Synthesis, Docking and Biological Activities of Novel Hybrids Celecoxib and Anthraquinone Analogs as Potent Cytotoxic Agents

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
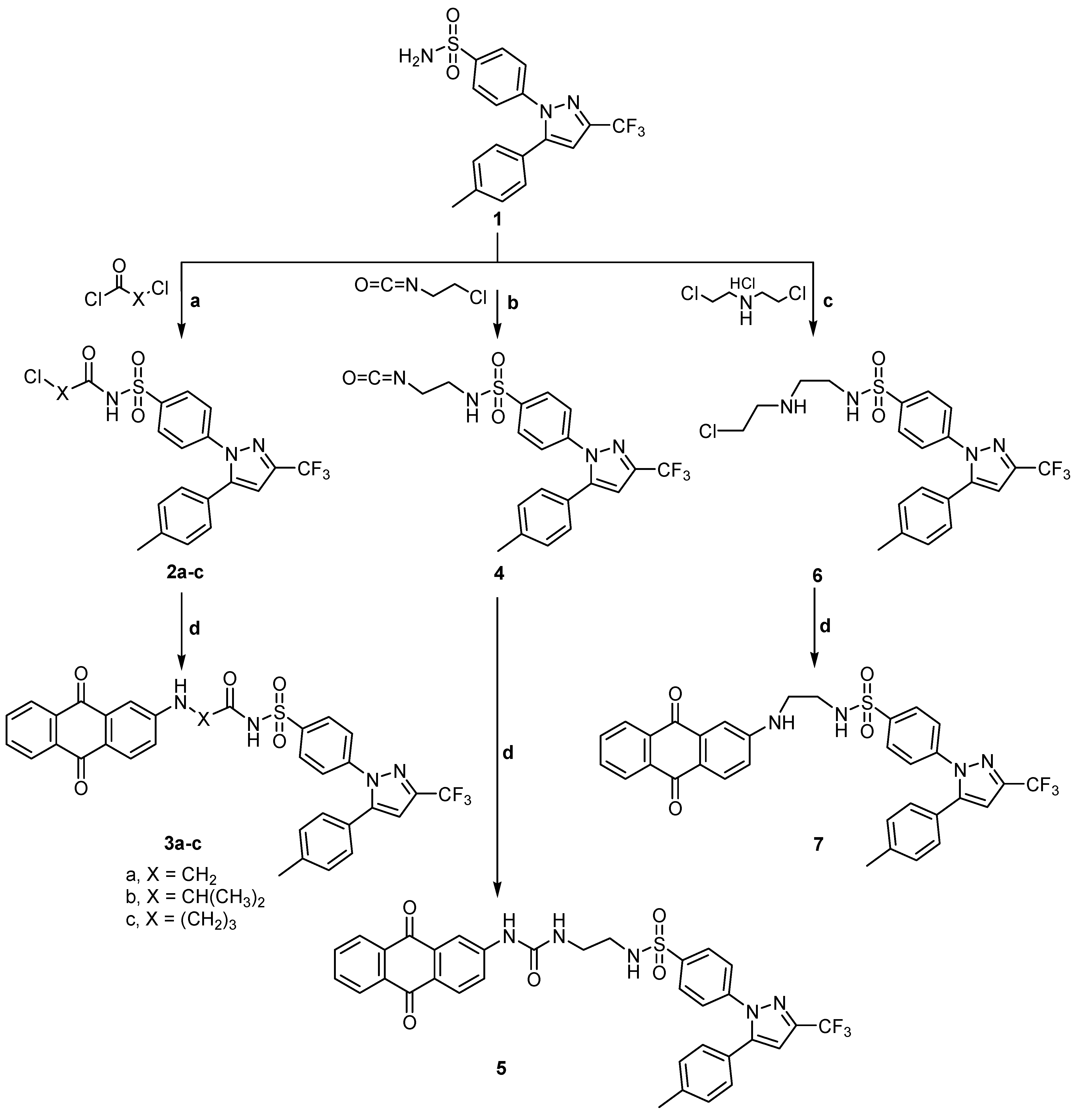
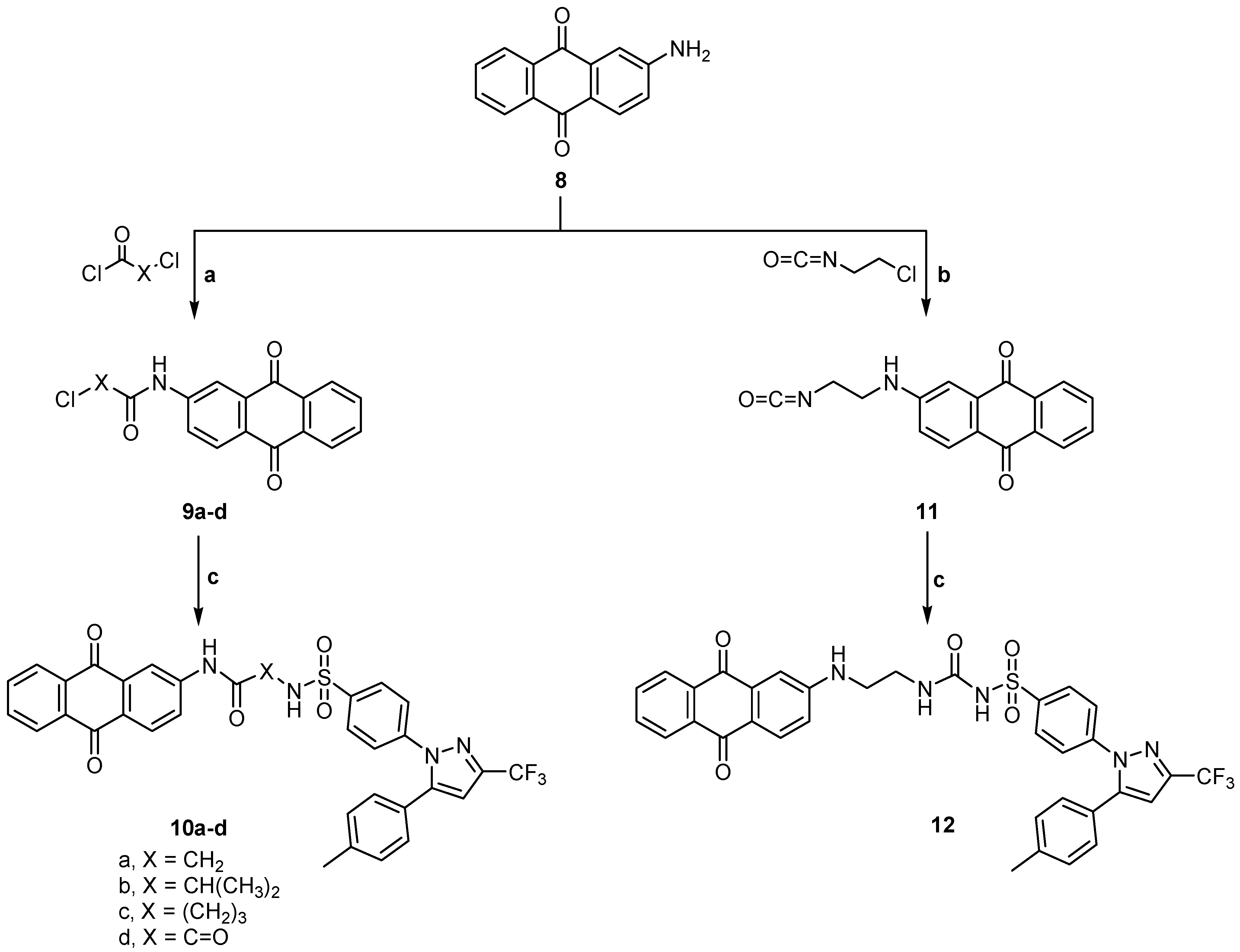
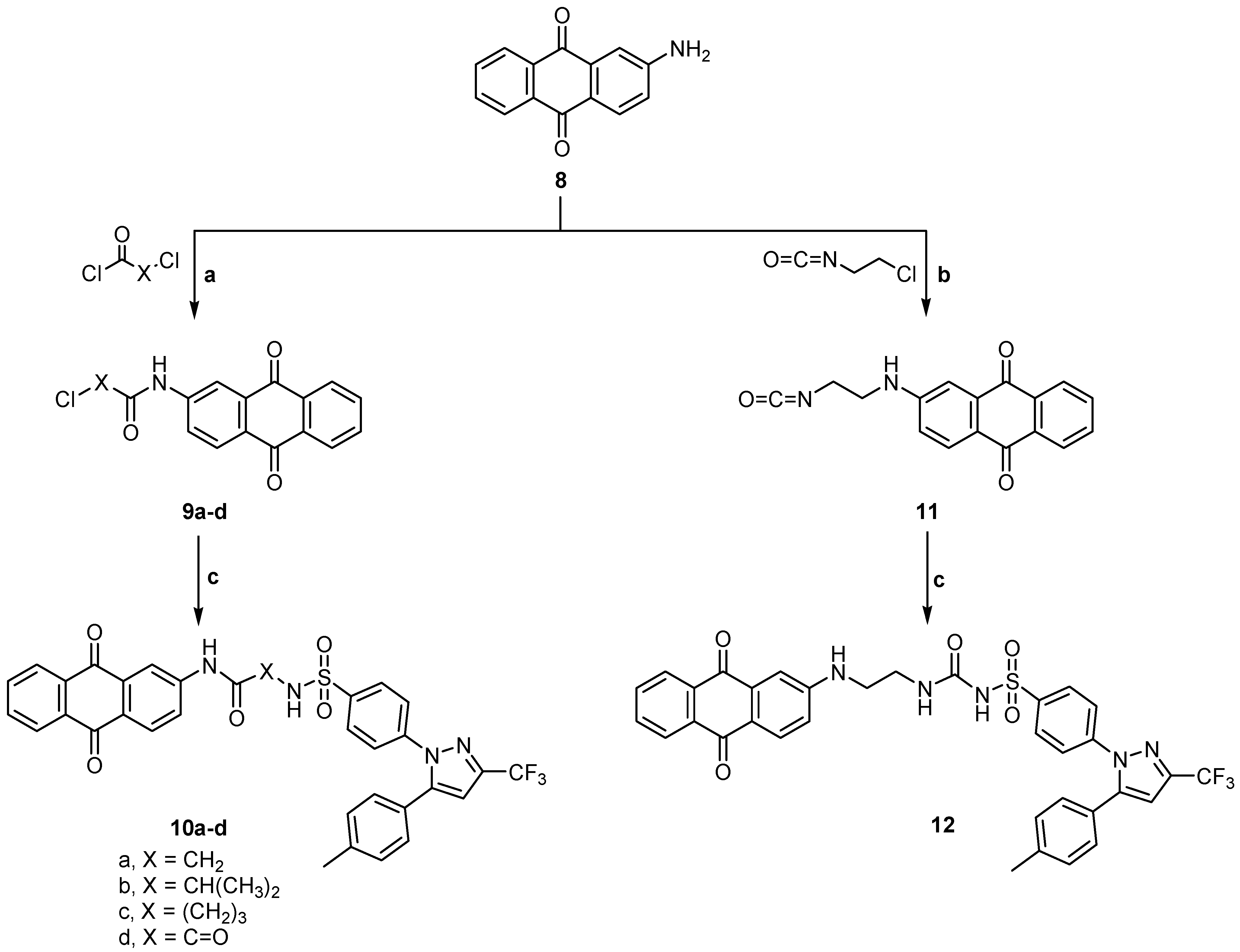
2.1. Chemistry
2.2. Pharmacological Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μg·mL−1) |
|---|---|
| 3a | 6.53 |
| 3b | 9.38 |
| 3c | 3.74 |
| 7 | 4.31 |
| 10a | 8.48 |
| 10b | 7.43 |
| 10c | 8.54 |
| 10d | 6.59 |
| 12 | 3.92 |
| 5-Flurouracil | 5.00 |
| Doxorubicin | 3.56 |
| Compounds | ALT (IU/mL) | AST (IU/mL) | ALP (k.k./dL) |
|---|---|---|---|
| 3a | 38.53 ± 6.50 *** | 111.59 ± 12.80 ** | 19.99 ± 4.39 *** |
| 3b | 60.80 ± 9.20 * | 157.28 ± 20.30 * | 43.25 ± 7.08 * |
| 3c | 67.39 ± 11.00 * | 146.40 ± 28.10 * | 36.90 ± 9.80 * |
| 7 | 39.10 ± 8.40 *** | 126.20 ± 12.10 ** | 18.76 ± 6.43 *** |
| 10a | 45.51 ± 4.23 *** | 108.66 ± 4.61 *** | 21.77 ± 3.50 ** |
| 10b | 54.2 ± 11.05 * | 146.50 ± 28.90 * | 46.47 ± 10.87 * |
| 10c | 78.10 ± 13.30 * | 141.20 ± 32.04 * | 31.46 ± 9.45 * |
| 10d | 48.07 ± 6.16 *** | 112.09 ± 8.83 *** | 17.79 ± 3.06 *** |
| 12 | 51.88 ± 11.50 ** | 113.00 ± 9.52 ** | 21.09 ± 3.48 ** |
| Control | 43.50 ± 2.03 | 108.32 ± 4.19 | 17.70 ± 1.10 |
| 5-Flurouracil | 51.47 ± 9.02 * | 130.43 ± 8.92 * | 25.49 ± 6.03 * |
| Doxorubicin | 59.26 ± 12.03 * | 147.23 ± 16.34 * | 30.32 ± 5.14 * |
| Compounds | Total Lipids (mg/dL) | Cholesterol (mg/dL) | Triglycerides (mg/dL) | Bilirubin (mg/dL) |
|---|---|---|---|---|
| 3a | 336.40 ± 19.10 *** | 97.20 ± 9.90 *** | 117.90 ± 18.40 ** | 0.67 ± 0.03 *** |
| 3b | 379.20 ± 37.80 * | 127.50 ± 25.10 * | 136.10 ± 27.09 * | 0.97 ± 0.05 * |
| 3c | 321.70 ± 18.90 *** | 92.80 ± 14.30 *** | 115.40 ± 8.70 *** | 0.63 ± 0.05 *** |
| 7 | 329.60 ± 14.40 *** | 97.40 ± 18.60 *** | 114.90 ± 10.70 *** | 0.64 ± 0.02 *** |
| 10a | 313.70 ± 31.20 *** | 94.80 ± 18.60 *** | 117.31 ± 21.60 *** | 0.55 ± 0.06 ** |
| 10b | 374.60 ± 36.80 * | 111.40 ± 16.50 ** | 97.40 ± 9.60 *** | 1.09 ± 0.60 * |
| 10c | 375.26 ± 27.80 * | 111.80 ± 31.40 ** | 157.90 ± 35.70 * | 0.74 ± 0.03 ** |
| 10d | 328.50 ± 22.70 *** | 96.39 ± 17.10 *** | 115.51 ± 18.10 *** | 0.54 ± 0.05 ** |
| 12 | 369.30 ± 26.30 * | 109.90 ± 18.60 ** | 114.20 ± 18.40 ** | 0.73 ± 0.16 ** |
| Control | 323.41 ± 27.10 | 94.32 ± 13.50 | 108.70 ± 16.80 | 0.63 ± 0.04 |
