Cancer Stem Cell Theory and the Warburg Effect, Two Sides of the Same Coin?

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Warburg Effect
3. Partial Pressure of O2
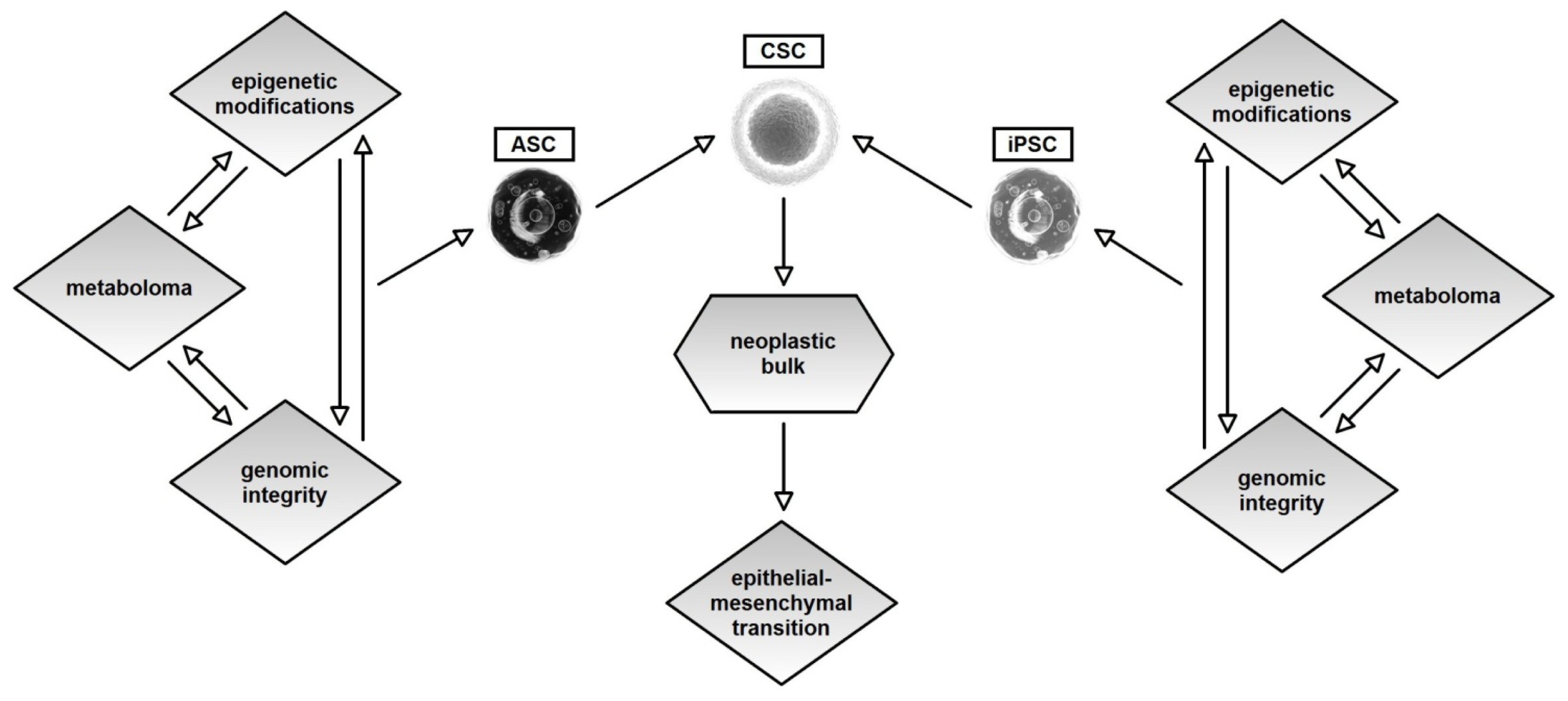
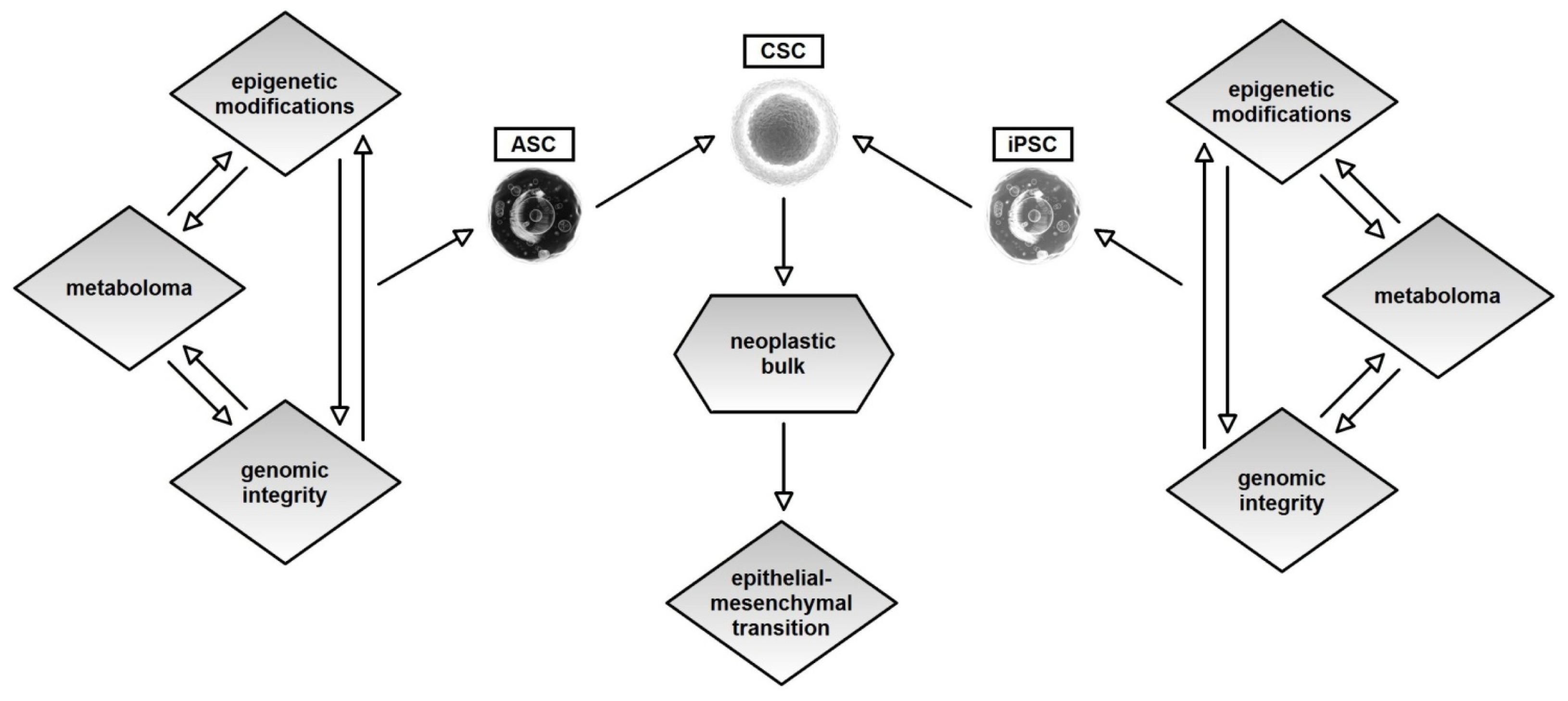
4. Genetics, Epigenetics and Metabolism
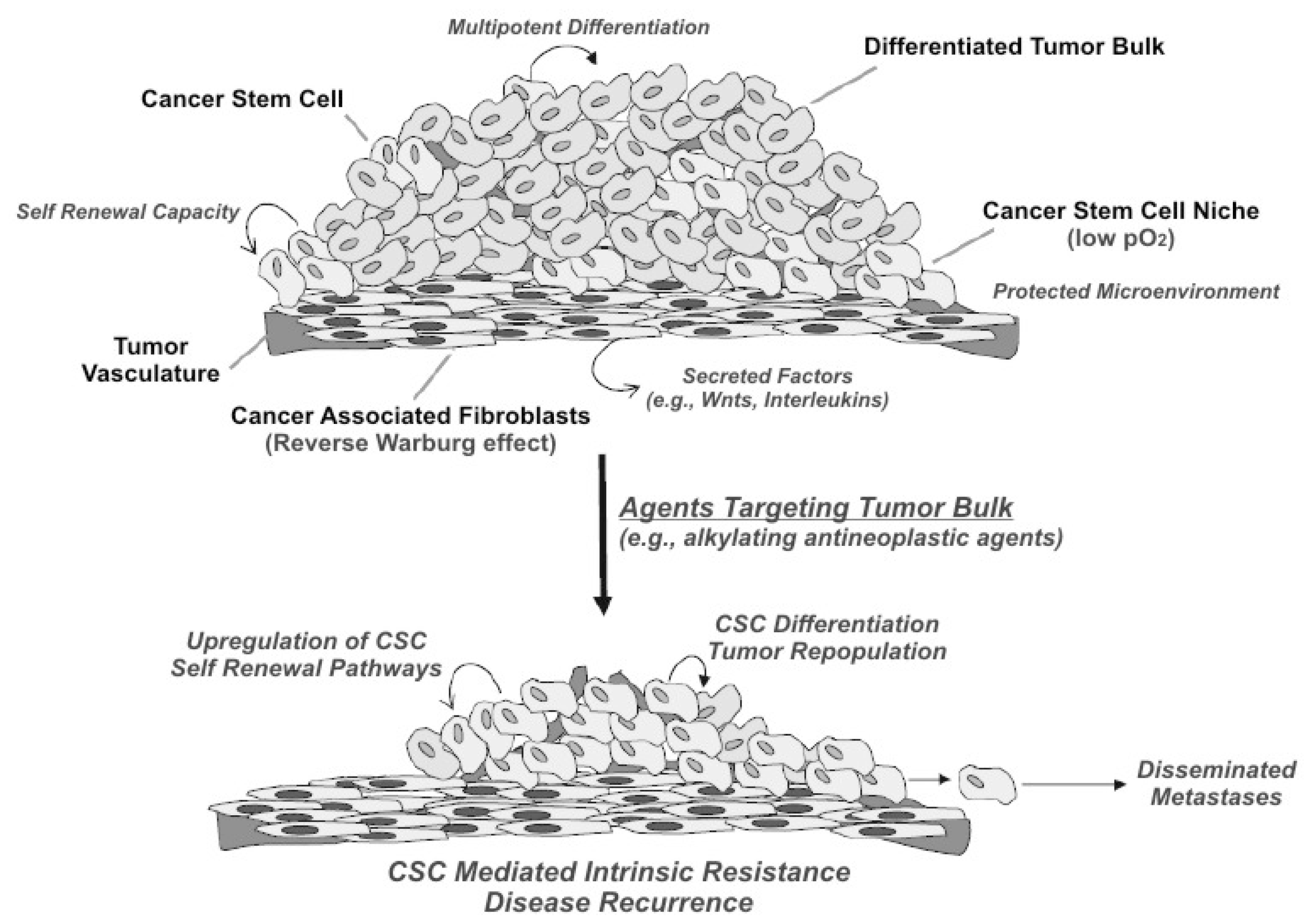
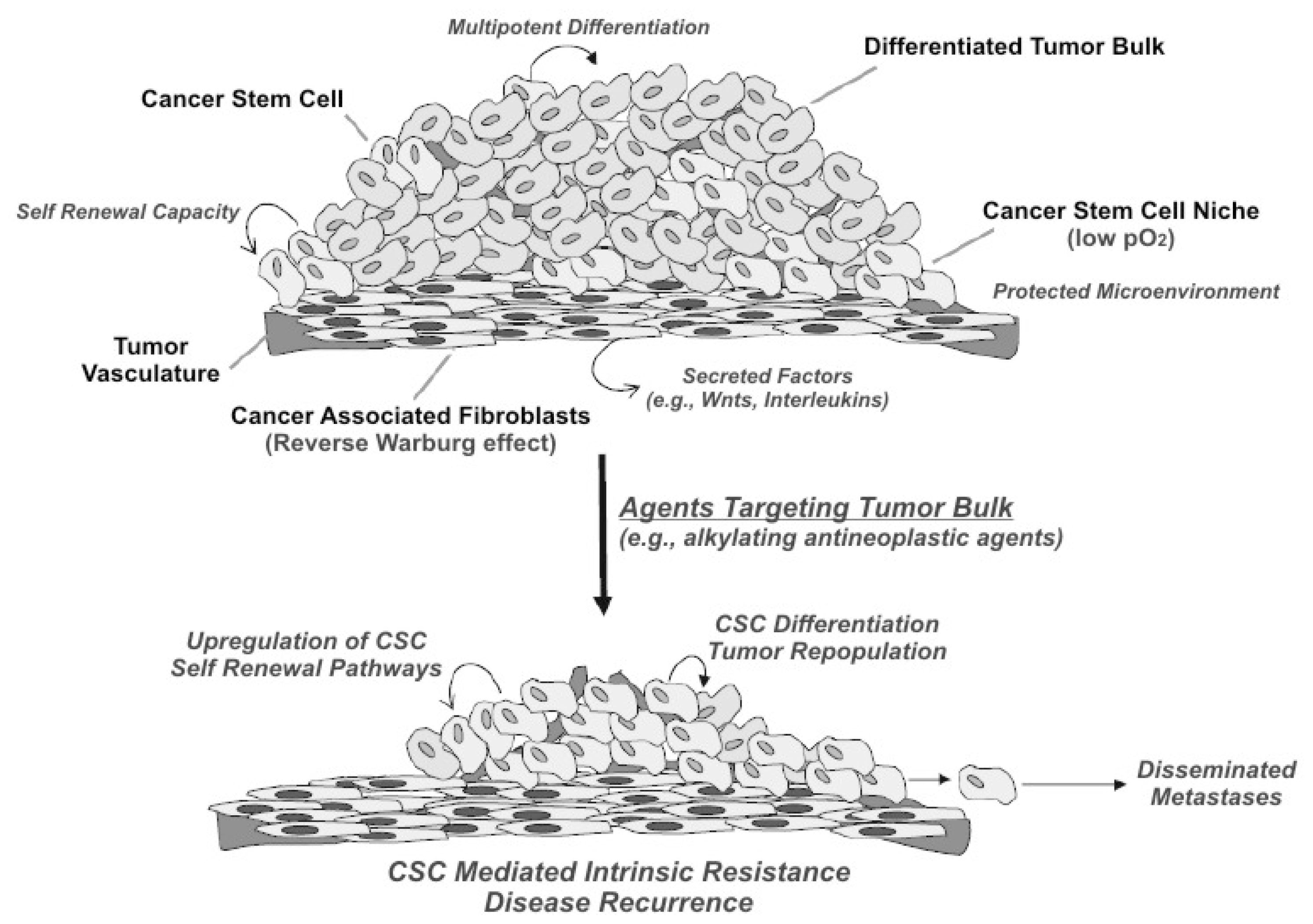
5. Population Dynamics and Microenvironment
6. Mitochondria
7. Metabolic State, Differentiation and Cell Cycle
8. Notes on Thermodynamics
- (a)
- Living systems show periodic structures with low (but not minimal) levels of entropy; these structures alternate with those with a disordered degree (greater randomness), such as the plasma membrane and chromosomes alternating with the cytoplasm;
- (b)
- Living systems use power sources from chemical reactions that far from the thermodynamic equilibrium to maintain order and ATP hydrolysis; even though there are systems with highly ordered structures (with low entropy), these systems also have low energy (in balance), such as crystals;
- (c)
- Living systems can replicate, and the cell split also represents a means of entropy decrease, thus a typical cell, as it finishes its cell loop, increases its order degree; through the split cells with a greater degree of randomness; the living systems then die or reach thermodynamic equilibrium (ΔG = 0);
- (d)
- In conclusion, living systems see their entropy and energy varying through time by maintaining certain parameters; the randomness is neither too high nor too low and oscillating between the maximum and minimum values occurs.
