PRRT2 Mutations Are Related to Febrile Seizures in Epileptic Patients


Abstract
:1. Introduction
2. Results and Discussion
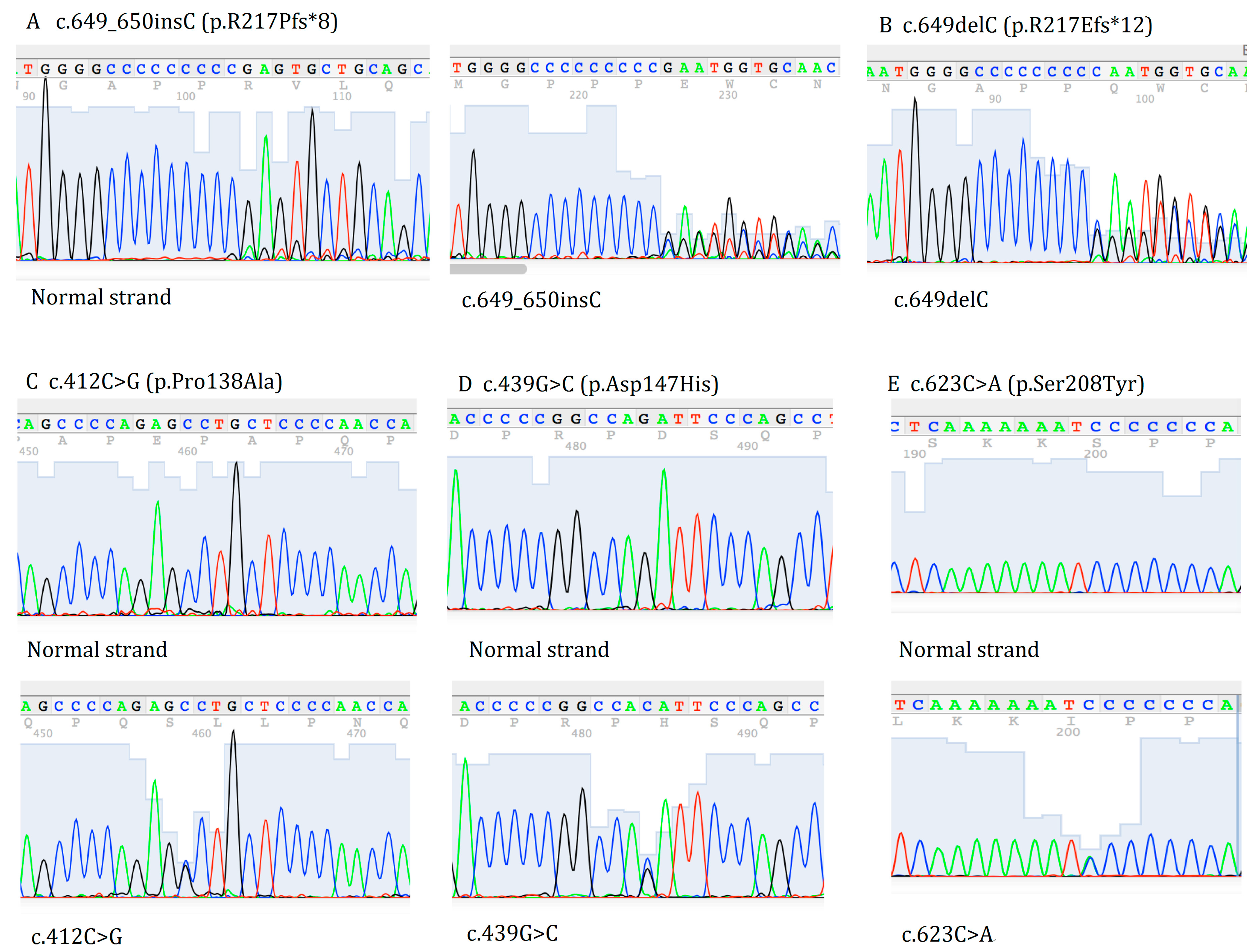

{kind=link}
| Case Number | Gender | Age (year) | Age at Onset | Subtypes | Familial/Sporadic | GGE/MRI/CT | Duration of Seizure | Frequency of Seizure | Current Medication | Nucleotide Changes | Amino Acid Changes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 89 | M | 7 | 2 y | FS+ | Sporadic | N/N/N | <20 s | 1–2 m | CBZ | c.412C>G | p.Pro138Ala |
| 123 | F | 18 | 13 y | FS+ | Sporadic | N/N/ND | 5–10 s | 1–2 m | VPA | c.623C>A | p.Ser208Tyr |
| 174 | M | 16 | 2 y | FS+ | Sporadic | AB/N/N | <30 s | 4–5 m | CBZ, PB | c.412C>G | p.Pro138Ala |
| 114 | M | 6 | 2 y | FS+ | Sporadic | N/ND/N | 1 min | 10–15 y | CBZ | c.412C>G | p.Pro138Ala |
| 192 | M | 13 | 1 y | FS+ | Sporadic | N/AB/N | 10 s | 2–3 y | VPA, LTG | c.439G>C | p.Asp147His |
| 311 | M | 6 | 4 y | FS+ | Sporadic | AB/N/N | 3–5 min | 3–5 m | LEV | c.412C>G | p.Pro138Ala |
| 313 | M | 6 | 3 y | FS+ | Sporadic | N/N/N | 10 s | 2–4 m | VPA, PHT | c.412C>G | p.Pro138Ala |
| SN540 | M | 18 | 5 m | DS | Familial | AB/ND/ND | 1–2 min | 1–2 m | CBZ, OXC | c.640_641insC | p.R217Pfs*8 |
| 363 | M | 33 | 9 y | FS+ | Sporadic | AB/N/N | 3–10 s | 1 m | CBZ,PHT | c.640_641insC | p.R217Pfs*8 |
| c.439G>C | p.Asp147His | ||||||||||
| 737 | M | 7 | 4 y | FS+ | Sporadic | N/ND/N | 2–5 s | 2 m | VPA | c.412C>G | p.Pro138Ala |
| 853 | M | 17 | 8 y | FS+ | Sporadic | N/ND/ND | 2–5 min | 4–8 y | CBZ | c.412C>G | p.Pro138Ala |
| HH69 | M | 15 | 3 y | FS+ | Sporadic | N/N/AB | 1 min | 1–2 m | VPA | c.439G>C | p.Asp147His |
| SN252 | M | 24 | 4 y | FS+ | Sporadic | AB/N/N | 2 min | 2–3 y | CBZ, VPA | c.439G>C | p.Asp147His |
| SN488 | F | 20 | 13 y | FS+ | Sporadic | AB/N/N | 2–3 min | 2–3 m | VPA, TPM | c.412C>G | p.Pro138Ala |
| SN275 | F | 12 | 3 y | FS+ | Sporadic | AB/N/N | 2–4 min | 1–2 y | CBZ | c.649delC | p.Arg217Glufs*12 |
| 812 | M | 32 | 10 y | GEFS+ | Familial | N/ND/ND | 5–6 min | 4–6 d | CBZ | c.412C>G | p.Pro138Ala |
| 1232 | M | 15 | 2 y | GEFS+ | Familial | N/ND/N | 1–3 min | 2 w | VPA | c.412C>G | p.Pro138Ala |
| SN854 | M | 36 | 8 y | GEFS+ | Familial | N/ND/ND | 1–2 min | 2 d | CBZ, PB | c.640_641insC | p.R217Pfs*8 |