| 5-Flurouracil | 378.20 ± 31.40 * | 105.90 ± 11.70 * | 126.50 ± 19.40 * | 0.75 ± 0.10 * |
| Doxorubicin | 366.70 ± 6.10 * | 109.30 ± 14.20 * | 137.80 ± 17.10 * | 0.81 ± 0.19 * |
| Biochemical Parameters | Albumin (mg/dL) | Globulin (mg/dL) | A/G Ratio | Creatinine (mg/dL) |
|---|---|---|---|---|
| 3a | 5.96 ± 0.4 *** | 4.3 ± 0.69 *** | 1.46 *** | 0.62 ± 0.08 *** |
| 3b | 6.43 ± 0.44 ** | 6.46 ± 0.8 ** | 1.001 * | 0.73 ± 0.06 ** |
| 3c | 5.65 ± 0.69 *** | 4.67 ± 1.09 *** | 1.13 *** | 0.66 ± 0.07 *** |
| 7 | 5.92 ± 0.86 *** | 4.73 ± 0.87 *** | 1.15 *** | 0.85 ± 0.08 ** |
| 10a | 5.95 ± 0.78 *** | 5.16 ± 0.7 *** | 1.15 *** | 0.72 ± 0.08 *** |
| 10b | 6.81 ± 0.47 ** | 6.79 ± 0.7 ** | 1.02 * | 1.62 ± 0.07 * |
| 10c | 10.26 ± 1.31 * | 8.97 ± 0.9 * | 1.14 ** | 0.73 ± 0.03 *** |
| 10d | 5.53 ± 0.71 *** | 4.88 ± 1.01 *** | 1.13 *** | 0.68 ± 0.04 *** |
| 12 | 7.4 ± 0.59 ** | 6.65 ± 0.81 ** | 1.006 * | 0.8 ± 0.09 ** |
| Control | 5.63 ± 0.51 | 4.32 ± 0.9 | 1.3 | 0.69 ± 0.03 |
| 5-FU | 6.49 ± 0.92 ** | 5.75 ± 0.8 ** | 1.13 ** | 0.81 ± 0.06 ** |
| DOX | 6.37 ± 0.85 ** | 5.91 ± 0.63 ** | 1.078 ** | 0.78 ± 0.04 ** |
2.3. Molecular Docking Study
2.3.1. Evaluation of Docking Performance and Accuracy into Different PTKs

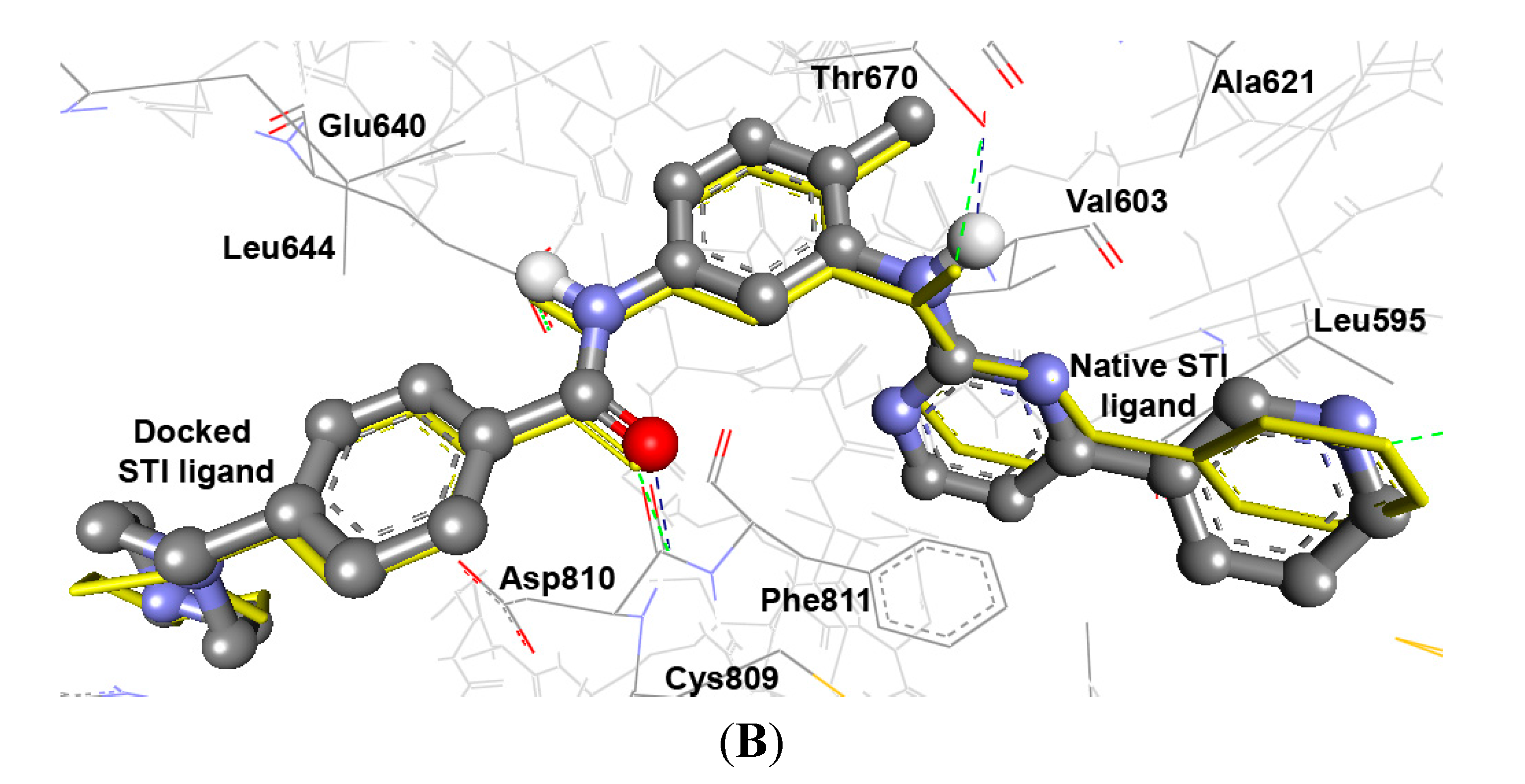
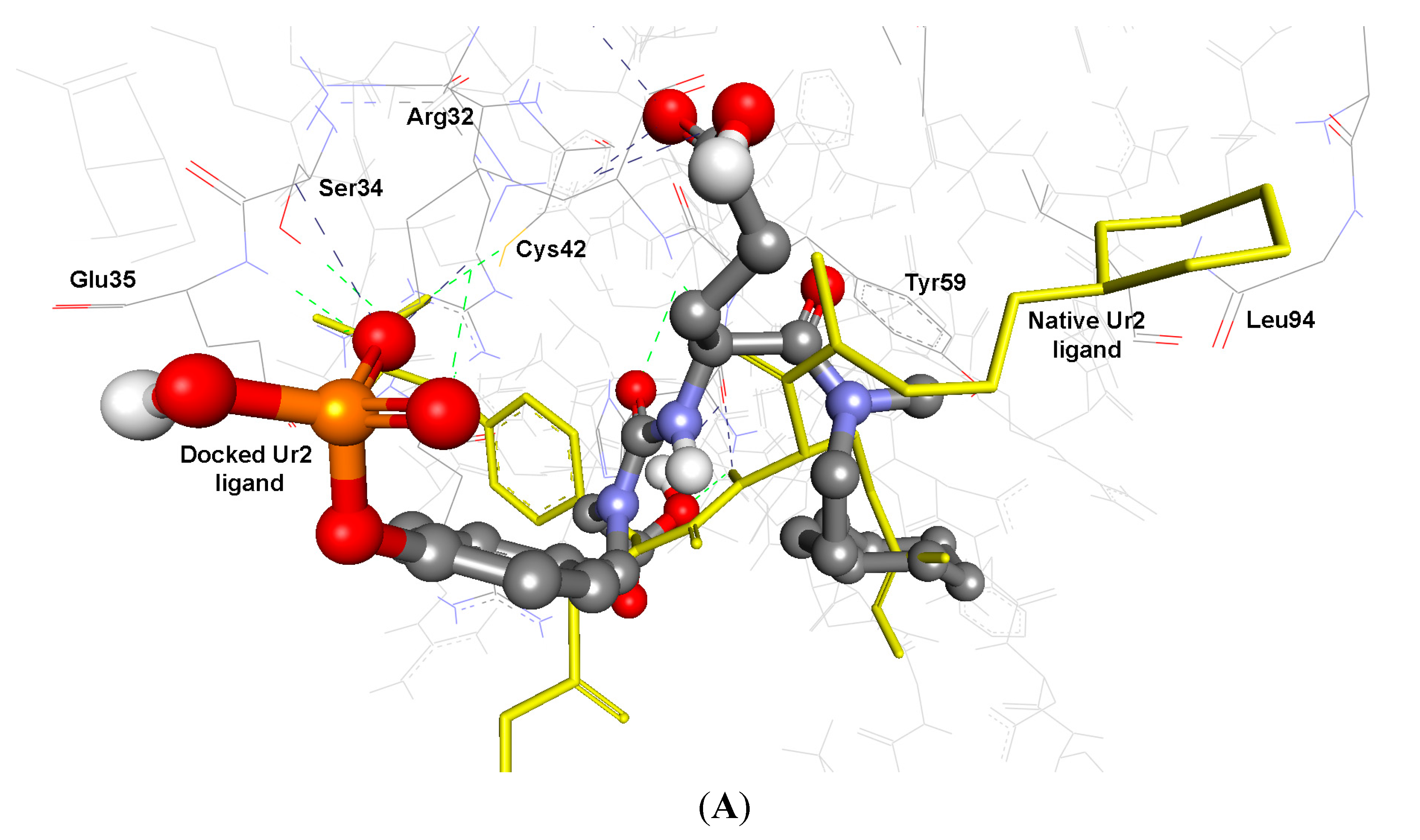
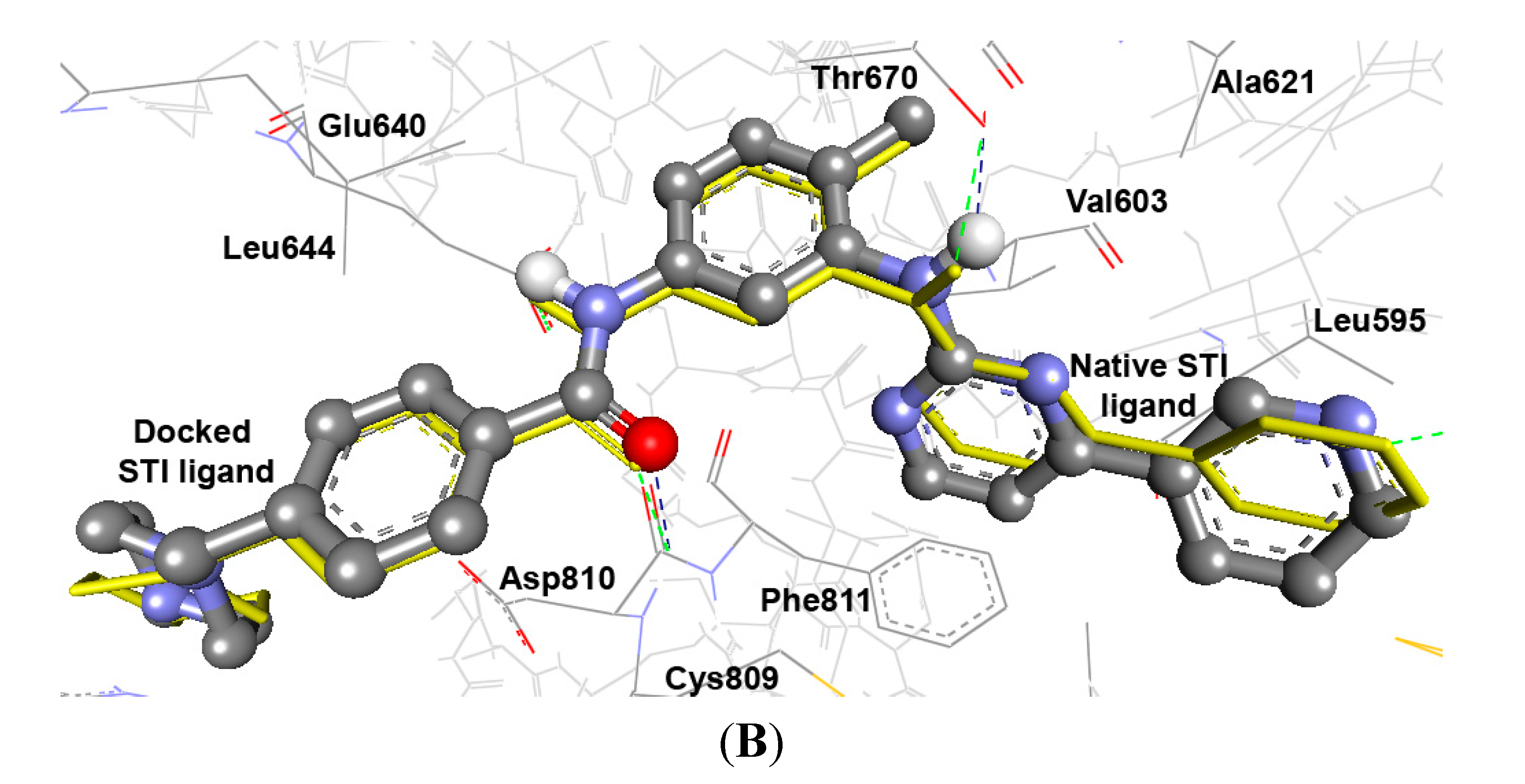
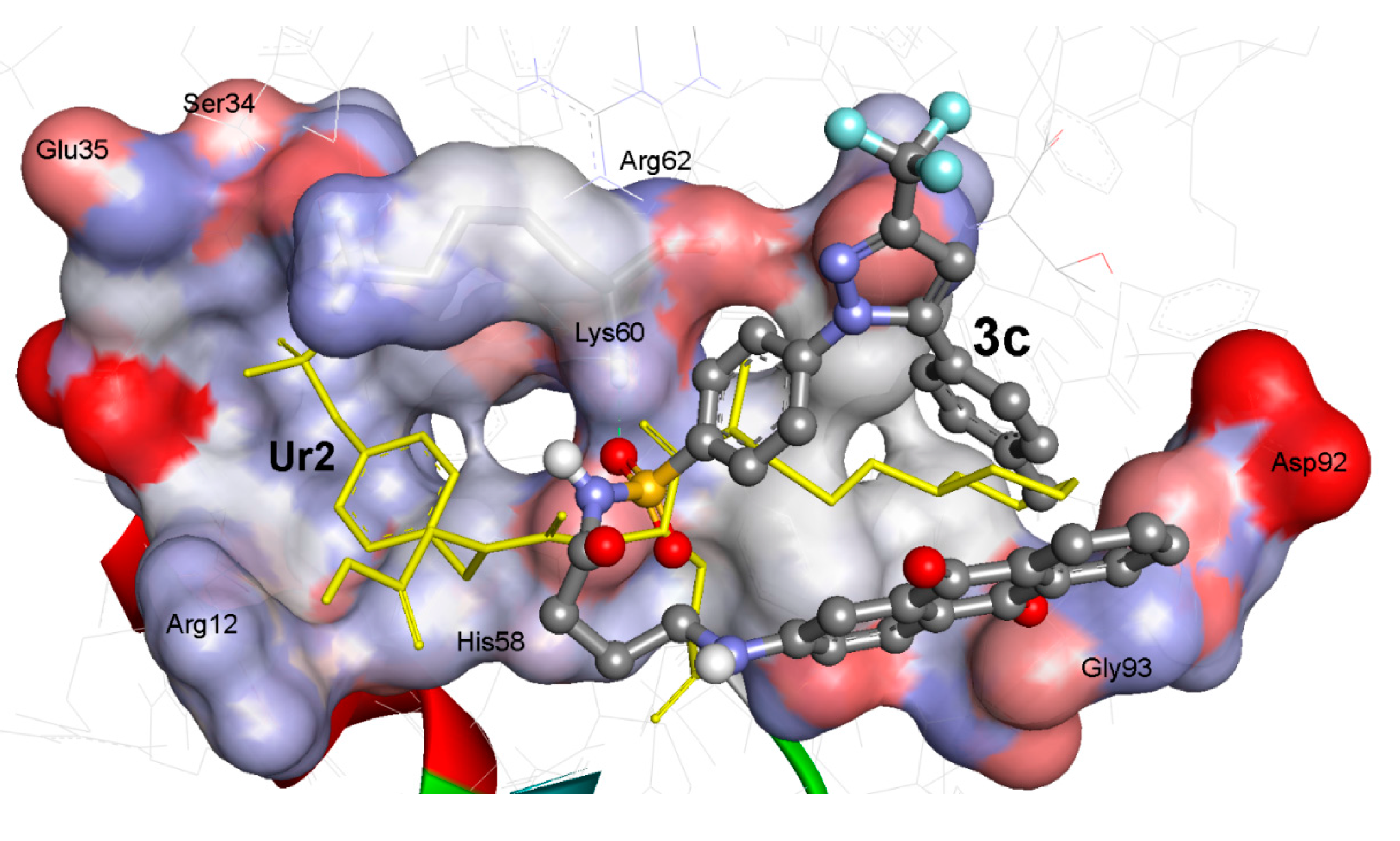
2.3.2. Docking Study of the Synthesized Compounds
| Comp. | ∆Gb a (kcal/mol) | Ki b (nM) | Hydrogen Bonds between Atoms of Compounds and Amino Acids of PTK (1skj) | RMSD c (Å) | |||
|---|---|---|---|---|---|---|---|
| Atoms of Compounds | Amino Acids | Distance (Å) | Angle (°) | ||||
| 3a | −11.57 | 3.31 | Ph–N | HN of Lys60 | 2.48 | 154.9 | 1.43 |
| 3b | −10.86 | 10.94 | Anthraquinone C=O | HN of His58 | 2.27 | 140.6 | 4.19 |
| Terminal C–F | HN of Arg12 | 2.29 | 168.3 | ||||
| 3c | −10.56 | 18.04 | –SO2NHC=O | HN of Lys60 | 1.90 | 172.1 | 5.60 |
| 7 | −9.86 | 59.49 | S=O 1 | 1HN of Arg12 | 2.39 | 134.4 | 5.72 |
| S=O 2 | 1HN of Arg12 | 2.24 | 134.9 | ||||
| S=O 2 | 2HN of Arg12 | 2.07 | 140.1 | ||||
| 10a | −10.89 | 10.40 | Anthraquinone C=O | HN of His58 | 2.31 | 123.8 | 4.20 |
| 10b | −11.17 | 6.45 | Anthraquinone C=O | HN of Arg12 | 1.91 | 135.8 | 2.01 |
| CONH | O=C of His58 | 1.91 | 142.0 | ||||
| SO2NH | O=C of His58 | 2.22 | 150.1 | ||||