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jacob, F.; Morton, K. The transmission of leukemia of mice with a single cell. Am. J. Cancer 1937, 31, 276–282. [Google Scholar]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic functions of the p53 tumor suppressor: Regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol 2009, 19, 32–41. [Google Scholar]
- Sonnenschein, C.; Soto, A.M. Somatic mutation theory of carcinogenesis: Why it should be dropped and replaced. Mol. Carcinog 2000, 29, 205–211. [Google Scholar]
- Loeb, L.A. A mutator phenotype in Cancer. Cancer Res 2001, 61, 3230–3239. [Google Scholar]
- Nowell, P.C. Tumor progression: A brief historical perspective. Semin. Cancer Biol 2002, 12, 261–266. [Google Scholar]
- Baker, S.G.; Kramer, B.S. Paradoxes in carcinogenesis: New opportunities for research directions. BMC Cancer 2007, 7, 151. [Google Scholar]
- Mori, H.; Colman, S.M.; Xiao, Z.; Ford, A.M.; Healy, L.E.; Donaldson, C.; Hows, J.M.; Navarrete, C.; Greaves, M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. USA 2002, 99, 8242–8247. [Google Scholar]
- Sengupta, A.; Cancelas, J.A. Cancer stem cells: A stride towards cancer cure? J. Cell Physiol 2010, 225, 7–14. [Google Scholar]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic. Cell. Nat. Med 1997, 3, 730–737. [Google Scholar]
- Al-Hajj, M. Cancer stem cells and oncology therapeutics. Curr. Opin. Oncol 2007, 19, 61–64. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63, 5821–5828. [Google Scholar]
- He, J.; Liu, Y.; Lubman, D.M. Targeting glioblastoma stem cells: Cell surface markers. Curr. Med. Chem 2012, 19, 6050–6055. [Google Scholar]
- O’Flaherty, J.D.; Barr, M.; Fennell, D.; Richard, D.; Reynolds, J.; O’Leary, J.; O’Byrne, K. The cancer stem-cell hypothesis: Its emerging role in lung cancer biology and its relevance for future therapy. J. Thorac. Oncol 2012, 7, 1880–1890. [Google Scholar]
- Dhawan, P.; Ahmad, R.; Srivastava, A.S.; Singh, A.B. Cancer stem cells and colorectal cancer: An overview. Curr. Top. Med. Chem 2011, 11, 1592–1598. [Google Scholar]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68, 4311–4320. [Google Scholar]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol. Aspects Med 2013, 2013. [Google Scholar] [CrossRef]
- Gou, S.; Liu, T.; Wang, C.; Yin, T.; Li, K.; Yang, M.; Zhou, J. Establishment of clonal colony-forming assay for propagation of pancreatic cancer cells with stem cell properties. Pancreas 2007, 34, 429–435. [Google Scholar]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun 2012, 421, 349–354. [Google Scholar]
- Lang, S.H.; Frame, F.M.; Collins, A.T. Prostate cancer stem cells. J. Pathol 2009, 217, 299–306. [Google Scholar]
- Malaguarnera, R.; Frasca, F.; Garozzo, A.; Gianì, F.; Pandini, G.; Vella, V.; Vigneri, R.; Belfiore, A. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J. Clin. Endocrinol. Metab 2010, 96, 766–774. [Google Scholar]
- Lorico, A.; Rappa, G. Phenotypic heterogeneity of breast cancer stem cells. J. Oncol 2011, 2011. [Google Scholar] [CrossRef]
- Curtin, J.C.; Lorenzi, M.V. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget 2010, 1, 552–566. [Google Scholar]
- Xia, T.; Jiang, H.; Li, C.; Tian, M.; Zhang, H. Molecular imaging in tracking tumor stem-like cells. J. Biomed. Biotechnol 2012, 2012. [Google Scholar] [CrossRef]
- Cukierman, E.; Bassi, D.E. The mesenchymal tumor microenvironment: A drug-resistant niche. Cell Adh. Migr 2012, 6, 285–296. [Google Scholar]
- Sell, S. On the stem cell origin of Cancer. Am. J. Pathol 2010, 176, 2584–2594. [Google Scholar]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol 2013, 108, 378–387. [Google Scholar]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res 2006, 66, 1883–1890. [Google Scholar]
- Siggins, R.W.; Zhang, P.; Welsh, D.; Lecapitaine, N.J.; Nelson, S. Stem cells, phenotypic inversion, and differentiation. Int. J. Clin. Exp. Med 2008, 1, 2–21. [Google Scholar]
- Kondoh, H.; Lleonart, M.E.; Bernard, D.; Gil, J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol. Histopathol 2007, 22, 85–90. [Google Scholar]
- Kondoh, H.; Lleonart, M.E.; Nakashima, Y.; Yokode, M.; Tanaka, M.; Bernard, D.; Gil, J.; Beach, D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid. Redox Signal 2007, 9, 293–299. [Google Scholar]
- Kraft, C.S.; LeMoine, C.M.; Lyons, C.N.; Michaud, D.; Mueller, C.R.; Moyes, C.D. Control of mitochondrial biogenesis during myogenesis. Am. J. Physiol. Cell Physiol 2006, 290, C1119–C1127. [Google Scholar]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by phasor fluorescence lifetime microscopy of NADH. Sci. Rep 2012, 2. [Google Scholar] [CrossRef]
- Wright, B.K.; Andrews, L.M.; Markham, J.; Jones, M.R.; Stringari, C.; Digman, M.A.; Gratton, E. NADH distribution in live progenitor stem cells by phasor-fluorescence lifetime image microscopy. Biophys. J 2012, 103, L7–L9. [Google Scholar]
- Sen, A.; Damm, V.T.; Cox, R.T. Drosophila clueless is highly expressed in larval neuroblasts, affects mitochondrial localization and suppresses mitochondrial oxidative damage. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Legname, A.H.; Salomón de Legname, H. Changes in the oxidative metabolism during maturation of amphibian oocytes. J. Embryol. Exp. Morphol 1980, 59, 175–186. [Google Scholar]
- Wales, R.G. Measurement of metabolic turnover in single mouse embryos. J. Reprod. Fertil 1986, 76, 717–725. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Nelson, T.J.; Terzic, A. Induced pluripotent stem cells: An emerging theranostics platform. Clin. Pharmacol. Ther 2011, 89, 648–650. [Google Scholar]
- Prigione, A.; Adjaye, J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int. J. Dev. Biol 2010, 54, 1729–1741. [Google Scholar]
- Vazquez-Martin, A.; Corominas-Faja, B.; Cufi, S.; Vellon, L.; Oliveras-Ferraros, C.; Menendez, O.J.; Joven, J.; Lupu, R.; Menendez, J.A. The mitochondrial H+-ATP synthase and the lipogenic switch: New core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle 2013, 12, 207–218. [Google Scholar]
- Folmes, C.D.; Nelson, T.J.; Terzic, A. Energy metabolism in nuclear reprogramming. Biomark. Med 2011, 5, 715–729. [Google Scholar]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res 2012, 22, 168–177. [Google Scholar]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol 2010, 11, 872–884. [Google Scholar]
- Varum, S.; Momcilović, O.; Castro, C.; Ben-Yehudah, A.; Ramalho-Santos, J.; Navara, C.S. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res 2009, 3, 142–156. [Google Scholar]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency—dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 2011, 14, 264–271. [Google Scholar]
- Ding, D.F.; Li, X.F.; Xu, H.; Wang, Z.; Liang, Q.Q.; Li, C.G.; Wang, Y.J. Mechanism of resveratrol on the promotion of induced pluripotent stem cells. J. Integr. Med 2013, 11, 389–396. [Google Scholar]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A., 4th; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Abu Dawud, R.; Schreiber, K.; Schomburg, D.; Adjaye, J. Human embryonic stem cells and embryonal carcinoma cells have overlapping and distinct metabolic signatures. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J. Cell Biochem 2014, 115, 368–379. [Google Scholar]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol 2005, 17, 548–558. [Google Scholar]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 2005, 65, 5996–6000. [Google Scholar]
- Smith, D.G.; Sturmey, R.G. Parallels between embryo and cancer cell metabolism. Biochem. Soc. Trans 2013, 41, 664–669. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol 1927, 8, 519–530. [Google Scholar]
- Tennant, D.A.; Durán, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab 2010, 7. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar]
