (FOXO1). Diabetologia 2010, 53, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Hehenberger, K.; Hansson, A.; Heilborn, J.D.; Abdel-Halim, S.M.; Ostensson, C.G.; Brismar, K. Impaired proliferation and increased l-lactate production of dermal fibroblasts in the GK-rat, a spontaneous model of non-insulin dependent diabetes mellitus. Wound Repair Regen. 1999, 7, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Xu, F.; Dong, G.; Li, S.; Tian, C.; Ponugoti, B.; Graves, D.T. Effect of bacteria on the wound healing behavior of oral epithelial cells. PLoS One 2014, 9, e89475. [Google Scholar] [CrossRef] [PubMed]
- Delima, A.J.; Oates, T.; Assuma, R.; Schwartz, Z.; Cochran, D.; Amar, S.; Graves, D.T. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J. Clin. Periodontol. 2001, 28, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Dong, G.; Moschidis, A.; Ortiz, J.; Benakanakere, M.R.; Kinane, D.F.; Graves, D.T. P. gingivalis modulates keratinocytes through FOXO transcription factors. PLoS One 2013, 8, e78541. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Segre, J.A. Interaction of the microbiome with the innate immune response in chronic wounds. Adv. Exp. Med. Biol. 2012, 946, 55–68. [Google Scholar] [PubMed]
- Al-Mulla, F.; Leibovich, S.J.; Francis, I.M.; Bitar, M.S. Impaired TGF-β signaling and a defect in resolution of inflammation contribute to delayed wound healing in a female rat model of type 2 diabetes. Mol. Biosyst. 2011, 7, 3006–3020. [Google Scholar]
- Kaiser, G.C.; Polk, D.B. Tumor necrosis factor alpha regulates proliferation in a mouse intestinal cell line. Gastroenterology 1997, 112, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Bal, H.S.; Desta, T.; Behl, Y.; Graves, D.T. Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am. J. Pathol. 2006, 168, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Hasnan, J.; Yusof, M.I.; Damitri, T.D.; Faridah, A.R.; Adenan, A.S.; Norbaini, T.H. Relationship between apoptotic markers (Bax and Bcl-2) and biochemical markers in type 2 diabetes mellitus. Singap. Med. J. 2010, 51, 50–55. [Google Scholar]
- Rai, N.K.; Suryabhan; Ansari, M.; Kumar, M.; Shukla, V.K.; Tripathi, K. Effect of glycaemic control on apoptosis in diabetic wounds. J. Wound Care 2005, 14, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Suttles, J.; Graves, D.T.; Martin, M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FOXO1. J. Biol. Chem. 2011, 286, 44295–42305. [Google Scholar] [CrossRef] [PubMed]
- Alblowi, J.; Tian, C.; Siqueira, M.F.; Kayal, R.A.; McKenzie, E.; Behl, Y.; Gerstenfeld, L.; Einhorn, T.A.; Graves, D.T. Chemokine expression is up-regulated in chondrocytes in diabetic fracture healing. Bone 2013, 53, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Kayal, R.A.; Siqueira, M.; Alblowi, J.; McLean, J.; Krothapalli, N.; Faibish, D.; Einhorn, T.A.; Gerstenfeld, L.C.; Graves, D.T. TNF-α mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J. Bone Miner. Res. 2010, 25, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hameedaldeen, A.; Liu, J.; Batres, A.; Graves, G.S.; Graves, D.T. FOXO1, TGF-β Regulation and Wound Healing. Int. J. Mol. Sci. 2014, 15, 16257-16269. https://doi.org/10.3390/ijms150916257
Hameedaldeen A, Liu J, Batres A, Graves GS, Graves DT. FOXO1, TGF-β Regulation and Wound Healing. International Journal of Molecular Sciences. 2014; 15(9):16257-16269. https://doi.org/10.3390/ijms150916257
Chicago/Turabian StyleHameedaldeen, Alhassan, Jian Liu, Angelika Batres, Gabrielle S. Graves, and Dana T. Graves. 2014. "FOXO1, TGF-β Regulation and Wound Healing" International Journal of Molecular Sciences 15, no. 9: 16257-16269. https://doi.org/10.3390/ijms150916257
APA StyleHameedaldeen, A., Liu, J., Batres, A., Graves, G. S., & Graves, D. T. (2014). FOXO1, TGF-β Regulation and Wound Healing. International Journal of Molecular Sciences, 15(9), 16257-16269. https://doi.org/10.3390/ijms150916257