| SO2HN | HN of Lys60 | 2.47 | 164.0 | ||||
| Pyrazole=N | HN of Arg62 | 2.47 | 100.8 | ||||
| 10c | −9.69 | 78.94 | Anthraquinone C=O | HN of Arg62 | 2.07 | 159.3 | 5.80 |
| –S=O 1 | HN of Lys60 | 1.92 | 144.1 | ||||
| –S=O 2 | HN of Lys60 | 1.91 | 126.4 | ||||
| SO2NH | O=C of His58 | 2.24 | 150.2 | ||||
| 10d | −11.73 | 2.53 | Pyrazole=N | HN of Lys60 | 1.97 | 155.1 | 1.67 |
| 12 | −9.85 | 59.82 | CONH | O=C of His58 | 1.74 | 142.4 | 3.87 |
| HNS=O | HN of Lys60 | 1.97 | 162.5 | ||||
| UR2 d | −11.61 | 3.11 | Terminal COO | HN of His58 | 2.27 | 146.4 | 1.84 |
| HNC=O | HN of Lys60 | 2.32 | 124.5 | ||||
| Terminal P=O 1 | HN of Arg62 | 2.44 | 122.4 | ||||
| Terminal P=O 2 | HN of Arg62 | 2.35 | 140.5 | ||||
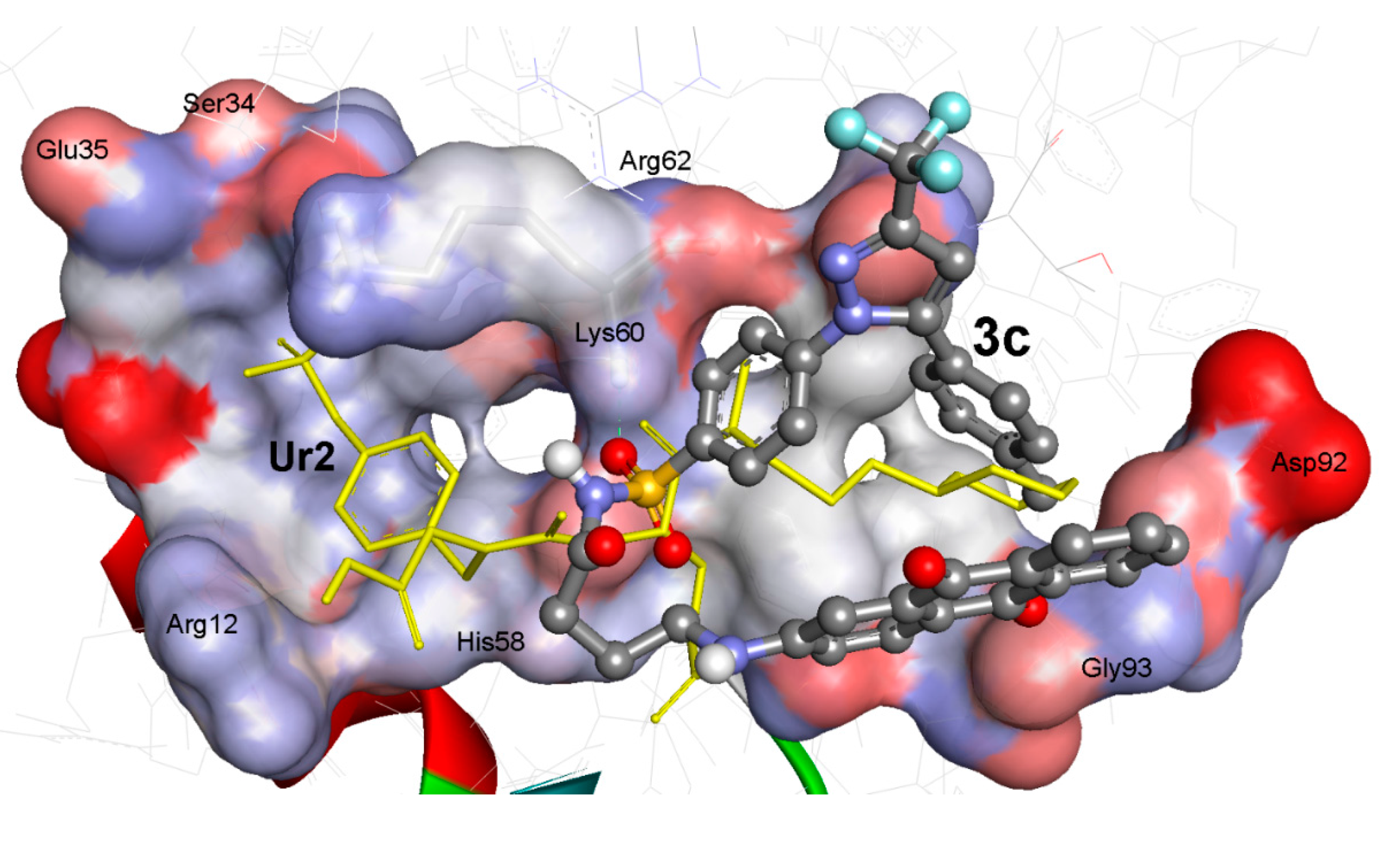
| Comp. | ∆Gb a (kcal/mol) | Ki b | Hydrogen Bonds between Atoms of Compounds and Amino Acids of PTK (1t46) | RMSD c (Å) | |||
|---|---|---|---|---|---|---|---|
| Atoms of Comp. | Amino Acids | Distance(Å) | Angle (°) | ||||
| 3a | −13.20 | 212.15 pM | –SO2NH | O=C of Glu640 | 2.46 | 175.8 | 5.98 |
| 3b | −9.91 | 54.76 nM | Ph–NH | O=C of Asp810 | 1.92 | 153.6 | 10.0 |
| 3c | −10.56 | 18.09 nM | d | 10.05 | |||
| 7 | −9.83 | 62.78 nM | Ph–NH | O=C of Asp810 | 2.34 | 114.2 | 9.46 |
| Ph–NHCH2NH | O=C of Asp81 | 2.26 | 112.1 | ||||
| Ph–NHCH2NH | O–C of Asp810 | 1.96 | 145.7 | ||||
| Terminal C–F | HN of Arg791 | 2.12 | 122.6 | ||||
| 10a | −13.37 | 157.86 pM | Ph–NH | O=C of Asp810 | 2.19 | 122.4 | 4.36 |
| Ph–NH | O=C of Glu640 | 1.75 | 105.4 | ||||
| 10b | −12.63 | 555.42 pM | Anthraquinone C=O | HN of Asp810 | 2.35 | 129.6 | 5.66 |
| Ph–NHC=O | HN of Lys623 | 2.41 | 103.0 | ||||
| 10c | −12.01 | 1.57 nM | Anthraquinone C=O a | HO of Thr670 | 2.39 | 100.6 | 5.31 |
| Anthraquinone C=O b | O=C of Glu640 | 2.02 | 144.5 | ||||
| Ph–NH | HN of Lys623 | 2.38 | 124.9 | ||||
| 10d | −14.22 | 37.78 pM | Anthraquinone C=O | HO of Thr670 | 2.22 | 110.1 | 4.29 |
| Ph–NH | O=C of Glu640 | 1.91 | 162.8 | ||||
| Ph–NHC=O | HN of Asp810 | 2.33 | 104.5 | ||||
| 12 | −11.86 | 2.02 nM | Ph–NH | CO of Asp792 | 2.05 | 130.2 | 10.84 |
| Terminal C–F | NH of Ile789 | 2.29 | |||||
| STI e | −15.85 | 2.41 pM | Ph–C=O | HN of Asp810 | 2.25 | 146.3 | 0.39 |
| Ph–NH | OH of Thr670 | 2.13 | 130.9 | ||||
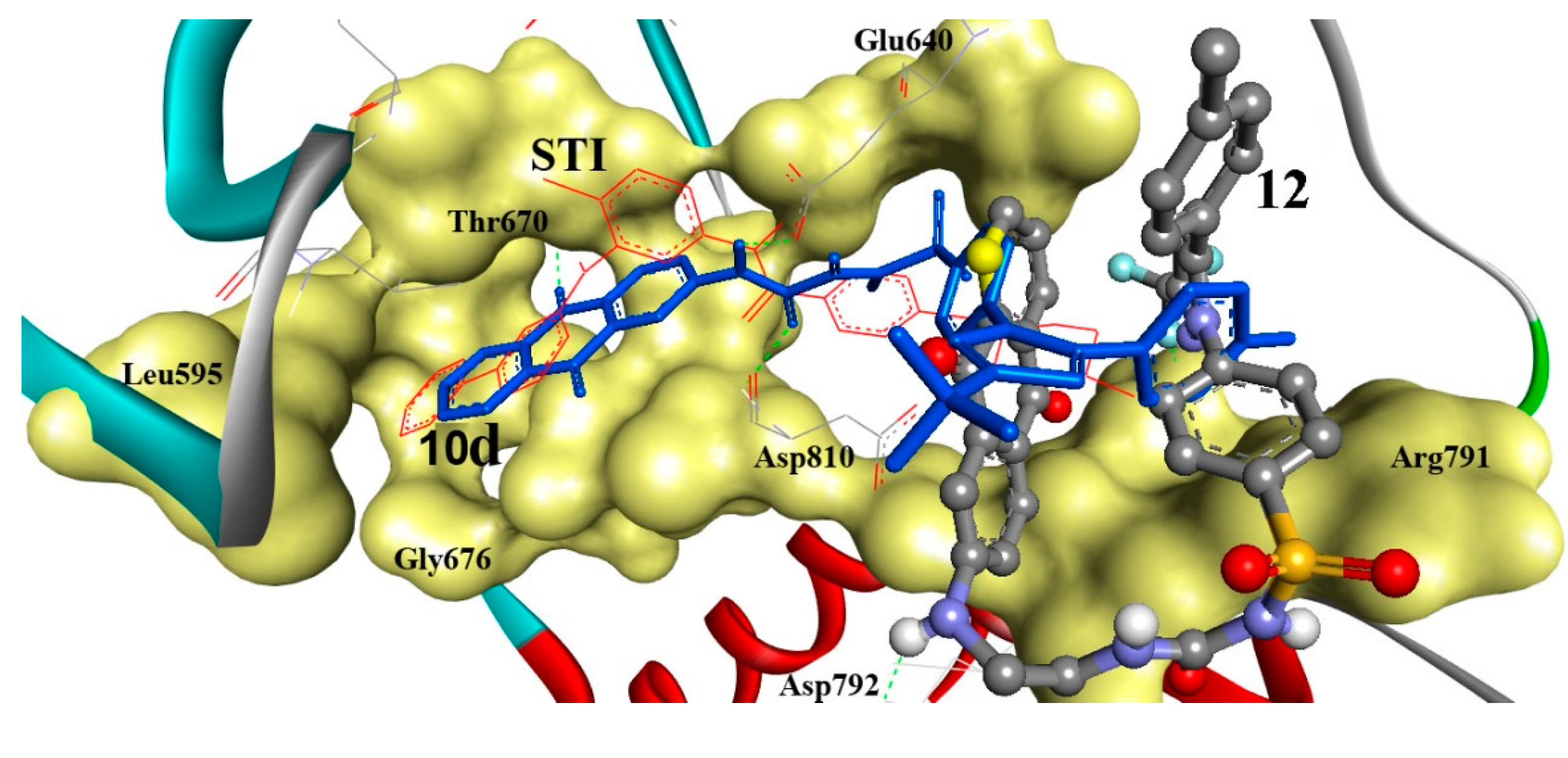
3. Experimental Section
3.1. Chemistry
3.1.1. N-2-Chloroacyl-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (2a)
