In Silico Discovery of Potential VEGFR-2 Inhibitors from Natural Derivatives for Anti-Angiogenesis Therapy

 ,
,
Abstract
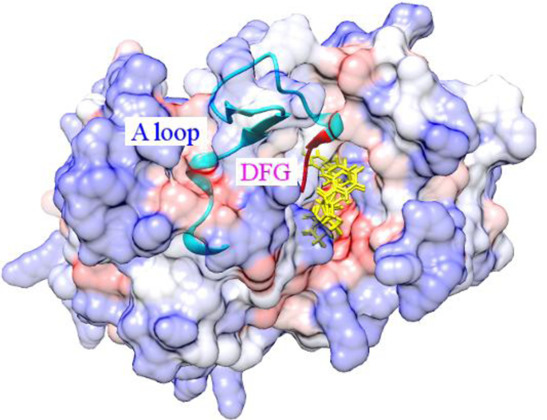
:
1. Introduction
2. Results and Discussion
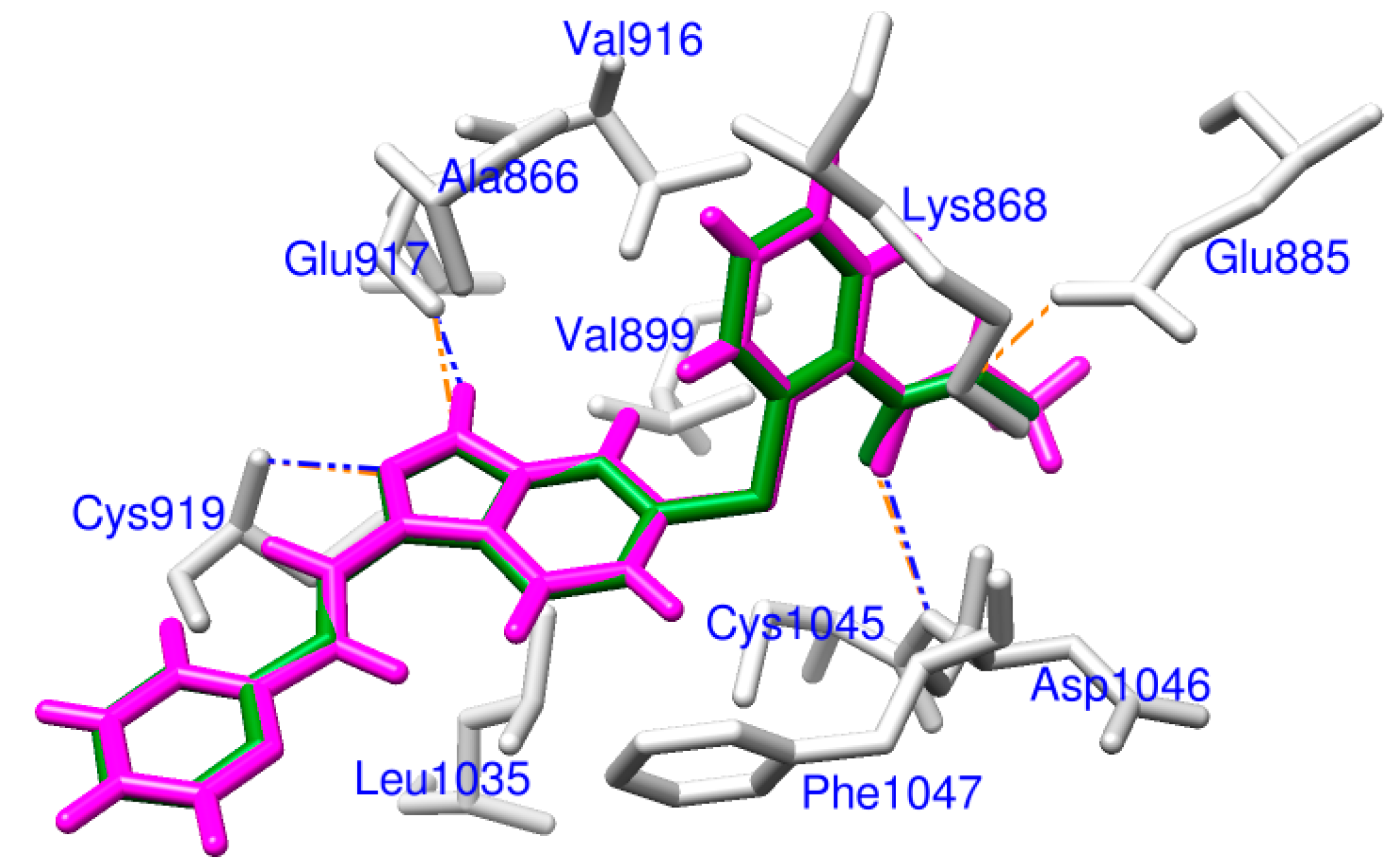
2.1. Docking Accuracy
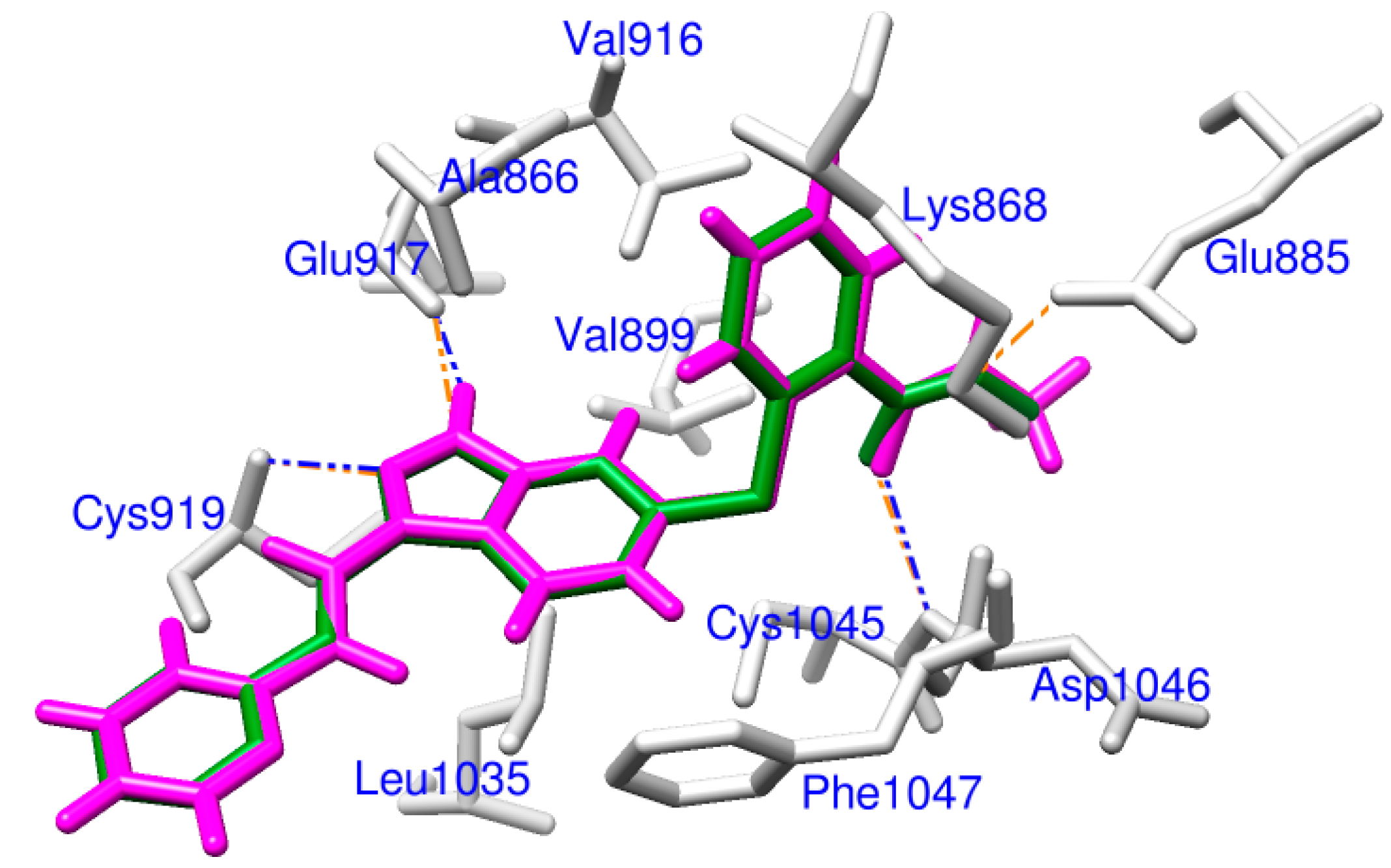
2.2. Potential VEGFR-2 Inhibitors for Anti-Angiogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Grid Score |
|---|---|---|
| ZINC08254217 |  | −63.06 |
| ZINC08254138 |  | −62.59 |
| ZINC03838680 |  | −62.34 |
| Axitinib |  | −62.11 |
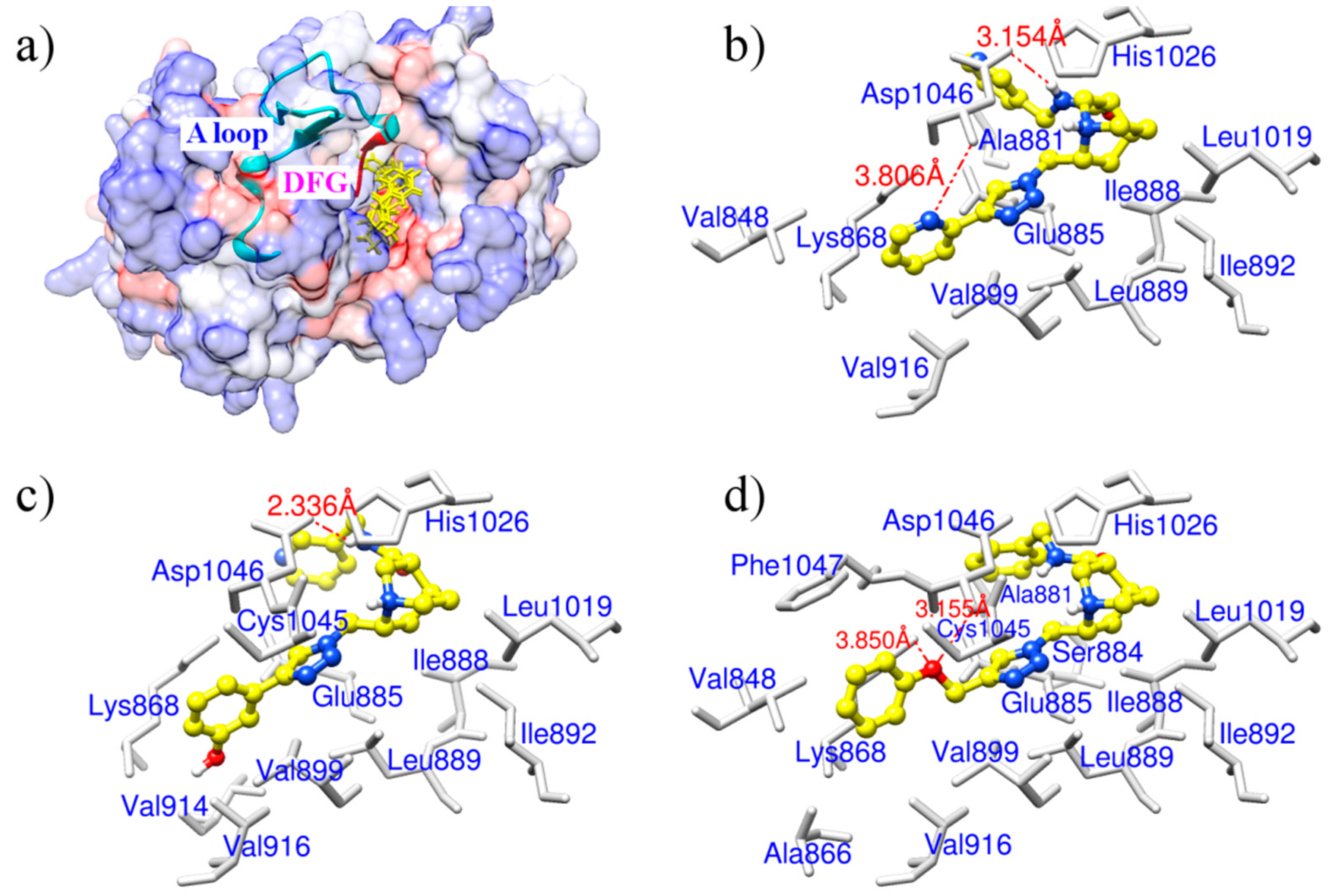
2.3. Molecular Dynamics Simulation Analysis
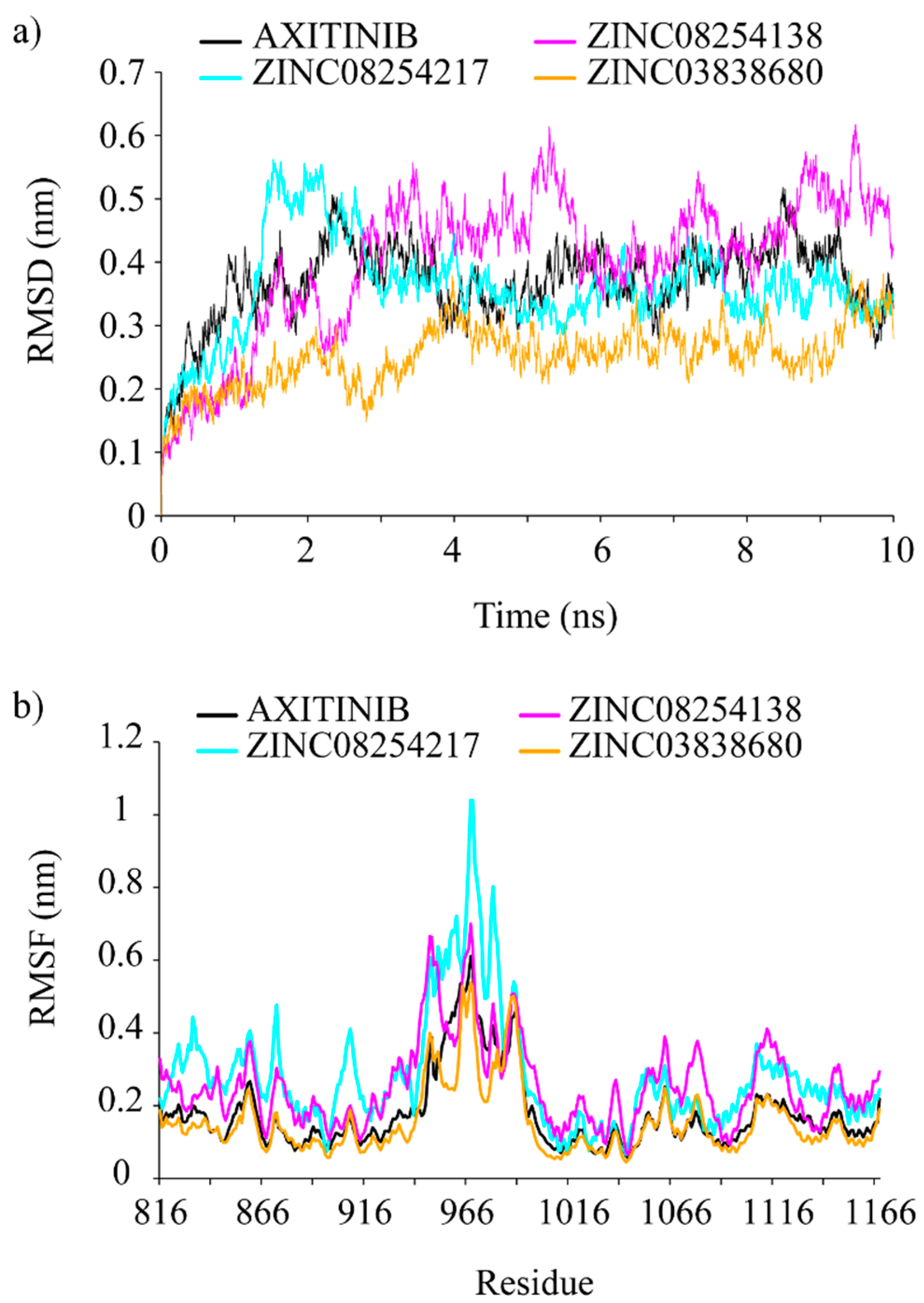
2.4. Binding Free Energy and Its Decomposition
| Terms a | AXITINIB | ZINC08254217 | ZINC08254138 | ZINC03838680 |
|---|---|---|---|---|
| ∆Eele | −36.32 ± 3.02 | −8.22 ± 13.26 | −16.24 ± 23.84 | −15 ± 10.28 |
