Comparative Transcriptome Analysis to Reveal Genes Involved in Wheat Hybrid Necrosis

Abstract
:1. Introduction
2. Results
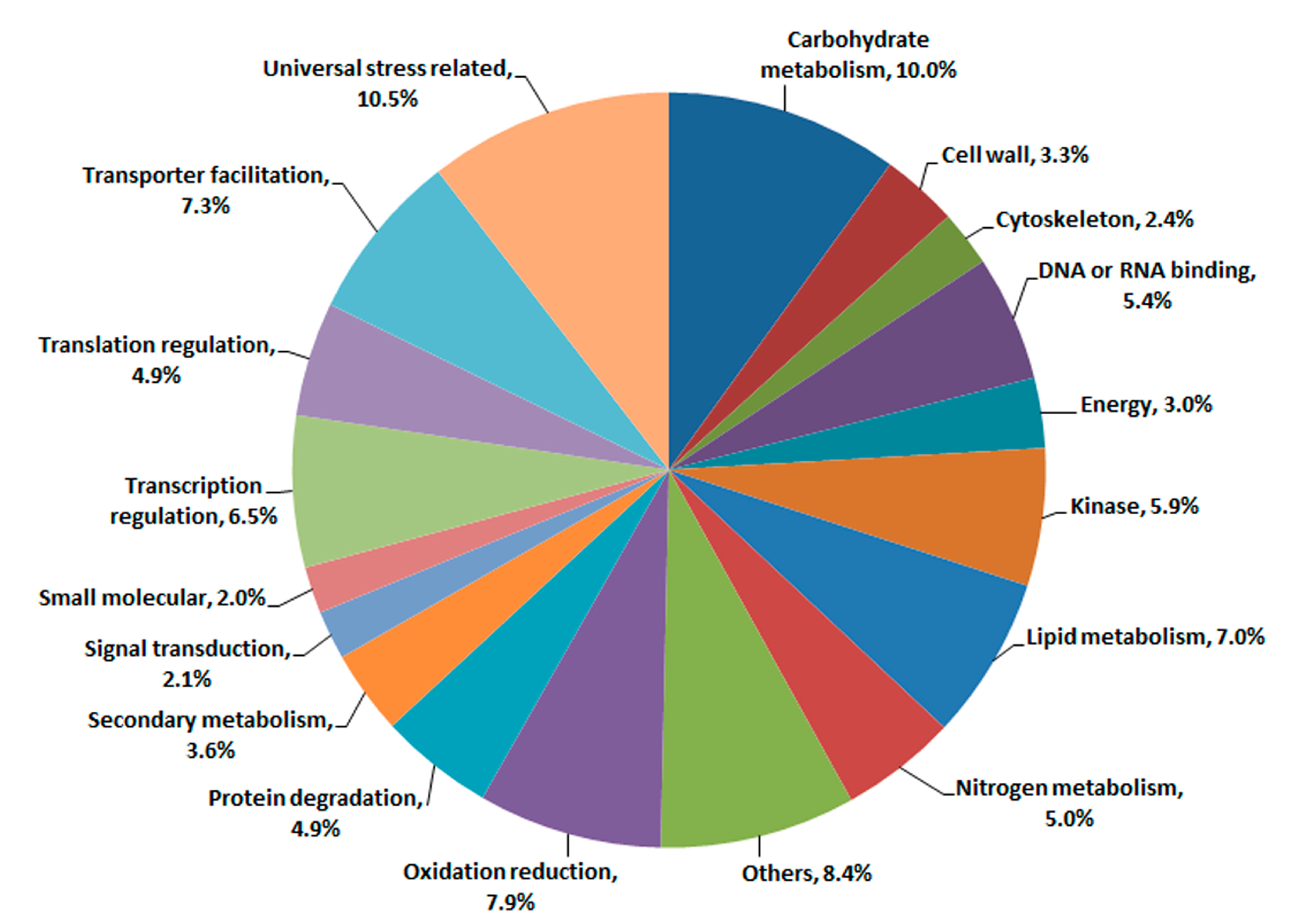
2.1. Illumina Sequencing and Gene Annotation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | High Quality Pair Reads | High Quality Single Reads | High Quality Nucleotides (bp) | Percent of Mapping Reads |
|---|---|---|---|---|---|
| F1 | 38,517,039 × 2 | 24,611,127 × 2 | 9,366,249 | 5,682,981,856 | 55.87% |
| II469 | 21,801,556 × 2 | 13,911,571 × 2 | 5,306,501 | 3,206,968,477 | 55.50% |
| N8 | 24,465,242 × 2 | 15,731,640 × 2 | 5,929,060 | 3,626,936,717 | 52.83% |
2.2. Changes in Global Gene Transcription under Hybrid Necrosis
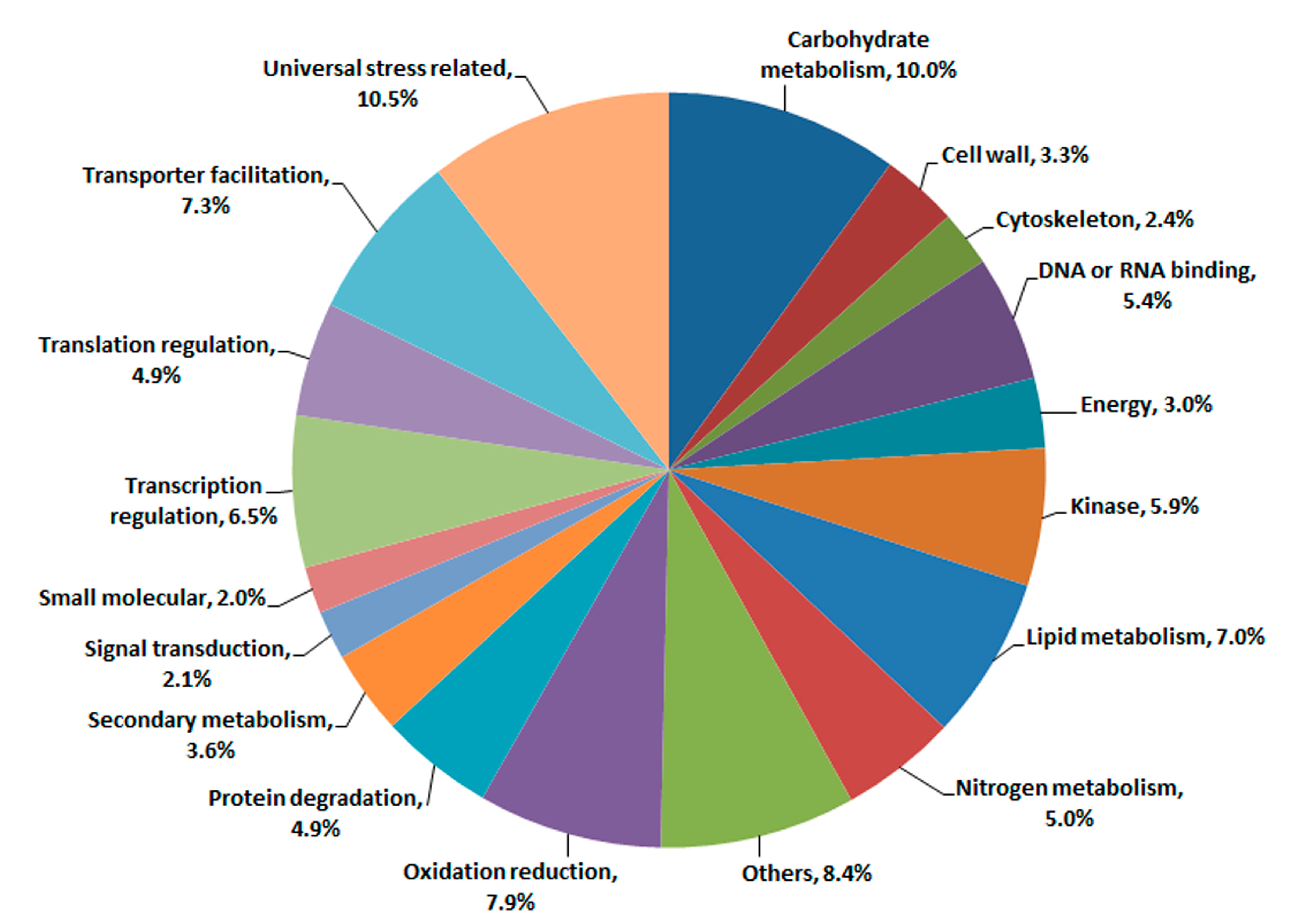
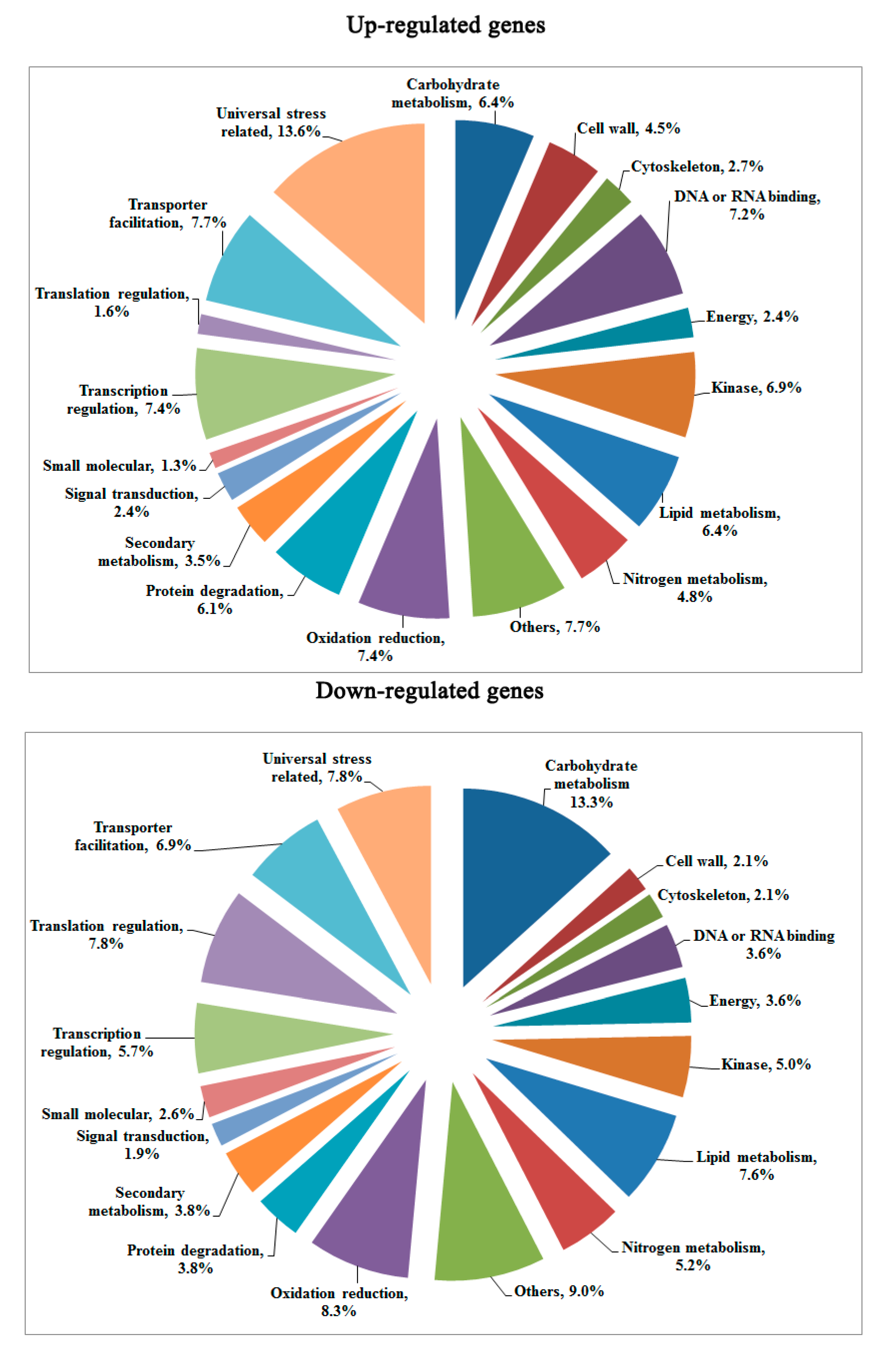
2.3. Transcriptomic Comparison of F1 and Its Parents Using Digital Gene Expression (DGE) Profiling
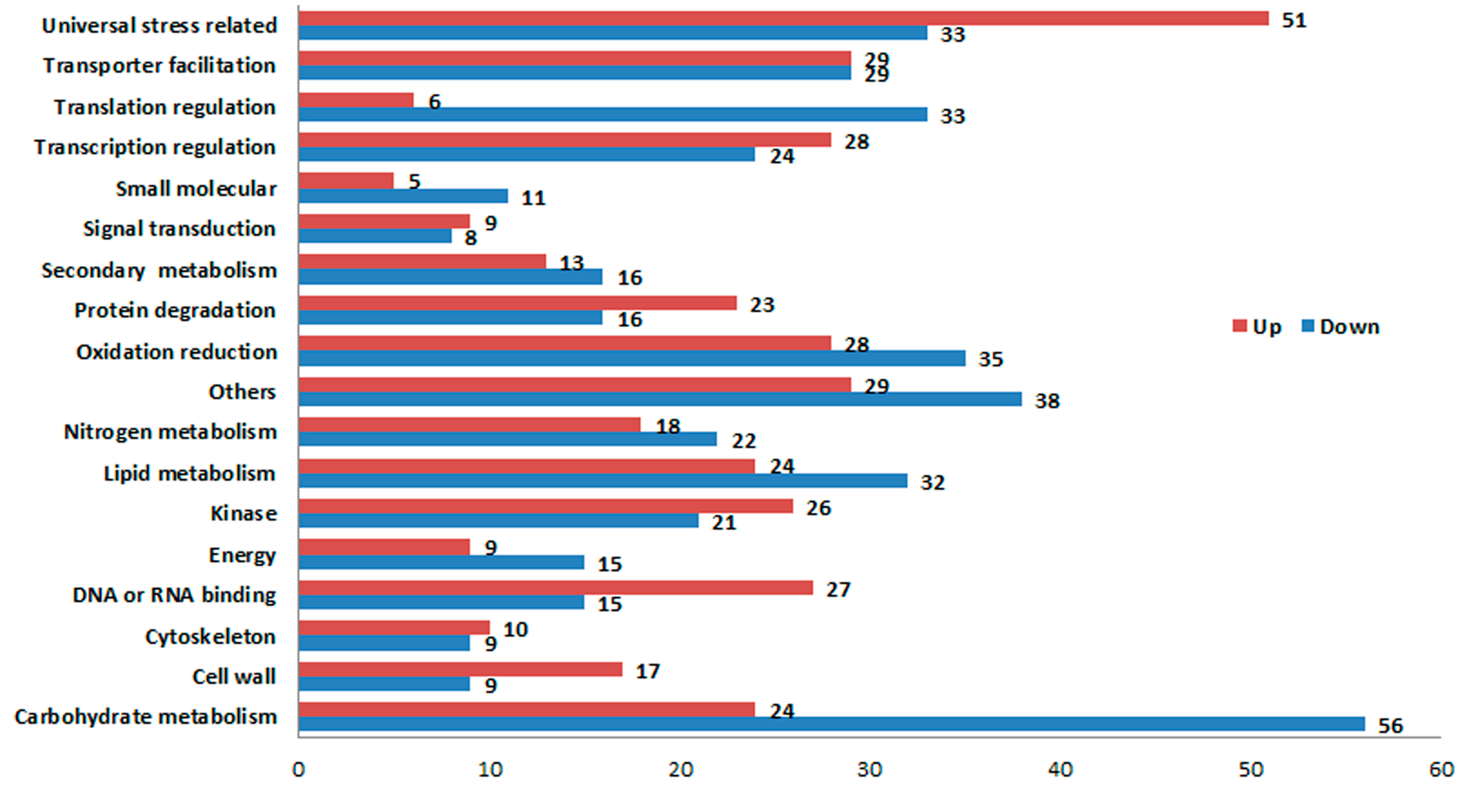
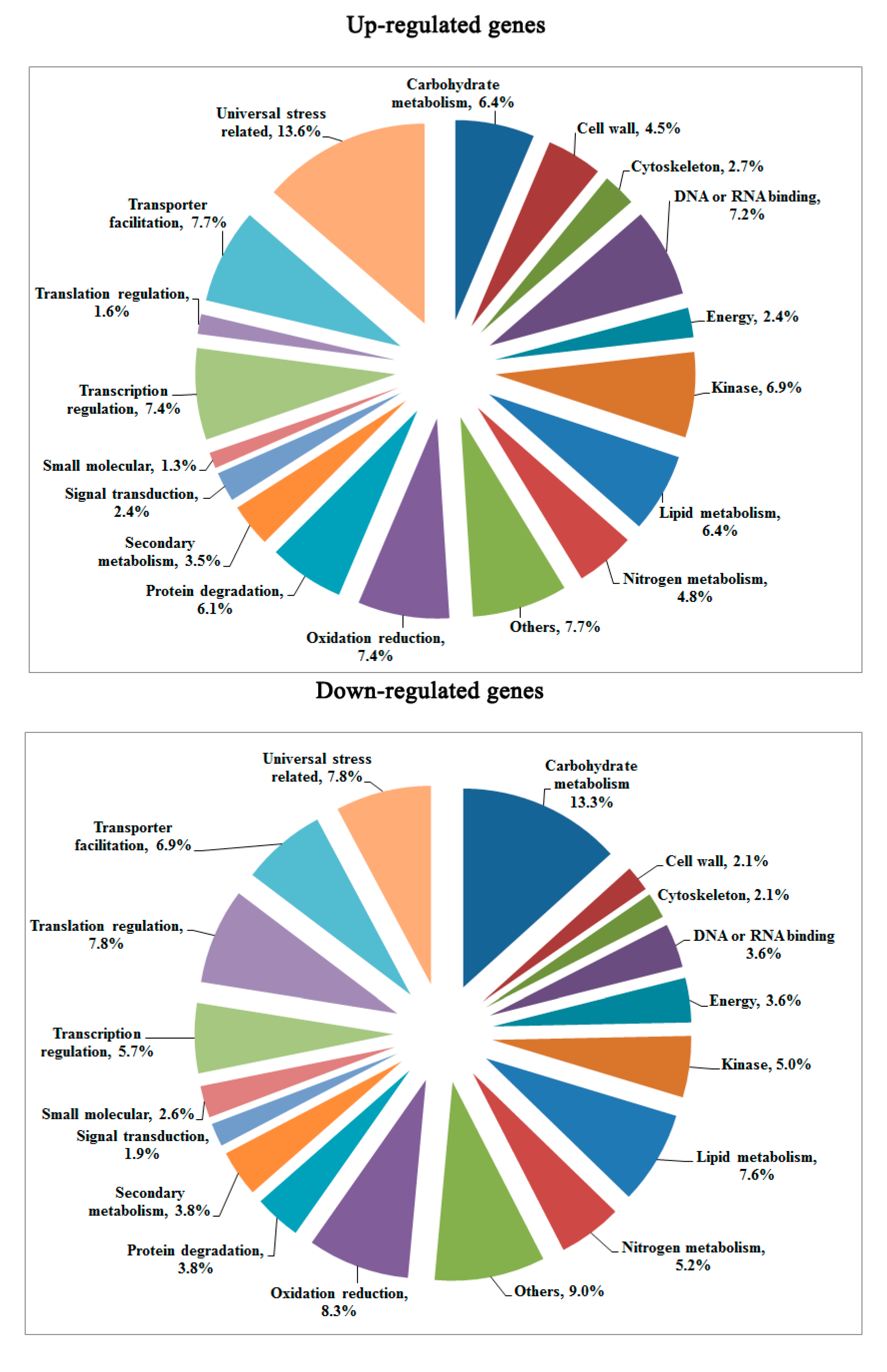
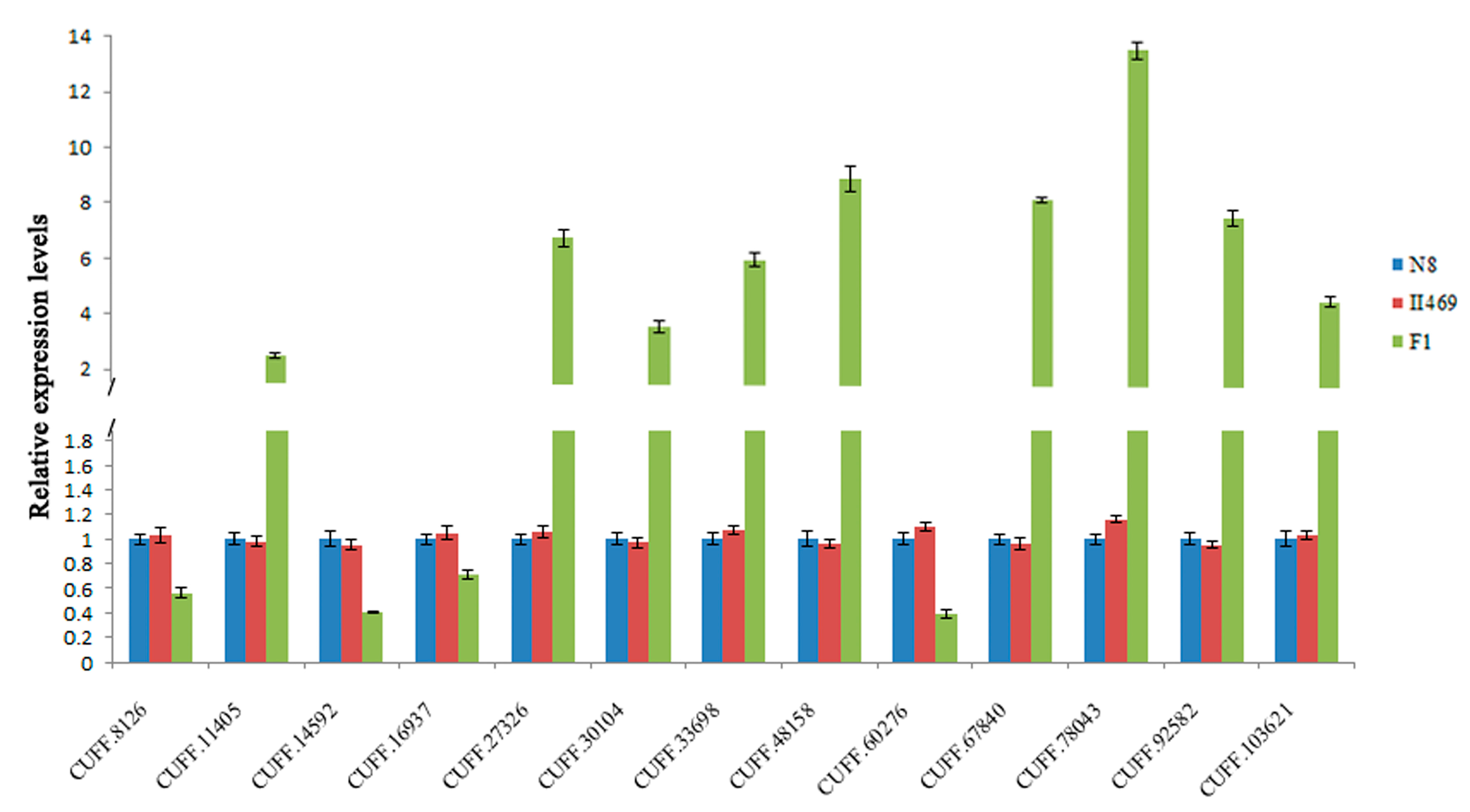
2.4. Validation of DGE Results Using Quantatitive RT-PCR (qRT-PCR)
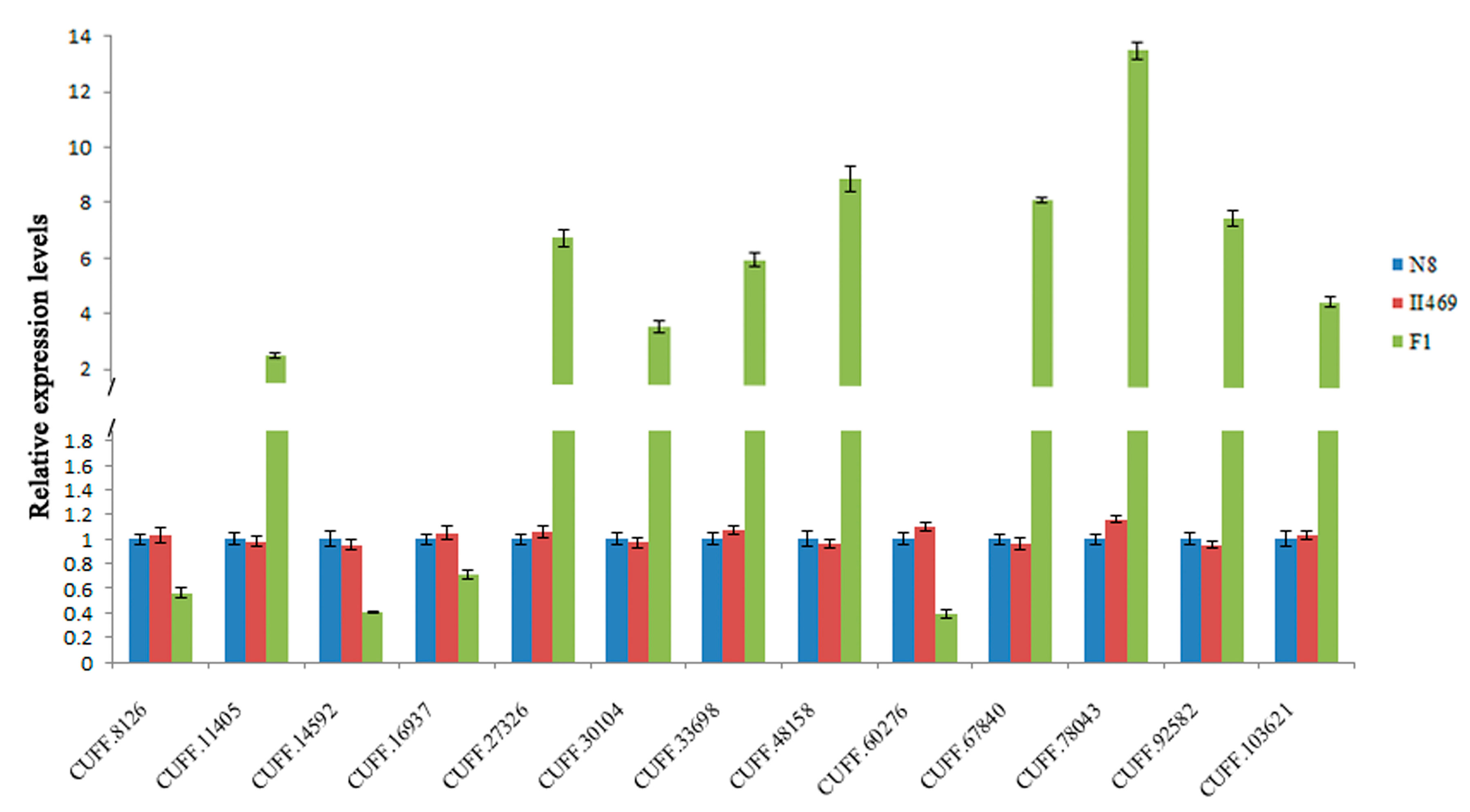
3. Discussion
4. Experimental Section
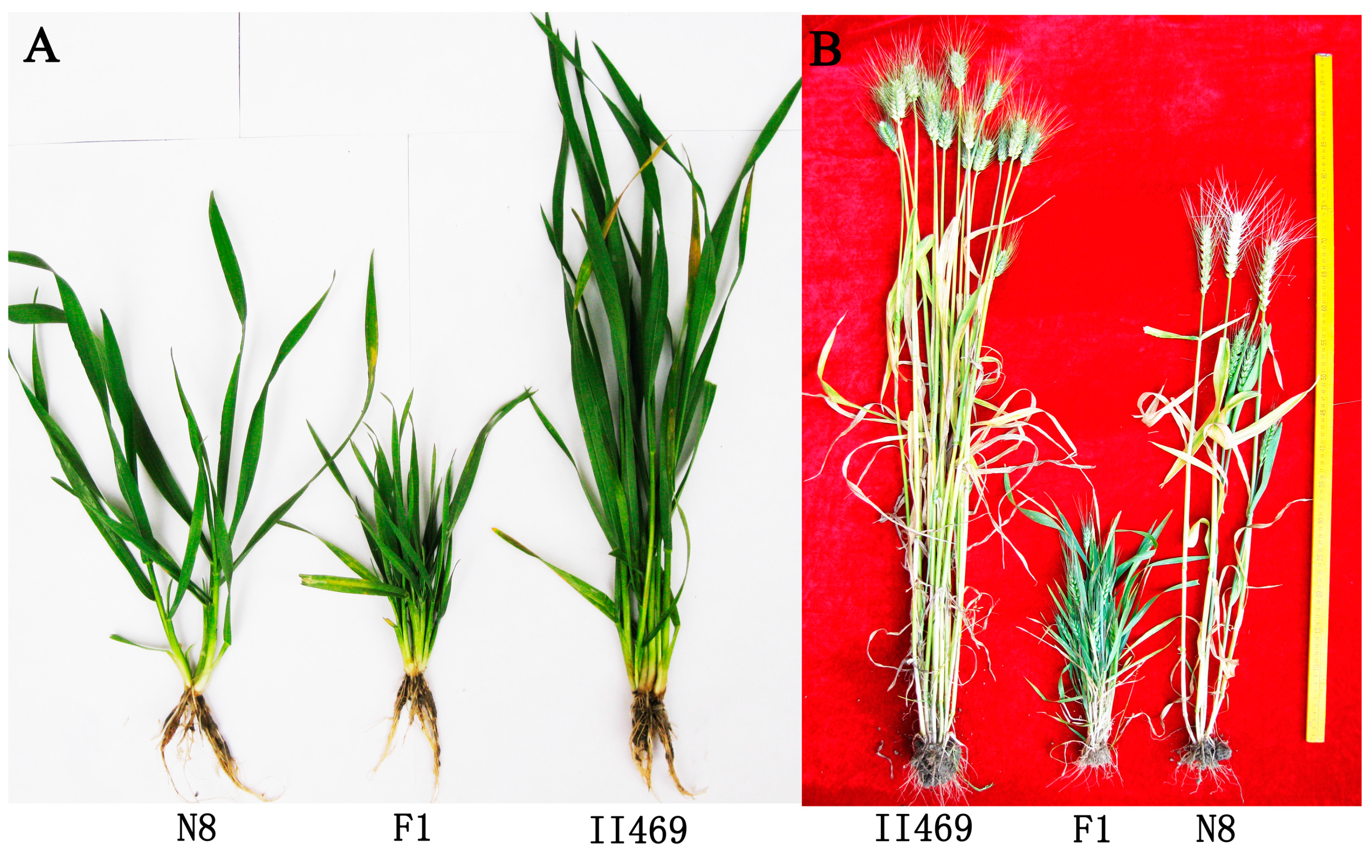
4.1. Plant Materials
4.2. RNA Extraction
4.3. cDNA Library Development and Sequencing
4.4. Data Filtering and Gene Annotation
4.5. Identification of Differentially Expressed Genes
4.6. Quantitative Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bomblies, K.; Weigel, D. Hybrid necrosis: Autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 2007, 8, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.M.S.; Kochumadhavan, M.; Nambisan, P.N.N. Hybrid weakness in Triticumdicoccum Schubl. Wheat Inf. Serv. 1991, 72, 9–11. [Google Scholar]