3.1.2. N-(2-Chloropropionyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (2b)
3.1.3. N-(4-Chlorobutyryl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (2c)
3.1.4. General Method for Preparation of N-(2- or 4-(9,10-Dihydro-9,10-dioxoanthracen-2-yl) aminoacyl) 4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (3a–c)
3.1.5. N-(2-(9,10-Dihydro-9,10-dioxoanthracen-2-yl) aminoacetyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (3a)
3.1.6. N-(2-(9,10-Dihydro-9,10-dioxoanthracen-2-yl)-aminopropionyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H pyrazol-1-yl] benzene sulfonamide (3b)
3.1.7. N-(4-(9,10-Dihydro-9,10-dioxoanthracen-2-yl-aminobutanoyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (3c)
3.1.8. N-(2-Isocyanatoethyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (4)
3.1.9. N-(2-(3-(9,10-Dihydro-9,10-dioxoanthracen-2-yl) ureido) ethyl)-4-(5-(p-tolyl)-3-(trifluoro methyl)-1H-pyrazol-1-yl) benzene sulfonamide (5)
3.1.10. N-[2-(2-Chloroethylamino) ethyl]-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (6)
3.1.11. N-[2-(2-(9,10-Dihydro-9,10-dioxoanthracen-2-yl) aminoethyl) aminoethyl]-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (7)
3.1.12. General Method for Preparation of 2- or 4-Chloro-N-(9,10-dihydro-9,10-dioxoanthracen-6-yl) alkanamide (9b,c)
3.1.13. 2-Chloro-N-(9,10-dihydro-9,10-dioxoanthracen-6-yl) propanamide (9b)
3.1.14. 4-Chloro-N-(9,10-dihydro-9,10-dioxoanthracen-6-yl) butanamide (9c)
3.1.15. N-(9,10-Dihydro-9,10-dioxoanthracen-2-yl-amino-carbonyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide derivatives (10a–d)
3.1.16. N-(9,10-Dihydro-9,10-dioxoanthracene-2-yl) aminoacetyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (10a)
3.1.17. N-[1-(9,10-Dihydro-9,10-dioxoanthracene-2-yl) aminocarbonyl) ethyl]-4-[5-(p-tolyl)-3-tri fluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (10b)
3.1.18. N-[3-(9,10-Dihydro-9,10-dioxoanthracene-2-yl) aminocarbonyl) propyl]-4-[5-(p-tolyl)-3-tri fluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (10c)
3.1.19. N-(9,10-Dihydro-9,10-dioxoanthracene-2-yl)aminooxalyl)-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (10d)
3.1.20. 2-(2-Isocyanatoethylamino)anthracene-9,10-dione (11)
3.1.21. N-[2-(9,10-Dihydro-9,10-dioxoanthracene-2-yl-amino) ethylcarbamoyl]-4-[5-(p-tolyl)-3-trifluoromethyl-1H-pyrazol-1-yl] benzene sulfonamide (12)