| ∆Evdw | −56.19 ± 2.87 | −54.91 ± 2.58 | −41.37 ± 4.6 | −59.54 ± 2.88 |
| ∆Ggas | −92.51 ± 2.95 | −63.14 ± 13.4 | −57.61 ± 25.96 | −74.54 ± 10.32 |
| ∆Esurf | −6.28 ± 0.11 | −6.95 ± 0.16 | −6.04 ± 0.35 | −7.5 ± 0.23 |
| ∆EGB | 44.11 ± 1.79 | 26.76 ± 12.38 | 30.91 ± 22.35 | 32.67 ± 10.06 |
| ∆Gsolv | 37.83 ± 1.8 | 19.81 ± 12.4 | 24.88 ± 22.18 | 25.17 ± 10.12 |
| ∆Gbind | −54.68 ± 2.62 | −43.32 ± 3.36 | −32.73 ± 5.81 | −49.37 ± 3.21 |
2.5. Anti-Angiogenic Use of these VEGFR-2 Inhibitors
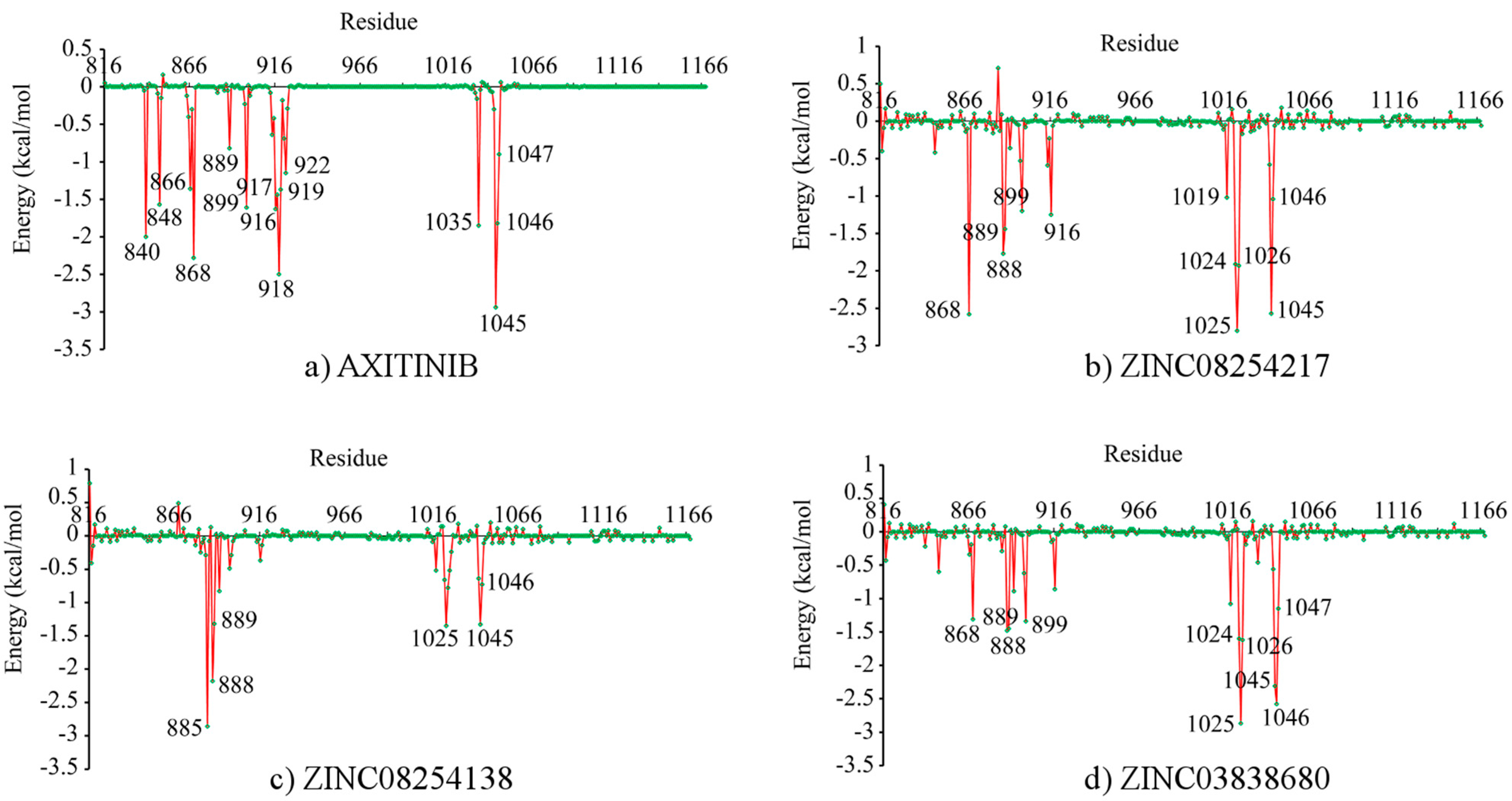
3. Experimental Section
3.1. Data Collection and Preparation
3.2. Virtual Screening
3.3. Molecular Docking
3.4. Molecular Dynamics Simulation
3.5. Energy Analysis via MM/GBSA Approach

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yoo, S.Y.; Kwon, S.M. Angiogenesis and its therapeutic opportunities. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. VEGF receptor protein–tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 375, 287–291. [Google Scholar] [CrossRef]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.-H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar]
- Seetharam, L.; Gotoh, N.; Maru, Y.; Neufeld, G.; Yamaguchi, S.; Shibuya, M. A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene 1995, 10, 135–147. [Google Scholar]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar]
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef]
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837. [Google Scholar] [CrossRef]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef]
- Hong, J. Role of natural product diversity in chemical biology. Curr. Opin. Chem. Biol. 2011, 15, 350–354. [Google Scholar] [CrossRef]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef]
- Jain, A.N. Virtual screening in lead discovery and optimization. Curr. Opin. Drug Discov. Dev. 2004, 7, 396. [Google Scholar]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Crawford, Y.; Ferrara, N. VEGF inhibition: Insights from preclinical and clinical studies. Cell Tissue Res. 2009, 335, 261–269. [Google Scholar] [CrossRef]
- Sharma, S.; Johnson, D.; Abouammoh, M.; Hollands, S.; Brissette, A. Rate of serious adverse effects in a series of bevacizumab and ranibizumab injections. Can. J. Ophthalmol. 2012, 47, 275–279. [Google Scholar] [CrossRef]
- Li, X.S.; Wu, X.; Zhao, P.J.; Huang, L.H.; Song, Y.; Gong, K.; Shen, C.; Yu, W.; Song, G.; Zhao, Z.; et al. Efficacy and safety of sunitinib in the treatment of metastatic renal cell carcinoma. Chin. Med. J. 2011, 124, 2920–2924. [Google Scholar]
- Byrne, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J. Cell. Mol. Med. 2005, 9, 777–794. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Durrant, J.D.; Amaro, R.E.; McCammon, J.A. AutoGrow: A novel algorithm for protein inhibitor design. Chem. Biol. Drug Des. 2009, 73, 168–178. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Agrawal, M.R.; Ozarkar, A.D.; Gupta, S.; Deobagkar, D.N.; Deobagkar, D.D. Comparative study of Plasmodium falciparum erythrocyte membrane protein 1-DBLα domain variants with respect to antigenic variations and docking interaction analysis with glycosaminoglycans. Mol. Biosyst. 2014, 10, 2466–2479. [Google Scholar] [CrossRef]
- Akrimah, D.; Musadad, A. Docking of potent anticancer agents; 4-(Pyrazol-4yl)-pyrimidine derivatives as selective cyclin-dependent kinase 4/6 inhibitors. Int. J. Chem. Eng. Appl. 2013, 4, 419–422. [Google Scholar]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast, C.V.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Kan, C.-C.; Villafranca, J.E.; et al. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef]
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef]
- Kumar, K.M.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol. Biosyst. 2014, 10, 891–900. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.; Jiao, Y.; Liu, H.; Yuan, H.; Lu, S.; Ran, T.; Yao, S.; Ke, Z.; Xu, J.; et al. An integrated virtual screening approach for VEGFR-2 inhibitors. J. Chem. Inf. Model. 2013, 53, 3163–3177. [Google Scholar] [CrossRef]
- Christophers, E. Psoriasis–epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320. [Google Scholar] [CrossRef]
- Papoutsaki, M.; Costanzo, A. Treatment of psoriasis and psoriatic arthritis. BioDrugs 2013, 27, 3–12. [Google Scholar] [CrossRef]
- Scottish Intercollegiate Guidelines Network. Treating Psoriasis and Psoriatic Arthritis: A Booklet for Patients and Carers. Available online: http://www.sign.ac.uk/pdf/pat121.pdf (accessed on 21 March 2014).