| 576 | F | 5 | 5 m | DS | Sporadic | N/ND/N | 1 min | 10 w | CBZ, VPA | c.439G>C | p.Asp147His |
| 1186 | F | 10 | 8 m | DS | Sporadic | N/N/ND | 1–3 min | 2–3 w | CBZ, VPA | c.412C>G | p.Pro138Ala |
| 872 | F | 3 | 9 m | DS | Sporadic | AB/N/N | 10 min | 1 w | CBZ, LEV | c.412C>G | p.Pro138Ala |
| SN676 | M | 32 | 5 m | DS | Sporadic | N/ND/ND | 2–3 min | 2–3 d | OXC, VPA | c.649delC | p.Arg217Glufs*12 |
| SN740 | F | 25 | 40 d | DS | Sporadic | AB/N/N | 1–2 min | 3 m | CBZ, VPA | c.412C>G | p.Pro138Ala |
| SN540 | M | 18 | 5 m | DS | Familial | AB/N/N | 1–2 min | 1–2 m | CBZ, OXC | c.640_641insC | p.R217Pfs*8 |
| 428 | M | 12 | 11 m | DS | Sporadic | AB/ND/N | 1 min | 1–2 w | LEV, VPA | c.412C>G | p.Pro138Ala |
3. Experimental Section
3.1. Subjects
3.2. Mutation Analysis of the Proline-Rich Transmembrane Protein 2 (PRRT2) Gene
| PRRT2 | Forward Primer | Reverse Primer |
|---|---|---|
| Exon2A | 5'-ctcctcctcttccagggttt-3' | 5'-tttttgagggtggtgagtga-3' |
| Exon2B | 5'-tctgagagtgtaggggaaaagc-3' | 5'-ctagggagaggcaaacaaagg-3' |
| Exon3–4 | 5'-tccacctgatcccttctgg-3' | 5'-caggctcccttggtccttag-3' |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gardiner, A.R.; Bhatia, K.P.; Stamelou, M.; Dale, R.C.; Kurian, M.A.; Schneider, S.A.; Wali, G.M.; Counihan, T.; Schapira, A.H.; Spacey, S.D.; et al. PRRT2 gene mutations from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology 2012, 79, 2115–2121. [Google Scholar]
- Chen, W.J.; Lin, Y.; Xiong, Z.Q.; Wei, W.; Ni, W.; Tan, G.H.; Guo, S.L.; He, J.; Chen, Y.F.; Zhang, Q.J.; et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 2011, 43, 1252–1255. [Google Scholar]
- Cloarec, R.; Bruneau, N.; Rudolf, G.; Massacrier, A.; Salmi, M.; Bataillard, M.; Boulay, C.; Caraballo, R.; Fejerman, N.; Genton, P.; et al. PRRT2 links infantile convulsions and paroxysmal dyskinesia with migraine. Neurology 2012, 79, 2097–2103. [Google Scholar]
- De Vries, B.; Callenbach, P.M.C.; Kamphorst, J.T.; Weller, C.M.; Koelewijn, S.C.; ten Houten, R.; de Coo, I.F.M.; Brouwer, O.F.; van den Maagdenberg, A.M.J.M. PRRT2 mutation causes benign familial infantile convulsions. Neurology 2012, 79, 2154–2155. [Google Scholar] [CrossRef]
- Friedman, J.; Olvera, J.; Silhavy, J.L.; Gabriel, S.B.; Gleeson, J.G. Mild paroxysmal kinesigenic dyskinesia caused by PRRT2 missense mutation with reduced penetrance. Neurology 2012, 79, 946–948. [Google Scholar] [CrossRef]