- Bizimungu, B.; Collin, J.; Comeau, A.; St-Pierre, C.A. Hybrid necrosis as a barrier to gene transfer in hexaploid winter wheat triticale crosses. Can. J. Plant Sci. 1998, 78, 239–244. [Google Scholar] [CrossRef]
- Orr, H.A. Dobzhansky, Bateson, and the genetics of speciation. Genetics 1996, 144, 1331–1335. [Google Scholar] [PubMed]
- Alcázar, R.; García, A.V.; Parker, J.E.; Reymond, M. Incremental steps toward incompatibility revealed by Arabidopsisepistatic interactions modulating salicylic acid pathway activation. Proc. Natl. Acad. Sci. USA 2009, 106, 334–339. [Google Scholar] [CrossRef]
- Dalal, M.; Khanna-Chopra, R. Lipid peroxidation is an early event in necrosis of wheat hybrid. Biochem. Biophys. Res. Commun. 1999, 262, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Dalal, M.; Khanna-Chopra, R. Differential response of antioxidant enzymes in leaves of necrotic wheat hybrids and their parents. Physiol. Plant. 2001, 111, 297–304. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; bBarker, G.L.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Gill, B.S.; Appels, R.; Botha-Oberholster, A.M.; Buell, C.R.; Bennetzen, J.L.; Chalhoub, B.; Chumley, F.; Dvorak, J.; Iwanaga, M.; Keller, B.; et al. Aworkshop report on wheatgenome sequencing: Interna-Tional Genome Researchon Wheat Consortium. Genetics 2004, 168, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Tsunewaki, K. Aneuploid analysis of hybrid necrosis and hybrid chlorosis in tetraploidwheats using the d-genome chromosome substitution lines of durum wheat. Genome 1992, 35, 594–601. [Google Scholar] [CrossRef]