3.2. Pharmacological Screening
3.2.1. Measurement of Potential Cytotoxicity by Sulforhodamine B (SRB) Assay
3.2.2. Biochemical Analysis
3.3. Molecular Docking
3.3.1. Preparing the Target Macromolecules Investigated
3.3.2. Preparing a Ligand File for AutoDock
3.3.3. Setting the GRID Box, Preparing the GRID Parameter File, Running AutoGrid4, and Preparation of the Flexible Residue File
3.3.4. Preparing the Docking Parameter File and Running AutoDock4
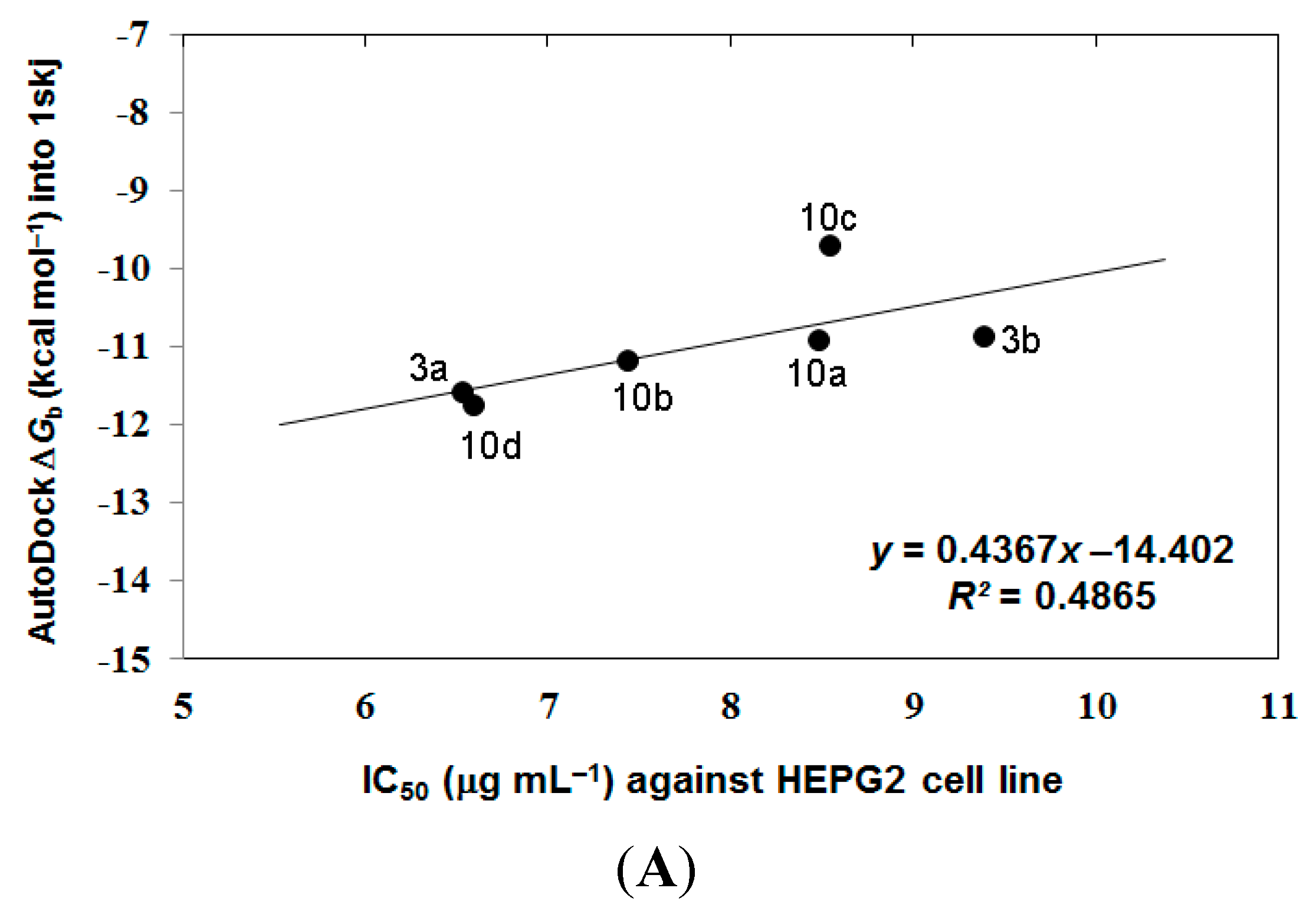
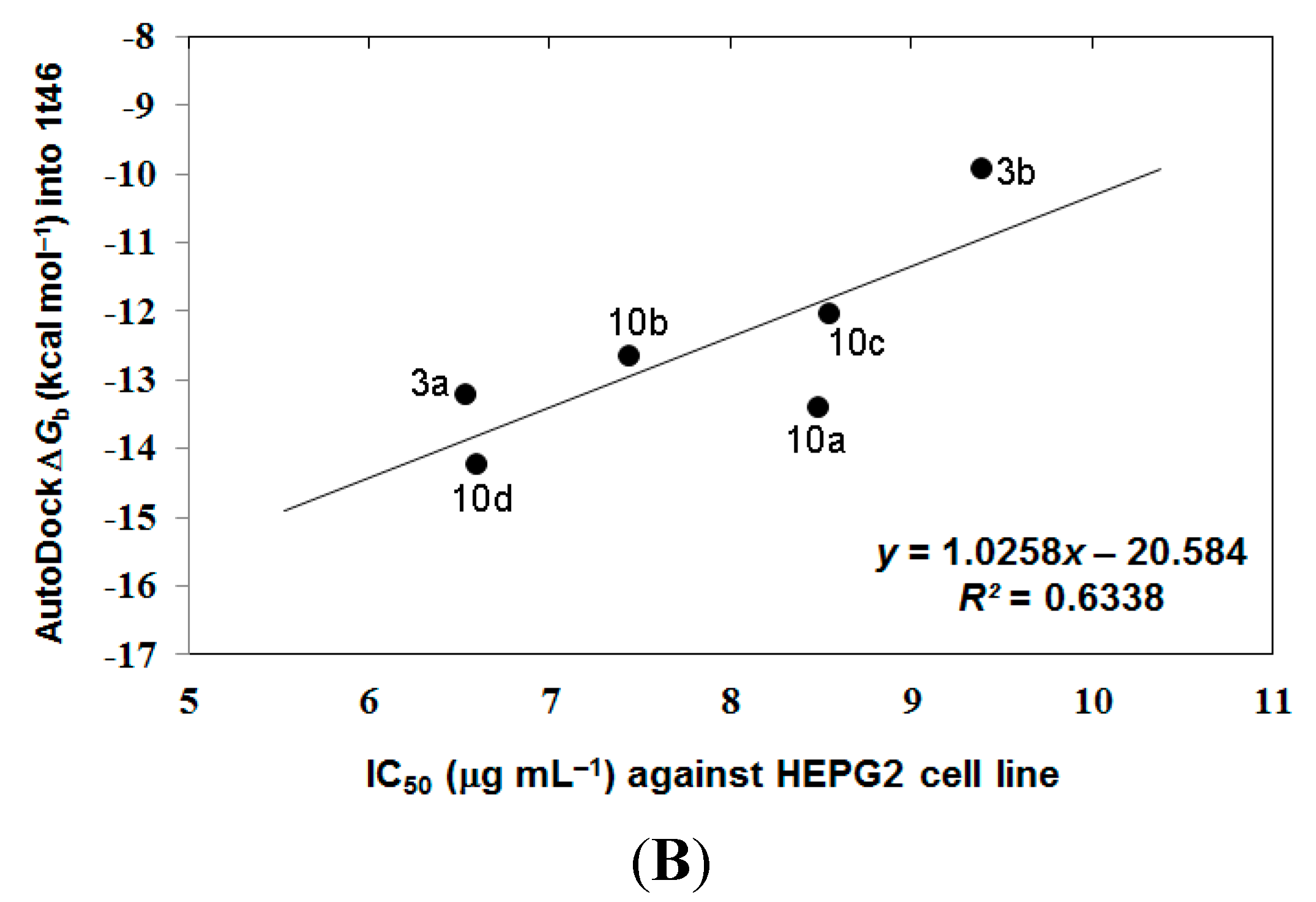
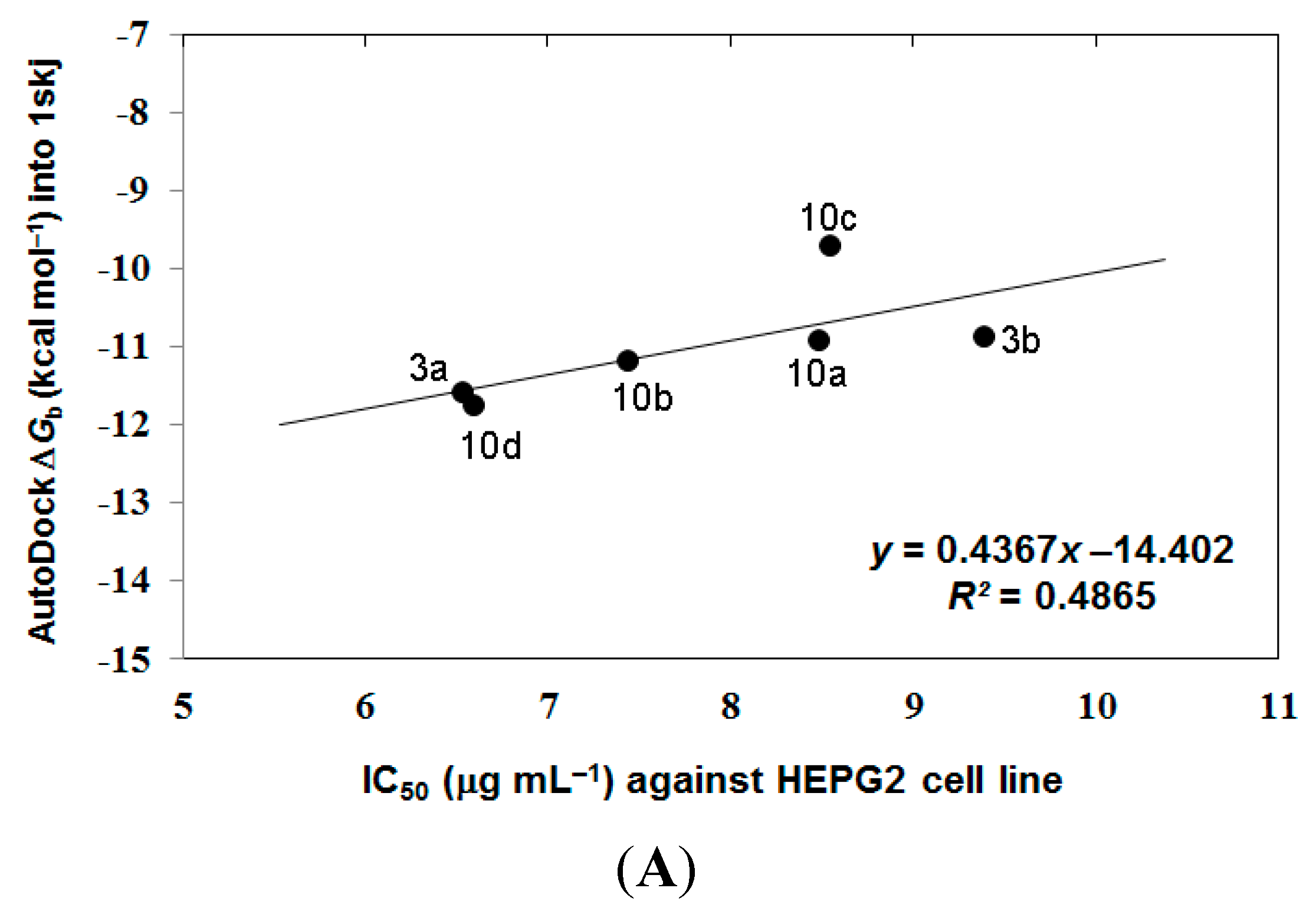
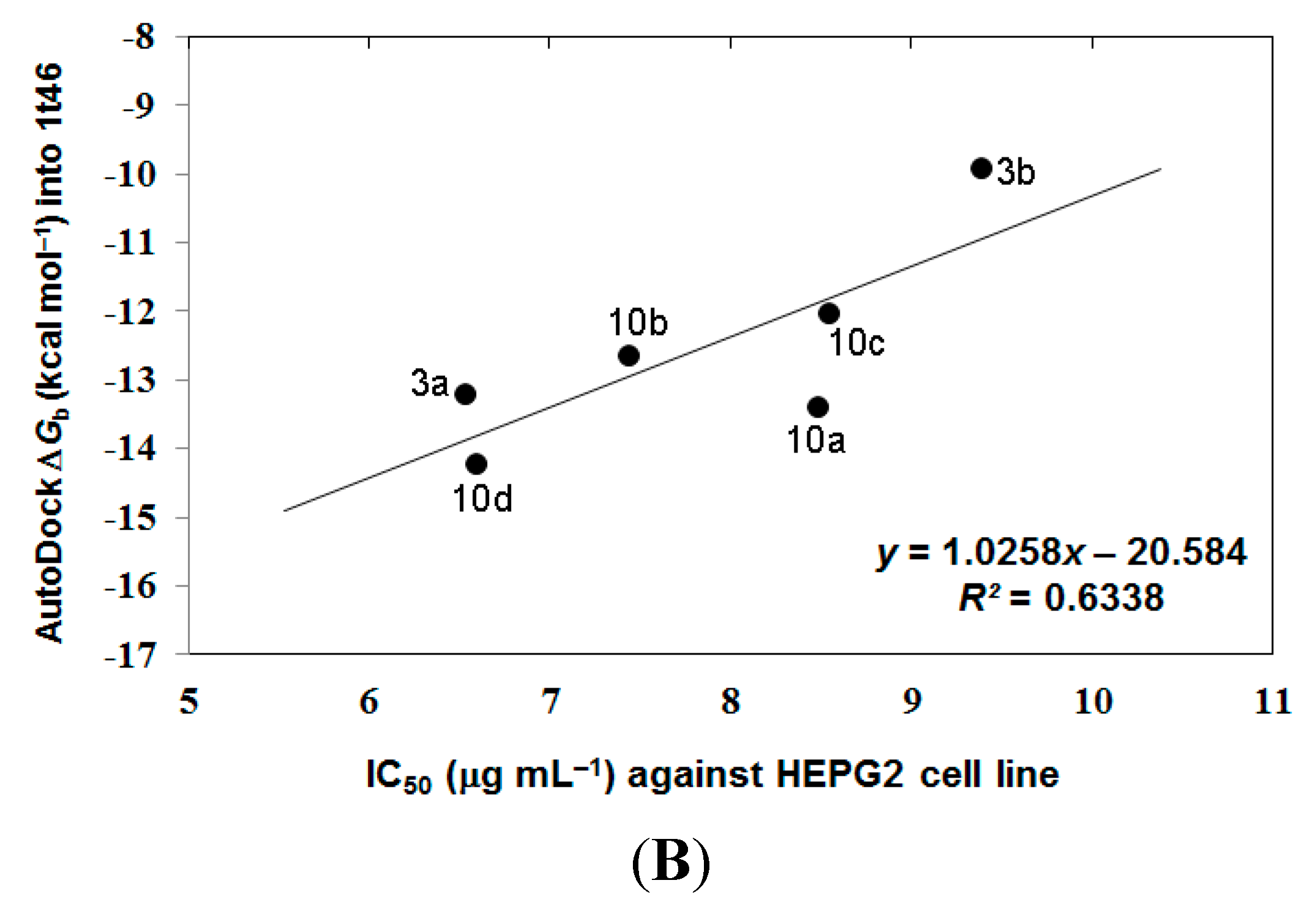
3.4. Analyzing the Docking Results
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aragon-Ching, B.J.; Dahut, W.L. Cancer treatment anti-angiogenesis approach to genitourinary. Update Cancer Ther. 2009, 3, 182–188. [Google Scholar]
- Koki, A.T.; Masferrer, J.L. Celecoxib: A specific Cox-2 inhibitor with anticancer properties. Cancer Control 2002, 9, 28–35. [Google Scholar]
- Bamba, H.; Ota, S.; Kato, A.; Kawamoto, C.; Fujiwara, K.; Matsuzaki, F. Effect of prostaglandin E1 on vascular endothelial growth factor production by human macrophages and colon cancer cells. J. Exp. Clin. Cancer Res. 2000, 19, 219–223. [Google Scholar]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Lrie, T.; Sujii, M.T.; Tsuji, S.; Yoshio, T.; Lshii, S.; Shinzaki, S.; Egawa, S.; Kakiuchi, Y.; Nishida, T.; Yasumaru, M.; et al. Synergistic antitumor effects of celecoxib with 5-fluorouracil depend on IFN-γ. Int. J. Cancer 2007, 121, 878–883. [Google Scholar]
- Saldivar, J.S.; Lopez, D.; Feldman, R.A.; Tharappel, R.J.; de-la Rosa, A.; Terreros, D.; Baldwin, W.S. Cox-2 overexpression as a biomarker of early cervical carcinogenesis: A pilot study. Gynecol. Oncol. 2007, 10, 155–162. [Google Scholar] [CrossRef]
- Dai, Z.; Ma, X.; Kang, H.; Gao, J.; Min, W.; Guan, H.; Diao, Y.; Lu, W.; Wang, X. Antitumor activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on breast cancer in vitro and in vivo. Cancer Cell Int. 2012, 12, 53. [Google Scholar] [CrossRef]
- Park, W.; Oh, T.; Han, J.; Pyo, H. Antitumor enhancement of celecoxib, a selective cyclooxygenase-2 inhibitor, in a Lewis lung carcinoma expressing cyclooxygenase-2. J. Exp. Clin. Cancer Res. 2008, 27, 66. [Google Scholar] [CrossRef]