- Lee, H.Y.; Huang, Y.; Bruneau, N.; Roll, P.; Roberson, E.D.O.; Hermann, M.; Quinn, E.; Maas, J.; Edwards, R.; Ashizawa, T.; et al. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. 2012, 1, 2–12. [Google Scholar]
- Marini, C.; Conti, V.; Mei, D.; Battaglia, D.; Lettori, D.; Losito, E.; Bruccini, G.; Tortorella, G.; Guerrini, R. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 2012, 79, 2109–2114. [Google Scholar] [CrossRef]
- Rochette, J.; Roll, P.; Szepetowski, P. Genetics of infantile seizures with paroxysmal dyskinesia: The infantile convulsions and choreoathetosis (ICCA) and icca-related syndromes. J. Med. Genet. 2008, 45, 773–779. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Grinton, B.E.; Heron, S.E.; Kivity, S.; Afawi, Z.; Iona, X.; Goldberg-Stern, H.; Kinali, M.; Andrews, I.; Guerrini, R.; et al. PRRT2 phenotypic spectrum includes sporadic and fever-related infantile seizures. Neurology 2012, 79, 2104–2108. [Google Scholar]
- Wang, J.L.; Cao, L.; Li, X.H.; Hu, Z.M.; Li, J.D.; Zhang, J.G.; Liang, Y.; San-A; Li, N.; Chen, S.Q.; et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011, 134, 3490–3498. [Google Scholar]
- Wang, J.L.; Mao, X.; Hu, Z.M.; Li, J.D.; Li, N.; Guo, J.F.; Jiang, H.; Shen, L.; Li, J.; Shi, Y.T.; et al. Mutation analysis of PRRT2 in two chinese BFIS families and nomenclature of PRRT2 related paroxysmal diseases. Neurosci. Lett. 2013, 552, 40–45. [Google Scholar]
- Wang, K.; Zhao, X.Y.; Du, Y.; He, F.P.; Peng, G.P.; Luo, B.Y. Phenotypic overlap among paroxysmal dyskinesia subtypes: Lesson from a family with PRRT2 gene mutation. Brain Dev. Jpn. 2013, 35, 664–666. [Google Scholar] [CrossRef]
- Rochette, J.; Roll, P.; Fu, Y.H.; Lemoing, A.G.; Royer, B.; Roubertie, A.; Berquin, P.; Motte, J.; Wong, S.W.; Hunter, A.; et al. Novel familial cases of ICCA (infantile convulsions with paroxysmal choreoathetosis) syndrome. Epileptic Disord. 2010, 12, 199–204. [Google Scholar]
- Schubert, J.; Paravidino, R.; Becker, F.; Berger, A.; Bebek, N.; Bianchi, A.; Brockmann, K.; Capovilla, G.; Bernardina, B.D.; Fukuyama, Y.; et al. PRRT2 mutations are the major cause of benign familial infantile seizures. Hum. Mutat. 2012, 33, 1439–1443. [Google Scholar]