- Tomar, S.M.S.; Singh, B. Hybrid chlorosis in wheat × ryecrosses. Euphytica 1998, 99, 1–4. [Google Scholar] [CrossRef]
- Chu, C.G.; Faris, J.D.; Friesen, T.L.; Xu, S.S. Molecular mapping of hybrid necrosis genes Ne1 and Ne2 in hexaploid wheat using microsatellite markers. Theor. Appl. Genet. 2006, 112, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.M.; Compton, L.E. Complementary lethal genes in wheat causing a progressive lethal necrosis of seedlings. J. Hered. 1943, 34, 67–70. [Google Scholar]
- Hermsen, J.G.T. Hybrid dwarfness in wheat. Euphytica 1967, 16, 134–162. [Google Scholar] [CrossRef]
- Nishikawa, K.; Mori, T.; Takami, N.; Furuta, Y. Mapping of progressive necrosis gene Ne1 and Ne2 of common wheat by the telocentric method. Jpn. J. Breed. 1974, 24, 277–281. [Google Scholar] [CrossRef]
- Singh, S.; Chaudhary, H.K.; Sethi, G.S. Distribution and allelic expressivity of genes for hybrid necrosis in some elite winter and spring wheat ecotypes. Euphytica 2000, 112, 95–100. [Google Scholar] [CrossRef]
- Zeven, A.C. Determination of the chromosome and its arm carrying the Ne1-locus of Triticumaestivum L., Chinese Spring and the Ne1-expressivity. Wheat Inf. Serv. 1972, 33–34, 4–6. [Google Scholar]
- Lee, J.H.; Choi, J.Y.; Tao, X.Y.; Kim, J.S.; Kim, W.; Je, Y.H. Transcriptome analysis of the small brownplanthopper, Laodelphaxstriatellus carrying rice stripe virus. Plant Pathol. J. 2013, 29, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Mao, D.; Xu, L.; Li, D.; Song, S.; Chen, C. Integrated analysis of miRNA and mRNA expression profiles in response to Cd exposure in rice seedlings. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Kakumanu, A.; Ambavaram, M.M.R.; Klumas, C.; Krishnan, A.; Batlang, U.; Myers, E.; Grene, R.; Pereira, A.; Grene, R. Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq. Plant Physiol. 2012, 160, 846–867. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.J.; Li, X.H.; Liu, Z.W.; Xu, Z.S.; Zhuang, J. De novo assembly and transcriptome characterization: Novel insights into catechins biosynthesis in Camellia sinensis. BMC Plant Biol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.L.; Qi, X.Q.; Wang, L.H.; Zhang, Y.X.; Hua, W.; Li, D.H.; Lv, H.X.; Zhang, X.R. Characterization of the sesame (Sesamumindicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.L.; Tan, X.Y.; Hu, C.W.; Zeng, L.; Lu, G.Y.; Fu, G.P.; Cheng, Y.; Zhang, X.K. The Transcriptome of Brassica napus L. Roots under water logging at the seedling stage. Int. J. Mol. Sci. 2013, 14, 2637–2651. [Google Scholar] [PubMed]
- Liu, D.F.; Sui, S.Z.; Ma, J.; Li, Z.N.; Guo, Y.L.; Luo, D.P.; Yang, J.F.; Li, M.Y. Transcriptomicanalysis of flower development in winter sweet (Chimonanthus praecox). PLoS One 2014, 9, e86976. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gupta, S.; Lindquist, I.E.; Cameron, C.T.; Mudge, J.; Rashotte, A.M. Transcriptome Analysis of cytokinin response in tomato leaves. PLoS One 2013, 8, e55090. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Pearce, S.P.; Distelfeld, A.; Christiansen, M.W.; Uauy, C.; Akhunov, E.; Fahima, T.; Dubcovsky, J. Effect of the down-regulation of the high Grain Protein Content (GPC) genes on the wheat transcriptome during monocarpic senescence. BMC Genomics 2011, 12, 492. [Google Scholar] [CrossRef] [PubMed]