- Huang, S.; Sinicrope, F. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 2010, 6, 256–269. [Google Scholar] [CrossRef]
- Victor, V.; Cristina, B.; David, C.; Denise, O.; Alejandro, F.; Mack, B.; William, G. Celecoxib inhibits cellular growth, decreases Ki-67 expression and modifies apoptosis in ovarian cancer cell lines. Arch. Med. Res. 2006, 37, 689–695. [Google Scholar] [CrossRef]
- Choe, M.; Chen, Z.; Klass, C.; Zhang, X.; Shin, D. Enhancement of docetaxel-induced cytotoxicity by blocking epidermal growth factor receptor and cyclooxygenase-2 pathways in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2007, 13, 3015–3023. [Google Scholar] [CrossRef]
- Taher, A.T.; Hegazy, G.H. Synthesis of novel bis-anthraquinone derivatives and their biological evaluation as antitumor agents. Arch. Pharm. Res. 2013, 36, 573–578. [Google Scholar] [CrossRef]
- Denny, W.; Wakelin, L. Kinetics of the binding of mitoxantrone, ametantrone and analogues to DNA: Relationship with binding mode and anti-tumour activity. Anticancer Drug Des. 1990, 5, 189–200. [Google Scholar]
- Kaneshiro, T.; Morioka, T.; Lnamine, M.; Kinjo, T.; Arakaki, J.; Chiba, L.; Sunagawa, N.; Suzui, M.; Yoshimi, N. Anthraquinone derivative emodin inhibits tumor-associated angiogenesis through inhibition of extracellular signal-regulated kinase 1/2 phosphorylation. Eur. J. Pharmcol. 2006, 533, 46. [Google Scholar] [CrossRef]
- Wei, D.; Jiang, X.; Zhou, L.; Chen, J.; Chen, Z.; He, C.; Yang, K.; Liu, Y.; Pei, J.; Lai, L. Discovery of multitarget inhibitors by combining molecular docking with common pharmacophore matching. J. Med. Chem. 2008, 51, 7882–7888. [Google Scholar] [CrossRef]
- Ali, H.I.; Nagamatsu, T.; Akaho, E. Structure-based drug design and AutoDock study of potential protein tyrosine kinase inhibitors. Bioinformation 2011, 5, 368–374. [Google Scholar] [CrossRef]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Molecular docking and pharmacophore filtering in the discovery of dual-inhibitors for human leukotriene A4 hydrolase and leukotriene C4 synthase. J. Chem. Inf. Model. 2011, 51, 33–44. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Qandil, M.A.; el-Mohtadi, F.H.; Tashtoush, B.M. Chemical and in vitro enzymatic stability of newly synthesized celecoxib lipophilic and hydrophilic amide. Int. J. Pharm. 2011, 416, 85–96. [Google Scholar] [CrossRef]