- Lynen, F. Die rolle der phosphorsaeure bei dehydrierungsvorgaengen und ihre biologische bedeutung. Naturwissenschaften 1951, 30, 398–406. (In German) [Google Scholar]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006, 66, 8927–8930. [Google Scholar]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH—A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar]
- Patel, M.S.; Korotchkina, L.G. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: Complexity of multiple phosphorylation sites and kinases. Exp. Mol. Med 2001, 33, 191–197. [Google Scholar]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol 2011, 39, 453–463. [Google Scholar]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res 2011, 71, 6921–6925. [Google Scholar]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 2011, 71, 2550–2560. [Google Scholar]
- Kennedy, K.M.; Dewhirst, M.W. Tumor metabolism of lactate: The influence and therapeutic potential for MCT and CD147 regulation. Futur. Oncol 2010, 6, 127–148. [Google Scholar]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol 2004, 14, 267–274. [Google Scholar]
- Eigentler, T.K.; Figl, A.; Krex, D.; Mohr, P.; Mauch, C.; Rass, K.; Bostroem, A.; Heese, O.; Koelbl, O.; Garbe, C.; et al. Number of metastases, serum lactate dehydrogenase level, and type of treatment are prognostic factors in patients with brain metastases of malignant melanoma. Cancer 2011, 117, 1697–1703. [Google Scholar]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Trarbach, T.; Folprecht, G.; Shi, M.M.; Lebwohl, D.; Jalava, T.; Laurent, D.; et al. Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin. Cancer Res 2011, 17, 4892–4900. [Google Scholar]
- Li, G.; Gao, J.; Tao, Y.L.; Xu, B.Q.; Tu, Z.W.; Liu, Z.G.; Zeng, M.S.; Xia, Y.F. Increased pretreatment levels of serum LDH and ALP as poor prognostic factors for nasopharyngeal carcinoma. Chin. J. Cancer 2012, 31, 197–206. [Google Scholar]
- Xie, H.; Valera, V.A.; Merino, M.J.; Amato, A.M.; Signoretti, S.; Linehan, W.M.; Sukhatme, V.P.; Seth, P. LDH–A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell Cancer. Mol. Cancer Ther 2009, 8, 626–635. [Google Scholar]
- Szanto, I.; Rubbia-Brandt, L.; Kiss, P.; Steger, K.; Banfi, B.; Kovari, E.; Herrmann, F.; Hadengue, A.; Krause, K.H. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol 2005, 207, 164–176. [Google Scholar]
- Kawahara, T.; Lambeth, J.D. Molecular evolution of Phox-related regulatory subunits for NADPH oxidase enzymes. BMC Evol. Biol 2007, 7, 178. [Google Scholar]
- Lu, W.; Hu, Y.; Chen, G.; Chen, Z.; Zhang, H.; Wang, F.; Feng, L.; Pelicano, H.; Wang, H.; Keating, M.J.; et al. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol 2012, 10. [Google Scholar] [CrossRef]
- Lin, C.C.; Cheng, T.L.; Tsai, W.H.; Tsai, H.J.; Hu, K.H.; Chang, H.C.; Yeh, C.W.; Chen, Y.C.; Liao, C.C.; Chang, W.T. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci. Rep 2012, 2. [Google Scholar] [CrossRef]
- Singh, M.; Singh, V.N.; August, J.T.; Horecker, B.L. Alterations in glucose metabolism in chick embryo cells transformed by Rous sarcoma virus. Transformation-specific changes in the activities of key enzymes of the glycolytic and hexose monophosphate shunt pathways. Arch. Biochem. Biophys. 1974, 165, 240–246. [Google Scholar]
- Darekar, S.; Georgiou, K.; Yurchenko, M.; Yenamandra, S.P.; Chachami, G.; Simos, G.; Klein, G.; Kashuba, E. Epstein-Barr virus immortalization of human B-cells leads to stabilization of hypoxia-induced factor 1 alpha, congruent with the Warburg effect. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Cole, M.A.; Crawford, D.W.; Warner, N.E.; Puffer, H.W. Correlation of regional disease and in vivo PO2 in rat mammary adenocarcinoma. Am. J. Pathol 1983, 112, 61–67. [Google Scholar]
- Joyce, R.M.; Vincent, P.C. Advantage of reduced oxygen tension in growth of human melanomas in semi-solid cultures: Quantitative analysis. Br. J. Cancer 1983, 48, 385–393. [Google Scholar]
- Ezashi, T.; Das, P.; Roberts, R.M. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4783–4788. [Google Scholar]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar]
- Brizel, D.M.; Sibley, G.S.; Prosnitz, L.R.; Scher, R.L.; Dewhirst, M.W. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int. J. Radiat. Oncol. Biol. Phys 1997, 38, 285–289. [Google Scholar]
- Nozue, M.; Lee, I.; Yuan, F.; Teicher, B.A.; Brizel, D.M.; Dewhirst, M.W.; Milross, C.G.; Milas, L.; Song, C.W.; Thomas, C.D.; et al. Interlaboratory variation in oxygen tension measurement by Eppendorf “Histograph” and comparison with hypoxic marker. J. Surg. Oncol 1997, 66, 30–38. [Google Scholar]
- Nordsmark, M.; Høyer, M.; Keller, J.; Nielsen, O.S.; Jensen, O.M.; Overgaard, J. The relationship between tumor oxygenation and cell proliferation in human soft tissue sarcomas. Int. J. Radiat. Oncol. Biol. Phys 1996, 35, 701–708. [Google Scholar]
- Sundfør, K.; Lyng, H.; Rofstad, E.K. Tumour hypoxia and vascular density as predictors of metastasis in squamous cell carcinoma of the uterine cervix. Br. J. Cancer 1998, 78, 822–827. [Google Scholar]
- Nordsmark, M.; Overgaard, J. A confirmatory prognostic study on oxygenation status and loco-regional control in advanced head and neck squamous cell carcinoma treated by radiation therapy. Radiother. Oncol 2000, 57, 39–43. [Google Scholar]
- Young, S.D.; Marshall, R.S.; Hill, R.P. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc. Natl. Acad. Sci. USA 1988, 85, 9533–9537. [Google Scholar]
- Young, S.D.; Hill, R.P. Effects of reoxygenation on cells from hypoxic regions of solid tumors: Anticancer drug sensitivity and metastatic potential. J. Natl. Cancer Inst 1990, 82, 371–380. [Google Scholar]
- Rofstad, E.K.; Måseide, K. Radiobiological and immunohistochemical assessment of hypoxia in human melanoma xenografts: Acute and chronic hypoxia in individual tumours. Int. J. Radiat. Biol 1999, 75, 1377–1393. [Google Scholar]
- Olsen, D.R.; Singstad, T.E.; Rofstad, E.K. Effects of hyperthermia on bioenergetic status and phosphorus T1S in human melanoma xenografts monitored by 31P-MRS. Magn. Reson. Imaging 1999, 17, 1049–1056. [Google Scholar]
- Stackpole, C.W.; Groszek, L.; Kalbag, S.S. Benign-to-malignant B16 melanoma progression induced in two stages in vitro by exposure to hypoxia. J. Natl. Cancer Inst 1994, 86, 361–367. [Google Scholar]
- Graham, C.H.; Forsdike, J.; Fitzgerald, C.J.; Macdonald-Goodfellow, S. Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int. J. Cancer 1999, 80, 617–623. [Google Scholar]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar]
- Cairns, R.A.; Kalliomaki, T.; Hill, R.P. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res 2001, 61, 8903–8908. [Google Scholar]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar]
- Bertram, J.S. The molecular biology of cancer. Mol. Aspects Med 2000, 21, 167–223. [Google Scholar]
- Roach, J.C.; Glusman, G.; Smit, A.F.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010, 328, 636–639. [Google Scholar]
- Hitchler, M.J.; Domann, F.E. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic. Biol. Med 2009, 47, 115–127. [Google Scholar]
- Hitchler, M.J.; Domann, F.E. Redox regulation of the epigenetic landscape in cancer: A role for metabolic reprogramming in remodeling the epigenome. Free Radic. Biol. Med 2012, 53, 2178–2187. [Google Scholar]
- Gambini, J.; Gomez-Cabrera, M.C.; Borras, C.; Valles, S.L.; Lopez-Grueso, R.; Martinez-Bello, V.E.; Herranz, D.; Pallardo, F.V.; Tresguerres, J.A.; Serrano, M.; et al. Free [NADH]/[NAD(+)] regulates sirtuin expression. Arch. Biochem. Biophys 2011, 512, 24–29. [Google Scholar]
- Van Horssen, R.; Willemse, M.; Haeger, A.; Attanasio, F.; Güneri, T.; Schwab, A.; Stock, C.M.; Buccione, R.; Fransen, J.A.; Wieringa, B. Intracellular NAD(H) levels control motility and invasion of glioma cells. Cell Mol. Life Sci. 2013, 70, 2175–2190. [Google Scholar]
- Mato, J.M.; Lu, S.C. The hepatocarcinogenic effect of methionine and choline deficient diets: An adaptation to the Warburg effect? Alcohol Clin. Exp. Res 2011, 35, 811–814. [Google Scholar]