- Schmidt, A.; Kumar, K.R.; Redyk, K.; Grunewald, A.; Leben, M.; Munchau, A.; Sue, C.M.; Hagenah, J.; Hartmann, H.; Lohmann, K.; et al. Two faces of the same coin: Benign familial infantile seizures and paroxysmal kinesigenic dyskinesia caused by PRRT2 mutations. Arch. Neurol. 2012, 69, 668–670. [Google Scholar]
- Djemie, T.; Weckhuysen, S.; Holmgren, P.; Hardies, K.; van Dyck, T.; Hendrickx, R.; Schoonjans, A.S.; van Paesschen, W.; Jansen, A.C.; de Meirleir, L.; et al. PRRT2 mutations: Exploring the phenotypical boundaries. J. Neurol. Neurosurg. Psychiatry 2014, 85, 462–465. [Google Scholar] [CrossRef]
- Igarashi, A.; Okumura, A.; Shimada, S.; Shimojima, K.; Abe, S.; Ikeno, M.; Shimizu, T.; Yamamoto, T. Phenotype of patients with a common c.649_650c PRRT2 mutation. Epilepsia 2013, 54, 198–198. [Google Scholar]
- Jing, X.Y.; Li, X.H.; Yuan, P.; Deng, J.; Hu, B.; Wang, Y.M. A novel mutation and functional implications of 5 variants in the PRRT2 gene in 20 paroxysmal kinesigenic dyskinesia pedigrees. Parkinsonism Relat. Disord. 2013, 19, 639–642. [Google Scholar] [CrossRef]
- Labate, A.; Tarantino, P.; Viri, M.; Mumoli, L.; Gagliardi, M.; Romeo, A.; Zara, F.; Annesi, G.; Gambardella, A. Homozygous c.649dupc mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia 2012, 53, e196–e199. [Google Scholar]
- Li, H.F.; Ni, W.; Xiong, Z.Q.; Xu, J.F.; Wu, Z.Y. PRRT2 c.649dupc mutation derived from de novo in paroxysmal kinesigenic dyskinesia. CNS Neurosc. Ther. 2013, 19, 61–65. [Google Scholar]
- Yang, X.; Zhang, Y.; Xu, X.; Wang, S.; Yang, Z.; Wu, Y.; Liu, X.; Wu, X. Phenotypes and PRRT2 mutations in chinese families with benign familial infantile epilepsy and infantile convulsions with paroxysmal choreoathetosis. BMC Neurol. 2013, 13, 209. [Google Scholar] [CrossRef]
- Youn, J.; Jeong, Y.; Ahn, J.Y.; Cho, J.W. PRRT2 gene mutation analysis in korean familial and sporadic patients with paroxysmal kinesigenic dyskinesia. Mov. Disord. 2013, 28, S401–S401. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lee, M.J.; Yu, H.Y.; Chen, C.; Hsu, C.H.; Lin, K.P.; Liao, K.K.; Chang, M.H.; Liao, Y.C.; Soong, B.W. PRRT2 mutations in paroxysmal kinesigenic dyskinesia with infantile convulsions in a taiwanese cohort. PLoS One 2012, 7, e38543. [Google Scholar] [CrossRef]
- Sift. Available online: http://sift.Jcvi.Org/ (accessed on 1 August 2014).