- Pont, C.; Murat, F.; Confolent, C.; Balzergue, S.; Salse, J. RNA-Seqin grain unveils fate of neo- and paleopolyploidization events in bread wheat (Triticumaestivum L.). Genome Biol. 2011, 12, R119. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, R.; Pan, L.; Tang, L.; Zhao, G.; Zhu, M.; Chu, J.; Sun, X.; Wei, B.; Zhang, X.; et al. Transcriptome analysis of H2O2-treated wheat seedlings reveals a H2O2-responsive fatty acid desaturase gene participating in powdery mildew resistance. PLoS One 2011, 6, e28810. [Google Scholar] [CrossRef] [PubMed]
- Oono, Y.; Kobayashi, F.; Kawahara, Y.; Yazawa, T.; Handa, H.; Itoh, T.; Matsumoto, T. Characterisation of the wheat (Triticumaestivum L.) transcriptome by de novo assembly for the discovery of phosphate starvation-responsive genes: Gene expression in Pi-stressed wheat. BMC Genomics 2013, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Song, G.; Gao, J.; Li, Y.; Guo, D.; Fan, Q.; Sui, X.; Chu, X.; Huang, C.; Liu, J.; et al. Transcriptome characterization and differential expression analysis of cold-responsive genes in young spikes of common wheat. J. Biotechnol. 2014, 189, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Mackey, D.; Belkhadir, Y.; Alonso, J.M.; Ecker, J.R.; Dangl, J.L. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 2003, 112, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Mackey, D.; Holt, B.F., 3rd.; Wiig, A.; Dangl, J.L. RIN4 interacts with Pseudomonas syringaetype III effector molecules and is required for for RPM1-mediated disease resistance in