- Mato, J.M.; Lu, S.C. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev 2012, 92, 1515–1542. [Google Scholar]
- Watson, W.H.; Song, Z.; Kirpich, I.A.; Deaciuc, I.V.; Chen, T.; McClain, C.J. Ethanol exposure modulates hepatic S-adenosylmethionine and S-adenosylhomocysteine levels in the isolated perfused rat liver through changes in the redox state of the NADH/NAD(+) system. Biochim. Biophys. Acta 2011, 1812, 613–618. [Google Scholar]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol 2012, 14, 1295–1304. [Google Scholar]
- Neary, C.L.; Pastorino, J.G. Nucleocytoplasmic shuttling of hexokinase II in a cancer cell. Biochem. Biophys. Res. Commun 2010, 394, 1075–1081. [Google Scholar]
- Peláez, R.; Herrero, P.; Moreno, F. Nuclear export of the yeast hexokinase 2 protein requires the Xpo1 Crm1-dependent pathway. J. Biol. Chem 2009, 284, 20548–20555. [Google Scholar]
- Friis, R.M.; Wu, B.P.; Reinke, S.N.; Hockman, D.J.; Sykes, B.D.; Schultz, M.C. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic. Acids Res 2009, 37, 3969–3980. [Google Scholar]
- Daran-Lapujade, P.; Rossell, S.; van Gulik, W.M.; Luttik, M.A.; de Groot, M.J.; Slijper, M.; Heck, A.J.; Daran, J.M.; de Winde, J.H.; Westerhoff, H.V.; et al. The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc. Natl. Acad. Sci. USA 2007, 104, 15753–15758. [Google Scholar]
- Chen, Z.; Odstrcil, E.A.; Tu, B.P.; McKnight, S.L. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science 2007, 316, 1916–1919. [Google Scholar]
- Hilton, J.; Walker, M.D. DNA strand scission and its repair following exposure of cells to inhibitors of oxidative phosphorylation. Biochem. Biophys. Res. Commun 1977, 75, 909–914. [Google Scholar]
- Fry, D.W. Cytotoxic synergism between trimetrexate and etoposide. Evidence that trimetrexate potentiates etoposide-induced protein-associated DNA strand breaks in L1210 leukemia cells through alterations in intracellular ATP concentrations. Biochem. Pharmacol 1990, 40, 1981–1988. [Google Scholar]
- Dalrymple, G.V.; Sanders, J.L.; Baker, M.L.; Wilkinson, K.P. The effect of 2,4-dinitrophenol on the repair of radiation injury by L-cells. Radiat. Res 1969, 37, 90–102. [Google Scholar]
- Moss, A.J., Jr.; Dalrymple, G.V.; Sanders, J.L.; Wilkinson, K.P.; Nash, J.C. Dinitrophenol inhibits the rejoining of radiation-induced DNA breaks by L-cells. Biophys. J 1971, 11, 158–174. [Google Scholar]
- Wang, Z.; Wu, X.; Friedberg, E.C. Nucleotide-excision repair of DNA in cell-free extracts of the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1993, 90, 4907–4911. [Google Scholar]
- Lagostera, M.; Guerrero, R.; Villaverde, A.; Barbé, J. Effect of adenine, cytidine and guanosine on the expression of the SOS system in Escherichia coli. J. Gen. Microbiol 1985, 131, 113–118. [Google Scholar]
- Thomas, D.C.; Roberts, J.D.; Kunkel, T.A. Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem 1991, 266, 3744–3751. [Google Scholar]
- Rapaport, E.; Garcia-Blanco, M.A.; Zamecnik, P.C. Regulation of DNA replication in S phase nuclei by ATP and ADP pools. Proc. Natl. Acad. Sci. USA 1979, 76, 1643–1647. [Google Scholar]
- Laureti, L.; Selva, M.; Dairou, J.; Matic, I. Reduction of dNTP levels enhances DNA replication fidelity in vivo. DNA Repair 2013, 12, 300–305. [Google Scholar]
- Wijker, J.E.; Jensen, P.R.; Snoep, J.L.; vaz Gomes, A.; Guiral, M.; Jongsma, A.P.; de Waal, A.; Hoving, S.; van Dooren, S.; van der Weijden, C.C.; et al. Energy, control and DNA structure in the living cell. Biophys. Chem 1995, 55, 153–165. [Google Scholar]
- Singleton, M.R.; Wigley, D.B. Multiple roles for ATP hydrolysis in nucleic acid modifying enzymes. EMBO J 2003, 22, 4579–4583. [Google Scholar]
- Okorokov, A.L.; Milner, J. An ATP/ADP-dependent molecular switch regulates the stability of p53-DNA complexes. Mol. Cell Biol 1999, 19, 7501–7510. [Google Scholar]
- Yang, J.G.; Narlikar, G.J. FRET-based methods to study ATP-dependent changes in chromatin structure. Methods 2007, 41, 291–295. [Google Scholar]
- Varga-Weisz, P. ATP-dependent chromatin remodeling factors: Nucleosome shufflers with many missions. Oncogene 2001, 20, 3076–3085. [Google Scholar]
- Laval, F. Effect of uncouplers on radiosensitivity and mutagenicity in x-irradiated mammalian cells. Proc. Natl. Acad. Sci. USA 1980, 77, 2702–2705. [Google Scholar]
- Tahanian, E.; Peiro, S.; Annabi, B. Low intracellular ATP levels exacerbate carcinogen-induced inflammatory stress response and inhibit in vitro tubulogenesis in human brain endothelial cells. J. Inflamm. Res 2011, 4, 1–10. [Google Scholar]
- Jonson, I.; Ougland, R.; Klungland, A.; Larsen, E. Oxidative stress causes DNA triplet expansion in Huntington’s disease mouse embryonic stem cells. Stem Cell Res 2013, 11, 1264–1271. [Google Scholar]
- Zhang, M.; Yang, C.; Liu, H.; Sun, Y. Induced pluripotent stem cells are sensitive to DNA damage. Genomics Proteomics Bioinform 2013, 11, 320–326. [Google Scholar]
- Tanori, M.; Pasquali, E.; Leonardi, S.; Casciati, A.; Giardullo, P.; de Stefano, I.; Mancuso, M.; Saran, A.; Pazzaglia, S. Developmental and oncogenic radiation effects on neural stem cells and their differentiating progeny in mouse cerebellum. Stem Cells 2013, 31, 2506–2516. [Google Scholar]
- Nouspikel, T. Genetic instability in human embryonic stem cells: Prospects and caveats. Future Oncol 2013, 9, 867–877. [Google Scholar]
- Kenyon, J.; Gerson, S.L. The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res 2007, 35, 7557–7565. [Google Scholar]
- Morley, A.; Seshadri, R.; Trainor, K.; Sorrell, J. Is aplastic anaemia due to abnormality of DNA? Lancet 1978, 2, 9–12. [Google Scholar]
- Gerson, S.L.; Trey, J.E.; Miller, K.; Benjamin, E. Repair of O6-alkylguanine during DNA synthesis in murine bone marrow hematopoietic precursors. Cancer Res 1987, 47, 89–95. [Google Scholar]
- Luo, L.Z.; Gopalakrishna-Pillai, S.; Nay, S.L.; Park, S.W.; Bates, S.E.; Zeng, X.; Iverson, L.E.; O’Connor, T.R. DNA repair in human pluripotent stem cells is distinct from that in non-pluripotent human cells. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Cousin, W.; Ho, M.L.; Desai, R.; Tham, A.; Chen, R.Y.; Kung, S.; Elabd, C.; Conboy, I.M. Regenerative capacity of old muscle stem cells declines without significant accumulation of DNA damage. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar]
- Sloan, E.K.; Ciocca, D.R.; Pouliot, N.; Natoli, A.; Restall, C.; Henderson, M.A.; Fanelli, M.A.; Cuello-Carrión, F.D.; Gago, F.E.; Anderson, R.L. Stromal cell expression of caveolin-1 predicts outcome in breast Cancer. Am. J. Pathol 2009, 174, 2035–2043. [Google Scholar]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar]
- Balliet, R.M.; Capparelli, C.; Guido, C.; Pestell, T.G.; Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Pestell, R.G.; Howell, A.; et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production: Promoting breast cancer tumor growth, understanding the aging and cancer connection. Cell Cycle 2011, 10, 4065–4073. [Google Scholar]
- Samudio, I.; Fiegl, M.; McQueen, T.; Clise-Dwyer, K.; Andreeff, M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res 2008, 68, 5198–5205. [Google Scholar]
- Konopleva, M.; Andreeff, M. Targeting the leukemia microenvironment. Curr. Drug Targets 2007, 8, 685–701. [Google Scholar]
- Wang, L.; O’Leary, H.; Fortney, J.; Gibson, L.F. Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood 2007, 110, 3334–3344. [Google Scholar]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem 2002, 277, 47129–47135. [Google Scholar]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res 2009, 69, 2163–2166. [Google Scholar]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J 2008, 22, 9–18. [Google Scholar]