- Polyphen SNP Data Collection. Available online: http://genetics.Bwh.Harvard.Edu/pph/data/ (accessed on 1 August 2014).
- Ali, H.; Urolagin, S.; Gurarslan, O.; Vihinen, M. Performance of protein disorder prediction programs on amino acid substitutions. Hum. Mutat. 2014, 35, 794–804. [Google Scholar] [CrossRef]
- Palamara, G.; Labate, A.; Mumoli, L.; Tarantino, P.; Ferlazzo, E.; Fratto, A.; Pantusa, M.; Annesi, G.; Aguglia, U.; Gambardella, A. Mutations in PRRT2 result in familial infantile seizures with heterogeneous phenotypes including febrile convulsions and probable sudep. Epilepsia 2013, 54, 194–194. [Google Scholar] [CrossRef]
- Heron, S.E.; Ong, Y.S.; Yendle, S.C.; McMahon, J.M.; Berkovic, S.F.; Scheffer, I.E.; Dibbens, L.M. Mutations in PRRT2 are not a common cause of infantile epileptic encephalopathies. Epilepsia 2013, 54, e86–e89. [Google Scholar] [CrossRef]
- Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989, 30, 389–399. [Google Scholar]
- Berg, A.T.; Scheffer, I.E. New concepts in classification of the epilepsies: Entering the 21st century. Epilepsia 2011, 52, 1058–1062. [Google Scholar] [CrossRef]
- Qu, J.; Zhou, B.T.; Yin, J.Y.; Xu, X.J.; Zhao, Y.C.; Lei, G.H.; Tang, Q.; Zhou, H.H.; Liu, Z.Q. ABCC2 polymorphisms and haplotype are associated with drug resistance in chinese epileptic patients. CNS Neurosci. Ther. 2012, 18, 647–651. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.-W.; Qu, J.; Zhang, Y.; Mao, C.-X.; Wang, Z.-B.; Mao, X.-Y.; Deng, Z.-Y.; Zhou, B.-T.; Yin, J.-Y.; Long, H.-Y.; et al. PRRT2 Mutations Are Related to Febrile Seizures in Epileptic Patients. Int. J. Mol. Sci. 2014, 15, 23408-23417. https://doi.org/10.3390/ijms151223408
He Z-W, Qu J, Zhang Y, Mao C-X, Wang Z-B, Mao X-Y, Deng Z-Y, Zhou B-T, Yin J-Y, Long H-Y, et al. PRRT2 Mutations Are Related to Febrile Seizures in Epileptic Patients. International Journal of Molecular Sciences. 2014; 15(12):23408-23417. https://doi.org/10.3390/ijms151223408
Chicago/Turabian StyleHe, Zheng-Wen, Jian Qu, Ying Zhang, Chen-Xue Mao, Zhi-Bin Wang, Xiao-Yuan Mao, Zhi-Yong Deng, Bo-Ting Zhou, Ji-Ye Yin, Hong-Yu Long, and et al. 2014. "PRRT2 Mutations Are Related to Febrile Seizures in Epileptic Patients" International Journal of Molecular Sciences 15, no. 12: 23408-23417. https://doi.org/10.3390/ijms151223408
APA StyleHe, Z.-W., Qu, J., Zhang, Y., Mao, C.-X., Wang, Z.-B., Mao, X.-Y., Deng, Z.-Y., Zhou, B.-T., Yin, J.-Y., Long, H.-Y., Xiao, B., Zhang, Y., Zhou, H.-H., & Liu, Z.-Q. (2014). PRRT2 Mutations Are Related to Febrile Seizures in Epileptic Patients. International Journal of Molecular Sciences, 15(12), 23408-23417. https://doi.org/10.3390/ijms151223408