Arabidopsis. Cell 2002, 108, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Staskawicz, B.J. Initiation of RPS2-specified disease resistance in Arabidopsisis coupled to the AvrRpt2-directed elimination of RIN4. Cell 2003, 112, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hua, J. A haplotype-specific Resistance gene regulated by BONZAI1 mediates temperature-dependent growth control in Arabidopsis. Plant Cell 2004, 16, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Liu, R.; Gallie, D.R. The unique evolution of the programmed cell death 4 protein in plants. BMC Evol. Biol. 2013, 13, 199. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cech, T.R. Pot1, the putative telomere end binding protein in fission yeast and humans. Science 2001, 292, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Shakirov, E.V.; Perroud, P.F.; Nelson, A.D.; Cannell, M.E.; Quatrano, R.S.; Shippen, D.E. Protection of Telomeres 1 is required for telomere integrity in the moss Physcomitrella patens. Plant Cell 2010, 22, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, H.; Gao, Q.; Wang, L.; Zhang, F.; Siva, V.S.; Zhou, Z.; Song, L.; Zhang, S. Transcriptome analysis of artificial hybrid pufferfish Jiyan-1 and its parental species: Implications for pufferfish heterosis. PLoS One 2013, 8, e58453. [Google Scholar] [CrossRef] [PubMed]
- Kornitzer, D.; Ciechanover, A. Modes of regulation of ubiquitin-mediated protein degradation. J. Cell. Physiol. 