- Harper, M.E.; Antoniou, A.; Villalobos-Menuey, E.; Russo, A.; Trauger, R.; Vendemelio, M.; George, A.; Bartholomew, R.; Carlo, D.; Shaikh, A.; et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J 2002, 16, 1550–1557. [Google Scholar]
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med 2003, 9, 1062–1068. [Google Scholar]
- Mintz, B.; Illmensee, K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1975, 72, 3585–3589. [Google Scholar]
- Ruckenstuhl, C.; Büttner, S.; Carmona-Gutierrez, D.; Eisenberg, T.; Kroemer, G.; Sigrist, S.J.; Fröhlich, K.U.; Madeo, F. The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for Cancer. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 1997, 57, 1835–1840. [Google Scholar]
- Eguchi, Y.; Srinivasan, A.; Tomaselli, K.J.; Shimizu, S.; Tsujimoto, Y. ATP-dependent steps in apoptotic signal transduction. Cancer Res 1999, 59, 2174–2181. [Google Scholar]
- Shen, J.; Liu, X.; Yu, W.M.; Liu, J.; Nibbelink, M.G.; Guo, C.; Finkel, T.; Qu, C.K. A critical role of mitochondrial phosphatase Ptpmt1 in embryogenesis reveals a mitochondrial metabolic stress-induced differentiation checkpoint in embryonic stem cells. Mol. Cell Biol 2011, 31, 4902–4916. [Google Scholar]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr 2009, 41, 517–521. [Google Scholar]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar]
- Lonergan, T.; Brenner, C.; Bavister, B. Differentiation-related changes in mitochondrial properties as indicators of stem cell competence. J. Cell Physiol 2006, 208, 149–153. [Google Scholar]
- Brown, G.C. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J 1992, 284, 1–13. [Google Scholar]
- Thundathil, J.; Filion, F.; Smith, L.C. Molecular control of mitochondrial function in preimplantation mouse embryos. Mol. Reprod. Dev 2005, 71, 405–413. [Google Scholar]
- Van Blerkom, J. Mitochondria in early mammalian development. Semin. Cell Dev. Biol 2009, 20, 354–364. [Google Scholar]
- St John, J.C.; Ramalho-Santos, J.; Gray, H.L.; Petrosko, P.; Rawe, V.Y.; Navara, C.S.; Simerly, C.R.; Schatten, G.P. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells 2005, 7, 141–153. [Google Scholar]
- St John, J. The control of mtDNA replication during differentiation and development. Biochim. Biophys. Acta 2014, 1840, 1345–1354. [Google Scholar]
- Holley, A.K.; Dhar, S.K.; Xu, Y.; St Clair, D.K. Manganese superoxide dismutase: Beyond life and death. Amino Acids 2012, 42, 139–158. [Google Scholar]
- Duttaroy, A.; Paul, A.; Kundu, M.; Belton, A. A SOD2 null mutation confers severely reduced adult life span in Drosophila. Genetics 2003, 165, 2295–2299. [Google Scholar]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genome 2003, 16, 29–37. [Google Scholar]
- Olivotto, M.; Arcangeli, A.; Caldini, R.; Chevanne, M.; Cipolleschi, M.G.; Dello Sbarba, P. Metabolic aspects of cell cycle regulation in normal and cancer cells. Toxicol. Pathol 1984, 12, 369–373. [Google Scholar]
- Li, W.; Nichols, K.; Nathan, C.A.; Zhao, Y. Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark 2013, 13, 377–383. [Google Scholar]
- Kuai, X.Y.; Ji, Z.Y.; Zhang, H.J. Mitochondrial uncoupling protein 2 expression in colon cancer and its clinical significance. World J. Gastroenterol 2010, 16, 5773–5778. [Google Scholar]
- Zhang, G.; Qu, Y.; Dang, S.; Yang, Q.; Shi, B.; Hou, P. Variable copy number of mitochondrial DNA (mtDNA) predicts worse prognosis in advanced gastric cancer patients. Diagn. Pathol 2013, 8, 173. [Google Scholar]
- Criscuolo, F.; Mozo, J.; Hurtaud, C.; Nübel, T.; Bouillaud, F. UCP2, UCP3, avUCP, what do they do when proton transport is not stimulated? Possible relevance to pyruvate and glutamine metabolism. Biochim. Biophys. Acta 2006, 1757, 1284–1291. [Google Scholar]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J 2011, 30, 4860–4873. [Google Scholar]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Béroud, C.; Demont, J.; Bouvier, R.; Schägger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar]
- Pokorný, J.; Jandová, A.; Nedbalová, M.; Jelínek, F.; Cifra, M.; Kučera, O.; Havelka, D.; Vrba, J.; Vrba, J., Jr.; Coček, A.; et al. Mitochondrial metabolism—Neglected link of cancer transformation and treatment. Prague Med. Rep 2012, 113, 81–94. [Google Scholar]
- Dang, C.V. Links between metabolism and Cancer. Genes Dev 2012, 26, 877–890. [Google Scholar]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar]
- Miceli, M.V.; Jazwinski, S.M. Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci 2005, 46, 1765–1773. [Google Scholar]
- Jazwinski, S.M. The retrograde response links metabolism with stress responses, chromatin-dependent gene activation, and genome stability in yeast aging. Gene 2005, 354, 22–27. [Google Scholar]
- Singh, K.K.; Kulawiec, M.; Still, I.; Desouki, M.M.; Geradts, J.; Matsui, S. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 2005, 354, 140–146. [Google Scholar]
- Traven, A.; Wong, J.M.; Xu, D.; Sopta, M.; Ingles, C.J. Interorganellar communication. Altered nuclear gene expression profiles in a yeast mitochondrial dna mutant. J. Biol. Chem 2001, 276, 4020–4027. [Google Scholar]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar]
- Erol, A. Retrograde regulation due to mitochondrial dysfunction may be an important mechanism for carcinogenesis. Med. Hypotheses 2005, 65, 525–529. [Google Scholar]
- Kyle, J.L.; Riesen, W.H. Stress and cigarette smoke effects on lung mitochondrial phosphorylation. Arch. Environ. Health 1970, 21, 492–497. [Google Scholar]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res 2013, 14, 97. [Google Scholar]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lin, Z.; Lamb, R.; Hulit, J.; Howell, A.; Sotgia, F.; Rubin, E.; Lisanti, M.P. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: Implications for breast cancer prevention. Cell Cycle 2013, 12, 289–301. [Google Scholar]
- Kiessling, K.H.; Pilström, L. Effect of ethanol on rat liver. I. Enzymatic and histological studies of liver mitochondria. Q. J. Stud. Alcohol 1966, 27, 189–200. [Google Scholar]
- Syed, M.; Skonberg, C.; Hansen, S.H. Effect of some organic solvents on oxidative phosphorylation in rat liver mitochondria: Choice of organic solvents. Toxicol. in Vitro 2013, 27, 2135–2141. [Google Scholar]
- Hadler, H.I.; Mueller, K.W. The disturbance of oxidative phosphorylation in rat liver mitochondria by the carcinogens 12-hydroxystearic acid and its methyl ester. J. Environ. Pathol. Toxicol 1978, 1, 75–85. [Google Scholar]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar]
- Koike, K. Hepatitis B virus X gene is implicated in liver carcinogenesis. Cancer Lett 2009, 286, 60–68. [Google Scholar]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol 2008, 82, 6798–6811. [Google Scholar]
- D’Agostino, D.M.; Bernardi, P.; Chieco-Bianchi, L.; Ciminale, V. Mitochondria as functional targets of proteins coded by human tumor viruses. Adv. Cancer Res 2005, 94, 87–142. [Google Scholar]
- Koura, M.; Isaka, H.; Yoshida, M.C.; Tosu, M.; Sekiguchi, T. Suppression of tumorigenicity in interspecific reconstituted cells and cybrids. Gann 1982, 73, 574–580. [Google Scholar]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic suppression of malignancy. In Vitro Cell Dev. Biol 1987, 23, 627–632. [Google Scholar]
- Howell, A.N.; Sager, R. Tumorigenicity and its suppression in cybrids of mouse and Chinese hamster cell lines. Proc. Natl. Acad. Sci. USA 1978, 75, 2358–2362. [Google Scholar]
- McKinnell, R.G.; Deggins, B.A.; Labat, D.D. Transplantation of pluripotential nuclei from triploid frog tumors. Science 1969, 165, 394–396. [Google Scholar]
- Li, L.; Connelly, M.C.; Wetmore, C.; Curran, T.; Morgan, J.I. Mouse embryos cloned from brain tumors. Cancer Res 2003, 63, 2733–2736. [Google Scholar]
- Hochedlinger, K.; Blelloch, R.; Brennan, C.; Yamada, Y.; Kim, M.; Chin, L.; Jaenisch, R. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev 2004, 1815, 1875–1885. [Google Scholar]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar]