- Berghot, M.A.; Hanna, M.A.; Girges, M.M. Synthesis and biological activity of some heterocyclic systems containing anthrquinone. Pharmazie 1992, 47, 340–343. [Google Scholar]
- Loskutov, V.A.; Savel’ev, V.A.; Konstantinova, A.V. Reaction of amino derivatives of 9,10-anthraquinone with oxalyl chloride. Izvestiya Sibirskogo Otdeleniya Akademii Nauk SSSR, Seriya Khimicheskikh Nauk 1985, 3, 114–118. [Google Scholar]
- Pautus, S.; Yee, S.W.; Jayne, M.; Coogan, M.P.; Simons, C. Synthesis and CYP26A1 inhibitory activity of 1-(benzofuran-2-yl-(4-alkyl/aryl-phenyl)-methyl)-1H-triazoles. Bioorg. Med. Chem. 2006, 14, 3643–3653. [Google Scholar] [CrossRef]
- Hayakawa, I.; Shioya, R.; Agatsuma, T.; Furukawa, H.; Naruto, S.; Sugano, Y. A library synthesis of 4-hydroxy-3-methyl-6-phenylbenzofuran-2-carboxylic acid ethyl ester derivatives as anti-tumor agents. Bioorg. Med. Chem. Lett. 2004, 14, 4383–4387. [Google Scholar]
- Attoub, S.; Rivat, C.; Rodrigues, S.; van, B.S.; Bedin, M.; Bruyneel, E.; Louvet, C.; Kornprobst, M.; Andre, T.; Mareel, M.; et al. The c-Kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy. Cancer Res. 2002, 62, 4879–4883. [Google Scholar]
- Plummer, M.S.; Holland, D.R.; Shahripour, A.E.; Lunney, A.; Fergus, J.H.; Marks, J.S.; McConnell, P.W.; Mueller, T.; Sawyer, T.K. Design, synthesis, and cocrystal structure of a nonpeptide Src SH2 domain ligand. J. Med. Chem. 1997, 40, 3719–3725. [Google Scholar] [CrossRef]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.C.; Wilson, K.P. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Anne Monks, A.; McMahon, J.; Vistica, D.; Warren, J.; Bokesch, H.; Kenney, S.; Boyd, M. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 829, 1107–1112. [Google Scholar] [CrossRef]
- Abo-Ghalia, M.H.; Soliman, A.M. Synthesis and study of the antischistosomal potency and induced biological parameters of a new 2-palmitoyl analogue of the universal antihelminthic praziquantel. Acta Pol. Pharm. Drug Res. 2000, 57, 53–59. [Google Scholar]
- Spencer, K.; Price, C.P. Kinetic immunoturbidimetry: The estimation of albumin. Clin. Chim. Acta 1979, 95, 263–276. [Google Scholar] [CrossRef]
- Mays, A. Baseline hematological and blood biochemical parameters of the Mongolian gerbil (Meriones unguiculatus). Lab. Anim. Sci. 1969, 19, 838–842. [Google Scholar]
- Joseph, V. Raptor hematology and chemistry evaluation. Vet. Clin. N. Am. Exot. Anim. Pract. 1999, 2, 689–699. [Google Scholar]