2000, 182, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, S.; Chen, F.; Chen, H.; Wang, J.; McCall, C.; Xiong, Y.; Deng, X.W. Arabidopsis DDB1-CUL4 ASSOCIATED FACTOR1 forms a nuclear E3 ubiquitin ligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant Cell 2008, 20, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Yi, J.; Yoon, J.; Cho, L.H.; Ping, J.; Jeong, H.J.; Cho, S.K.; Kim, W.T.; An, G. OsPUB15, an E3 ubiquitin ligase, functions to reduce cellular oxidative stress during seedling establishment. Plant J. 2011, 65, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.B.; Wang, J.M.; Yang, F.X.; Yang, L.; Yue, Y.F.; Xiang, J.B.; Gao, M.; Xiong, F.J.; Lv, D.; Wu, X.J.; et al. A novel membrane-bound E3 ubiquitin ligase enhances the thermal resistance in plants. Plant Biotechnol. J. 2014, 12, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, F.; Zhang, H.; He, H.; Ma, L.; Deng, X.W. Functional characterization of the Arabidopsis ubiquitin-specific protease gene family reveals specific role and redundancy of individual members in development. Plant J. 2008, 55, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Bai, X.; Luo, X.; Chen, Q.; Cai, H.; Ji, W.; Zhu, Y. Identification of wild soybean (Glycine soja) TIFY family genes and their expression profiling analysis under bicarbonate stress. Plant Cell Rep. 2013, 32, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.G.; Laloraya, M.; Wang, C.Y.; Ruan, Q.G.; Davoodi-Semiromi, A.; Kao, K.J.; She, J.X. The autoimmune regulator (AIRE) is a DNA-binding protein. J. Biol. Chem. 2001, 276, 41357–41364. [Google Scholar] [CrossRef] [PubMed]