- Wang, T.; Marquardt, C.; Foker, J. Aerobic glycolysis during lymphocyte proliferation. Nature 1976, 261, 702–705. [Google Scholar]
- Brand, K.; Aichinger, S.; Forster, S.; Kupper, S.; Neumann, B.; Nürnberg, W.; Ohrisch, G. Cell–cycle-related metabolic and enzymatic events in proliferating rat thymocytes. Eur. J. Biochem 1988, 172, 695–702. [Google Scholar]
- Siverio, J.M.; Torres, N.V.; Meléndez-Hevia, E. Activities of l-lactate and glycerol phosphate production rates in vitro from glucose 6-phosphate in regenerating rat liver. Int. J. Biochem 1985, 17, 1015–1017. [Google Scholar]
- Forni, E.; Filipazzi, A. Study of some aspects of liver regeneration after partial hepatectomy. I. Oxigen consumption and glycolysis of regenerating tissue. Chir. Patol. Sper 1964, 12, 263–267. [Google Scholar]
- Menyhárt, J.; Horváth, A.; Rosta, A. A reliable index of tissue metabolic activity during the initial phase of rat liver regeneration. Acta Biochim. Biophys. Acad. Sci. Hung 1971, 6, 139–144. [Google Scholar]
- Vihersaari, T.; Kivisaari, J.; Ninikoski, J. Effect of changes in inspired oxygen tension on wound metabolism. Ann. Surg 1979, 179, 889–895. [Google Scholar]
- Braskén, P.; Renvall, S. Local energy metabolism in healing colon anastomosis. An enzyme-histochemical study in rats. Acta Chir. Scand 1990, 156, 565–570. [Google Scholar]
- O’Connor, R.J. The effect on cell division of inhibiting aerobic glycolysis. Br. J. Exp. Pathol 1950, 31, 449–453. [Google Scholar]
- Bax, B.E.; Bloxam, D.L. Energy metabolism and glycolysis in human placental trophoblast cells during differentiation. Biochim. Biophys. Acta 1997, 1319, 283–292. [Google Scholar]
- Kwon, H.J. ATP oscillations mediate inductive action of FGF and SHH signalling on prechondrogenic condensation. Cell Biochem. Funct 2013, 31, 75–81. [Google Scholar]
- Tsuchiya, M.; Ross, J. Advantages of external periodic events to the evolution of biochemical oscillatory reactions. Proc. Natl. Acad. Sci. USA 2003, 100, 9691–9695. [Google Scholar]
- Boiteux, A.; Goldbeter, A.; Hess, B. Control of oscillating glycolysis of yeast by stochastic, periodic, and steady source of substrate: A model and experimental study. Proc. Natl. Acad. Sci. USA 1975, 72, 3829–3833. [Google Scholar]
- Yang, J.H.; Yang, L.; Qu, Z.; Weiss, J.N. Glycolytic oscillations in isolated rabbit ventricular myocytes. J. Biol. Chem 2008, 283, 36321–36327. [Google Scholar]
- Agathocleous, M.; Love, N.K.; Randlett, O.; Harris, J.J.; Liu, J.; Murray, A.J.; Harris, W.A. Metabolic differentiation in the embryonic retina. Nat. Cell Biol 2012, 14, 859–864. [Google Scholar]
- Ho, J.; de Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; van Houten, B.; et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar]
- Maglietta, R.; Liuzzi, V.C.; Cattaneo, E.; Laczko, E.; Piepoli, A.; Panza, A.; Carella, M.; Palumbo, O.; Staiano, T.; Buffoli, F.; et al. Molecular pathways undergoing dramatic transcriptomic changes during tumor development in the human colon. BMC Cancer 2012, 12, 608. [Google Scholar] [Green Version]
- Brown, N.J.; Higham, S.E.; Perunovic, B.; Arafa, M.; Balasubramanian, S.; Rehman, I. Lactate dehydrogenase-B is silenced by promoter methylation in a high frequency of human breast cancers. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Kim, S.; Kim do, H.; Jung, W.H.; Koo, J.S. Metabolic phenotypes in triple-negative breast Cancer. Tumour Biol 2012, 34, 1699–1712. [Google Scholar]
- Yadava, N.; Schneider, S.S.; Jerry, D.J.; Kim, C. Impaired mitochondrial metabolism and mammary carcinogenesis. J. Mammary Gland Biol. Neoplasia 2013, 18, 75–87. [Google Scholar]
- Buravkova, L.B.; Rylova, Y.V.; Andreeva, E.R.; Kulikov, A.V.; Pogodina, M.V.; Zhivotovsky, B.; Gogvadze, V. Low ATP level is sufficient to maintain the uncommitted state of multipotent mesenchymal stem cells. Biochim. Biophys. Acta 2013, 1830, 4418–4425. [Google Scholar]
- Xun, Z.; Lee, D.Y.; Lim, J.; Canaria, C.A.; Barnebey, A.; Yanonne, S.M.; McMurray, C.T. Retinoic acid-induced differentiation increases the rate of oxygen consumption and enhances the spare respiratory capacity of mitochondria in SH-SY5Y cells. Mech. Ageing Dev 2012, 133, 176–185. [Google Scholar]
- Saumet, A.; Vetter, G.; Bouttier, M.; Antoine, E.; Roubert, C.; Orsetti, B.; Theillet, C.; Lecellier, C.H. Estrogen and retinoic acid antagonistically regulate several microRNA genes to control aerobic glycolysis in breast cancer cells. Mol. Biosyst 2012, 8, 3242–3253. [Google Scholar]
- Heinrich, R.; Meléndez-Hevia, E.; Montero, F.; Nuño, J.C.; Stephani, A.; Waddell, T.G. The structural design of glycolysis: An evolutionary approach. Biochem. Soc. Trans 1999, 27, 294–298. [Google Scholar]
- Van Dijken, J.P.; Weusthuis, R.A.; Pronk, J.T. Kinetics of growth and sugar consumption in yeasts. Antonie Van Leeuwenhoek 1993, 63, 343–352. [Google Scholar]
- Inderlied, C.B.; Sypherd, P.S. Glucose metabolism and dimorphism in Mucor. J. Bacteriol 1978, 133, 1282–1286. [Google Scholar]
- Poolman, B. Energy transduction in lactic acid bacteria. FEMS Microbiol. Rev 1993, 12, 125–147. [Google Scholar]
- Veech, R.L.; Kashiwaya, Y.; Gates, D.N.; King, M.T.; Clarke, K. The energetics of ion distribution: The origin of the resting electric potential of cells. IUBMB Life 2002, 54, 241–252. [Google Scholar]
- Veech, R.L.; Kashiwaya, Y.; King, M.T. The resting membrane potential of cells are measures of electrical work, not of ionic currents. Integr. Physiol. Behav. Sci 1995, 30, 283–307. [Google Scholar]
- Masuda, T.; Dobson, G.P.; Veech, R.L. The Gibbs-Donnan near-equilibrium system of heart. J. Biol. Chem 1990, 265, 20321–20334. [Google Scholar]
- Racker, E. Why do tumor cells have a high aerobic glycolysis? J. Cell Physiol 1976, 89, 697–700. [Google Scholar]
- Schmidt, H.; Siems, W.; Müller, M.; Dumdey, R.; Rapoport, S.M. ATP-producing and consuming processes of Ehrlich mouse ascites tumor cells in proliferating and resting phases. Exp. Cell Res 1991, 194, 122–127. [Google Scholar]
- Termonia, Y.; Ross, J. Oscillations and control features in glycolysis: Numerical analysis of a comprehensive model. Proc. Natl. Acad. Sci. USA 1981, 78, 2952–2956. [Google Scholar]
- Ganitkevich, V.; Mattea, V.; Benndorf, K. Glycolytic oscillations in single ischemic cardiomyocytes at near anoxia. J. Gen. Physiol 2010, 135, 307–319. [Google Scholar]
- Ahsan, H.; Halpern, J.; Kibriya, M.G.; Pierce, B.L.; Tong, L.; Gamazon, E.; McGuire, V.; Felberg, A.; Shi, J.; Jasmine, F.; et al. A Genome-wide association study of early-onset breast cancer identifies PFKM as a novel breast cancer gene and supports a common genetic spectrum for breast cancer at any age. Cancer Epidemiol. Biomark. Prev 2014, 23, 658–669. [Google Scholar]
- Klarer, A.C.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab 2014, 23, 2. [Google Scholar]
- Goldbeter, A. Oscillatory enzyme reactions and Michaelis-Menten kinetics. FEBS Lett 2013, 587, 2778–2784. [Google Scholar]
- Mahnensmith, R.L.; Aronson, P.S. The plasma membrane sodium-hydrogen exchanger and its role in physiological and pathophysiological processes. Circ. Res 1985, 56, 773–788. [Google Scholar]
- Sennoune, S.R.; Bakunts, K.; Martínez, G.M.; Chua-Tuan, J.L.; Kebir, Y.; Attaya, M.N.; Martínez-Zaguilán, R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: Distribution and functional activity. Am. J. Physiol. Cell Physiol 2004, 286, C1443–C1452. [Google Scholar]
- Baluch, S.; Midwood, C.J.; Griffiths, J.R.; Stubbs, M.; Coombes, R.C. Monitoring therapeutic response to tamoxifen in NMU-induced rat mammary tumours by 31P MRS. Br. J. Cancer 1991, 63, 901–904. [Google Scholar]
- Navon, G.; Ogawa, S.; Shulman, R.G.; Yamane, T. 31P nuclear magnetic resonance studies of Ehrlich ascites tumor cells. Proc. Natl. Acad. Sci. USA 1977, 74, 87–91. [Google Scholar]