- Soliman, A.M.; Faddah, L.M. Screening of two pyrozolpyrimidines schistosomicidal activity and their effect on serum transaminases of albino mice. Egypt. J. Bilh. 1994, 16, 73. [Google Scholar]
- Garde, A.H.; Hansen, A.M.; Skovgaard, L.T.; Christensen, J.M. Seasonal and biological variation of blood concentrations of total cholesterol, dehydroepiandrosterone sulfate, hemoglobin A(1c), IgA, prolactin, and free testosterone in healthy women. Clin. Chem. 2000, 46, 551–559. [Google Scholar] [PubMed]
- Rietz, E.B.; Guilbault, G.G. Fluorometric estimation of triglycerides in serum by a modification of the method of Bucolo and David. Clin. Chem. 1977, 23, 286–288. [Google Scholar]
- Guilbaud, N.L.; Kraus-Berthier, F.; Meyer-Losic, V.; Malivet, C.; Chacun, M.; Jan, F.; Tillequin, S.; Michel, M.; Koch, B. Marked antitumor activity of a new potent acronycine derivative in orthotopic models of human solid tumors. Clin. Cancer Res. 2001, 7, 2573–2580. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almutairi, M.S.; Hegazy, G.H.; Haiba, M.E.; Ali, H.I.; Khalifa, N.M.; Soliman, A.E.-m.M. Synthesis, Docking and Biological Activities of Novel Hybrids Celecoxib and Anthraquinone Analogs as Potent Cytotoxic Agents. Int. J. Mol. Sci. 2014, 15, 22580-22603. https://doi.org/10.3390/ijms151222580
Almutairi MS, Hegazy GH, Haiba ME, Ali HI, Khalifa NM, Soliman AE-mM. Synthesis, Docking and Biological Activities of Novel Hybrids Celecoxib and Anthraquinone Analogs as Potent Cytotoxic Agents. International Journal of Molecular Sciences. 2014; 15(12):22580-22603. https://doi.org/10.3390/ijms151222580
Chicago/Turabian StyleAlmutairi, Maha S., Gehan H. Hegazy, Mogedda E. Haiba, Hamed I. Ali, Nagy M. Khalifa, and Abd El-mohsen M. Soliman. 2014. "Synthesis, Docking and Biological Activities of Novel Hybrids Celecoxib and Anthraquinone Analogs as Potent Cytotoxic Agents" International Journal of Molecular Sciences 15, no. 12: 22580-22603. https://doi.org/10.3390/ijms151222580
APA StyleAlmutairi, M. S., Hegazy, G. H., Haiba, M. E., Ali, H. I., Khalifa, N. M., & Soliman, A. E.-m. M. (2014). Synthesis, Docking and Biological Activities of Novel Hybrids Celecoxib and Anthraquinone Analogs as Potent Cytotoxic Agents. International Journal of Molecular Sciences, 15(12), 22580-22603. https://doi.org/10.3390/ijms151222580