- Pion, S.; Fontaine, P.; Desaulniers, M.; Jutras, J.; Filep, J.G.; Perreault, C. On the mechanisms of immunodominance in cytotoxic T lymphocyte responses to minor histocompatibility antigens. Eur. J. Immunol. 1997, 27, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Defraia, C.T.; Wang, Y.; Yao, J.; Mou, Z. Elongator subunit 3 positively regulates plant immunity through its histone acetyltransferase and radical S-adenosylmethionine domains. BMC Plant Biol. 2013, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Smeds, L.; Künstner, A. ConDeTri—A content dependent read trimmer for Illumina data. PLoS One 2011, 6, e26314. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomesusing RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Cheng, Y.; Guo, J.; Yang, E.; Liu, C.; Zheng, X.; Deng, K.; Zhou, J. Comparative Transcriptome Analysis to Reveal Genes Involved in Wheat Hybrid Necrosis. Int. J. Mol. Sci. 2014, 15, 23332-23344. https://doi.org/10.3390/ijms151223332
Zhang Y, Cheng Y, Guo J, Yang E, Liu C, Zheng X, Deng K, Zhou J. Comparative Transcriptome Analysis to Reveal Genes Involved in Wheat Hybrid Necrosis. International Journal of Molecular Sciences. 2014; 15(12):23332-23344. https://doi.org/10.3390/ijms151223332
Chicago/Turabian StyleZhang, Yong, Yan Cheng, Jiahui Guo, Ennian Yang, Cheng Liu, Xuelian Zheng, Kejun Deng, and Jianping Zhou. 2014. "Comparative Transcriptome Analysis to Reveal Genes Involved in Wheat Hybrid Necrosis" International Journal of Molecular Sciences 15, no. 12: 23332-23344. https://doi.org/10.3390/ijms151223332
APA StyleZhang, Y., Cheng, Y., Guo, J., Yang, E., Liu, C., Zheng, X., Deng, K., & Zhou, J. (2014). Comparative Transcriptome Analysis to Reveal Genes Involved in Wheat Hybrid Necrosis. International Journal of Molecular Sciences, 15(12), 23332-23344. https://doi.org/10.3390/ijms151223332