- Ross, B.D.; Higgins, R.J.; Boggan, J.E.; Knittel, B.; Garwood, M. 31P NMR spectroscopy of the in vivo metabolism of an intracerebral glioma in the rat. Magn. Reson. Med 1988, 6, 403–417. [Google Scholar]
- Bradbury, D.A.; Simmons, T.D.; Slater, K.J.; Crouch, S.P. Measurement of the ADP:ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 2000, 240, 79–92. [Google Scholar]
- Singer, S.; Souza, K.; Thilly, W.G. Pyruvate utilization, phosphocholine and adenosine triphosphate (ATP) are markers of human breast tumor progression: A 31P- and 13C- nuclear magnetic resonance (NMR) spectroscopy study. Cancer Res 1995, 55, 5140–5145. [Google Scholar]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res 2013, 19, 5158–5169. [Google Scholar]
- Park, J.M.; Park, J.H. Human in vivo 31P MR spectroscopy of benign and malignant breast tumors. Korean J. Radiol 2001, 2, 80–86. [Google Scholar]
- Golinska, M.; Troy, H.; Chung, Y.L.; McSheehy, P.M.; Mayr, M.; Yin, X.; Ly, L.; Williams, K.J.; Airley, R.E.; Harris, A.L.; et al. Adaptation to HIF-1 deficiency by upregulation of the AMP/ATP ratio and phosphofructokinase activation in hepatomas. BMC Cancer 2011, 11, 198. [Google Scholar]
- Cho, Y.M.; Kwon, S.; Pak, Y.K.; Seol, H.W.; Choi, Y.M.; Park, D.J.; Park, K.S.; Lee, H.K. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun 2006, 348, 1472–1478. [Google Scholar]
- Lonergan, T.; Bavister, B.; Brenner, C. Mitochondria in stem cells. Mitochondrion 2007, 7, 289–296. [Google Scholar]
- Hoffer, F.A.; Taylor, G.A.; Spevak, M.; Ingber, D.; Fenton, T. Metabolism of tumor regression from angiogenesis inhibition: 31P magnetic resonance spectroscopy. Magn. Reson. Med 1989, 11, 202–208. [Google Scholar]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar]
- Frieden, B.R.; Gatenby, R.A. Information dynamics in living systems, prokaryotes: Eukaryotes, and cancer. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Severini, S. Increased entropy of signal transduction in the cancer metastasis phenotype. BMC Syst. Biol 2010, 4, 104. [Google Scholar]
- West, J.; Lacasa, L.; Severini, S.; Teschendorff, A. Approximate entropy of network parameters. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 2012, 85. [Google Scholar] [CrossRef]
- Banerji, C.R.; Severini, S.; Teschendorff, A.E. Network transfer entropy and metric space for causality inference. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 2013, 87. [Google Scholar] [CrossRef]
- Banerji, C.R.; Miranda-Saavedra, D.; Severini, S.; Widschwendter, M.; Enver, T.; Zhou, J.X.; Teschendorff, A.E. Cellular network entropy as the energy potential in Waddington's differentiation landscape. Sci. Rep 2013, 3, 3039. [Google Scholar]
- Ozernyuk, N.D.; Zotin, A.I.; Yurowitzky, Y.G. Deviation of the living system from the stationary state during oogenesis. Wilhelm Roux Archiv 1973, 172, 66–74. [Google Scholar]
- Zotin, A.A.; Zotin, A.I. Phenomenological theory of ontogenesis. Int. J. Dev. Biol 1997, 41, 917–921. [Google Scholar]
- Martyushev, L.M. Entropy and entropy production: Old misconceptions and new breakthroughs. Entropy 2013, 151, 152–170. [Google Scholar]
- Kumar, P.; Rajput, S.; Verma, A.; De, S.; Datta, T.K. Expression pattern of glucose metabolism genes in relation to development rate of buffalo (Bubalus bubalis) oocytes and in vitro-produced embryos. Theriogenology 2013, 80, 914–922. [Google Scholar]
- Sugimura, S.; Matoba, S.; Hashiyada, Y.; Aikawa, Y.; Ohtake, M.; Matsuda, H.; Kobayashi, S.; Konishi, K.; Imai, K. Oxidative phosphorylation-linked respiration in individual bovine oocytes. J. Reprod. Dev 2012, 58, 636–641. [Google Scholar]
- Abele, D. Toxic oxygen: The radical life-giver. Nature 2002, 420, 27. [Google Scholar]
- Houghton, J.; Morozov, A.; Smirnova, I.; Wang, T.C. Stem cells and Cancer. Semin. Cancer Biol 2007, 17, 191–203. [Google Scholar]
- DePinho, R.A. The age of cancer. Nature 2000, 408, 248–254. [Google Scholar]
- Armitage, P.; Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 2004, 91, 1983–1989. [Google Scholar]
- Fanali, C.; Lucchetti, D.; Farina, M.; Corbi, M.; Cufino, V.; Cittadini, A.; Sgambato, A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: Controversies and perspectives. World J. Gastroenterol 2014, 20, 923–942. [Google Scholar]
- Trosko, J.E. Induction of iPS cells and of cancer stem cells: The stem cell or reprogramming hypothesis of cancer? Anat. Rec 2014, 297, 161–173. [Google Scholar]
- Agathocleous, M.; Harris, W.A. Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol 2013, 23, 484–492. [Google Scholar]
- Smith-Vikos, T. A report of the James Watson lecture at Yale University. Yale J. Biol. Med 2012, 85, 417–419. [Google Scholar]
- Pierce, G.B. Neoplasms, differentiations and mutations. Am. J. Pathol 1974, 77, 103–118. [Google Scholar]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar]
- Santin, G.; Paulis, M.; Vezzoni, P.; Pacchiana, G.; Bottiroli, G.; Croce, A.C. Autofluorescence properties of murine embryonic stem cells during spontaneous differentiation phases. Lasers Surg. Med 2013, 45, 597–607. [Google Scholar]
- Warburg, O.; Gawehn, K.; Geissler, A.W.; Lorenz, S. On the transformation of embryonal metabolism into cancer metabolism. Hoppe. Seylers. Z. Physiol. Chem 1960, 321, 252–257. [Google Scholar]
- Fliedner, S.M.; Kaludercic, N.; Jiang, X.S.; Hansikova, H.; Hajkova, Z.; Sladkova, J.; Limpuangthip, A.; Backlund, P.S.; Wesley, R.; Martiniova, L.; et al. Warburg effect’s manifestation in aggressive pheochromocytomas and paragangliomas: Insights from a mouse cell model applied to human tumor tissue. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Seppet, E.; Gruno, M.; Peetsalu, A.; Gizatullina, Z.; Nguyen, H.P.; Vielhaber, S.; Wussling, M.H.; Trumbeckaite, S.; Arandarcikaite, O.; Jerzembeck, D.; et al. Mitochondria and energetic depression in cell pathophysiology. Int. J. Mol. Sci 2009, 10, 2252–2303. [Google Scholar]
- Xu, T.; Zhao, J.; Hu, P.; Dong, Z.; Li, J.; Zhang, H.; Yin, D.; Zhao, Q. Pentachlorophenol exposure causes Warburg-like effects in zebrafish embryos at gastrulation stage. Toxicol. Appl. Pharmacol 2014, 277, 183–191. [Google Scholar]
- Campos, B.; Gal, Z.; Baader, A. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J. Pathol 2014, 2014. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, X.; Chen, X.; Yang, G.; Wang, Q.; Rao, K.; Xiong, W.; Yuan, J. Benzo(a)pyrene induced mitochondrial dysfunction and cell death in p53-null Hep3B Cells. Mutat. Res 2011, 726, 75–83. [Google Scholar]
- Pavanello, S.; Dioni, L.; Hoxha, M.; Fedeli, U.; Mielzynska-Svach, D.; Baccarelli, A.A. Mitochondrial DNA copy number and exposure to polycyclic aromatic hydrocarbons. Cancer Epidemiol. Biomark. Prev 2013, 22, 1722–1729. [Google Scholar]
- Xie, Y.; Zhong, C.; Zeng, M.; Guan, L.; Luo, L. Effect of hexavalent chromium on electron leakage of respiratory chain in mitochondria isolated from rat liver. Cell Physiol. Biochem 2013, 31, 473–485. [Google Scholar]
- Liu, G.; Cheresh, P.; Kamp, D.W. Molecular basis of asbestos-induced lung disease. Annu. Rev. Pathol 2013, 8, 161–187. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pacini, N.; Borziani, F. Cancer Stem Cell Theory and the Warburg Effect, Two Sides of the Same Coin? Int. J. Mol. Sci. 2014, 15, 8893-8930. https://doi.org/10.3390/ijms15058893
Pacini N, Borziani F. Cancer Stem Cell Theory and the Warburg Effect, Two Sides of the Same Coin? International Journal of Molecular Sciences. 2014; 15(5):8893-8930. https://doi.org/10.3390/ijms15058893
Chicago/Turabian StylePacini, Nicola, and Fabio Borziani. 2014. "Cancer Stem Cell Theory and the Warburg Effect, Two Sides of the Same Coin?" International Journal of Molecular Sciences 15, no. 5: 8893-8930. https://doi.org/10.3390/ijms15058893
APA StylePacini, N., & Borziani, F. (2014). Cancer Stem Cell Theory and the Warburg Effect, Two Sides of the Same Coin? International Journal of Molecular Sciences, 15(5), 8893-8930. https://doi.org/10.3390/ijms15058893