- Wolfe, F.; Michaud, K. Lymphoma in rheumatoid arthritis: The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheumatol. 2004, 50, 1740–1751. [Google Scholar] [CrossRef]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis drives psoriasis pathogenesis. Int. J. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef]
- Narayanan, S.; Callis-Duffin, K.; Batten, J.; Agarwal, N. Improvement of psoriasis during sunitinib therapy for renal cell carcinoma. Am. J. Med. Sci. 2010, 339, 580–581. [Google Scholar]
- Fournier, C.; Tisman, G. Sorafenib-associated remission of psoriasis in hypernephroma: Case report. Dermatol. Online J. 2010, 16, 17. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Patson, B.; B Cohen, R.; Olszanski, A.J. Pharmacokinetic evaluation of axitinib. Expert Opin. Drug Metab. Toxicol. 2011, 8, 259–270. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.-L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci.USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Axitinib. Available online: http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm289439.htm (accessed on 21 March 2014).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Cerqueira, N.; Ribeiro, J.; Fernandes, P.A.; Ramos, M.J. vsLab–An implementation for virtual high-throughput screening using AutoDock and VMD. Int. J. Quantum Chem. 2011, 111, 1208–1212. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA–small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Liu, L.; Ma, Y.; Wang, R.-L.; Xu, W.-R.; Wang, S.-Q.; Chou, K.-C. Find novel dual-agonist drugs for treating type 2 diabetes by means of cheminformatics. Drug Des. Dev. Ther. 2013, 7, 279. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar]
- Berendsen, H.J.C.; Marrink, S.-J. Molecular dynamics of water transport through membranes: Water from solvent to solute. Pure Appl. Chem. 1993, 65, 2513–2520. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000, 303, 567–582. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2010, 51, 69–82. [Google Scholar]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Pearlman, D.A.; Crowley, M.; et al. AMBER 9; University of California, San Francisco: San Francisco, CA, USA, 2006. [Google Scholar]
- Wang, J.; Kang, X.; Kuntz, I.D.; Kollman, P.A. Hierarchical database screenings for HIV-1 reverse transcriptase using a pharmacophore model, rigid docking, solvation docking, and MM-PB/SA. J. Med. Chem. 2005, 48, 2432–2444. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Zoete, V.; Meuwly, M.; Karplus, M. Study of the insulin dimerization: Binding free energy calculations and per-residue free energy decomposition. Proteins Struct. Funct. Bioinform. 2005, 61, 79–93. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Liang, X.; Li, X.; Shi, Z.; Zhou, N.; Bao, J. In silico identification of novel kinase inhibitors targeting wild-type and T315I mutant ABL1 from FDA-approved drugs. Mol. Biosyst. 2014, 10, 1524–1537. [Google Scholar] [CrossRef]
- Zhong, H.; Huang, W.; He, G.; Peng, C.; Wu, F.; Ouyang, L. Molecular dynamics simulation of tryptophan hydroxylase-1: Binding modes and free energy analysis to phenylalanine derivative inhibitors. Int. J. Mol. Sci. 2013, 14, 9947–9962. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In Silico Discovery of Potential VEGFR-2 Inhibitors from Natural Derivatives for Anti-Angiogenesis Therapy. Int. J. Mol. Sci. 2014, 15, 15994-16011. https://doi.org/10.3390/ijms150915994
Li J, Zhou N, Luo K, Zhang W, Li X, Wu C, Bao J. In Silico Discovery of Potential VEGFR-2 Inhibitors from Natural Derivatives for Anti-Angiogenesis Therapy. International Journal of Molecular Sciences. 2014; 15(9):15994-16011. https://doi.org/10.3390/ijms150915994
Chicago/Turabian StyleLi, Jing, Nan Zhou, Kun Luo, Wei Zhang, Xinru Li, Chuanfang Wu, and Jinku Bao. 2014. "In Silico Discovery of Potential VEGFR-2 Inhibitors from Natural Derivatives for Anti-Angiogenesis Therapy" International Journal of Molecular Sciences 15, no. 9: 15994-16011. https://doi.org/10.3390/ijms150915994
APA StyleLi, J., Zhou, N., Luo, K., Zhang, W., Li, X., Wu, C., & Bao, J. (2014). In Silico Discovery of Potential VEGFR-2 Inhibitors from Natural Derivatives for Anti-Angiogenesis Therapy. International Journal of Molecular Sciences, 15(9), 15994-16011. https://doi.org/10.3390/ijms150915994