The Stability of G6PD Is Affected by Mutations with Different Clinical Phenotypes

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
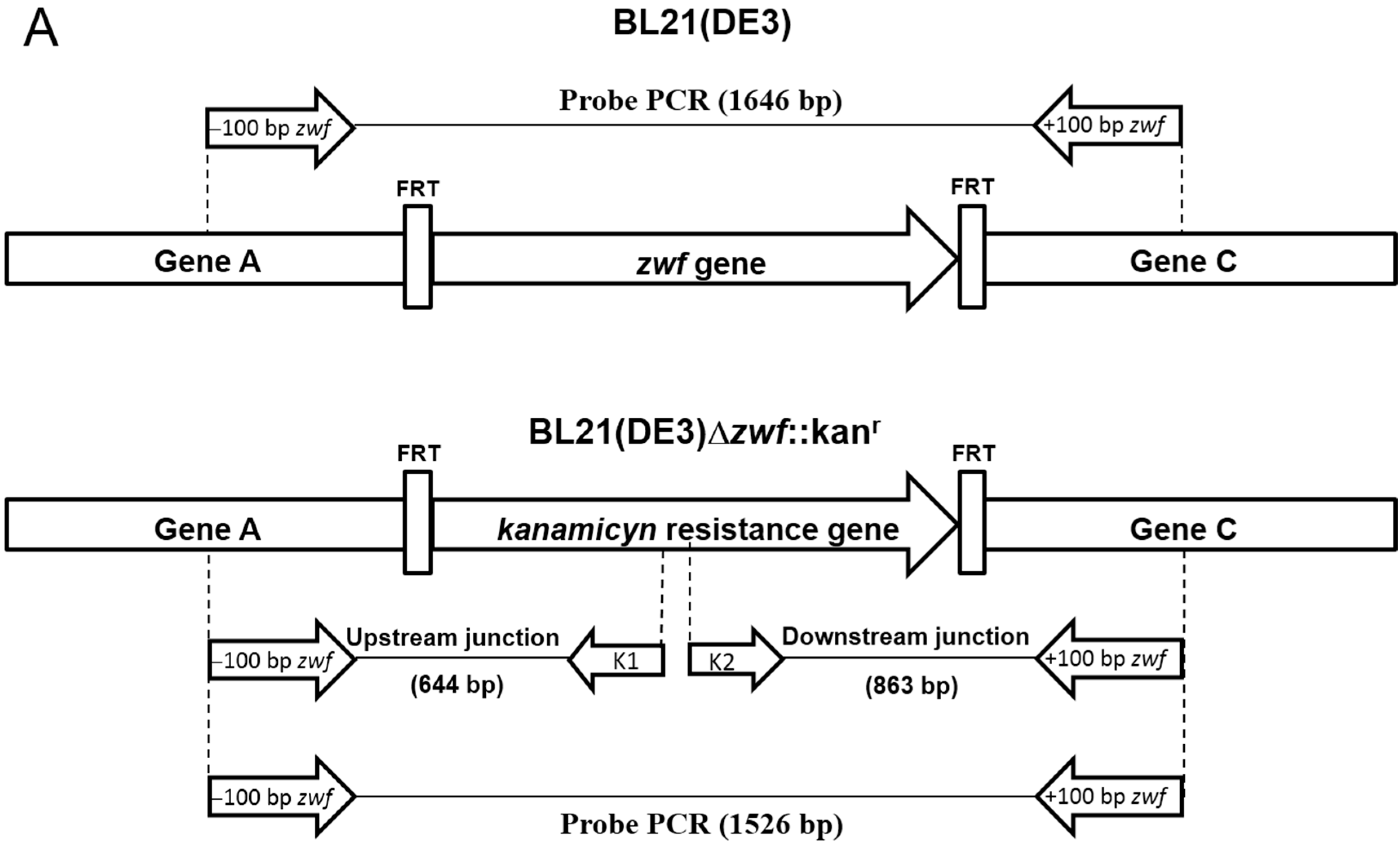
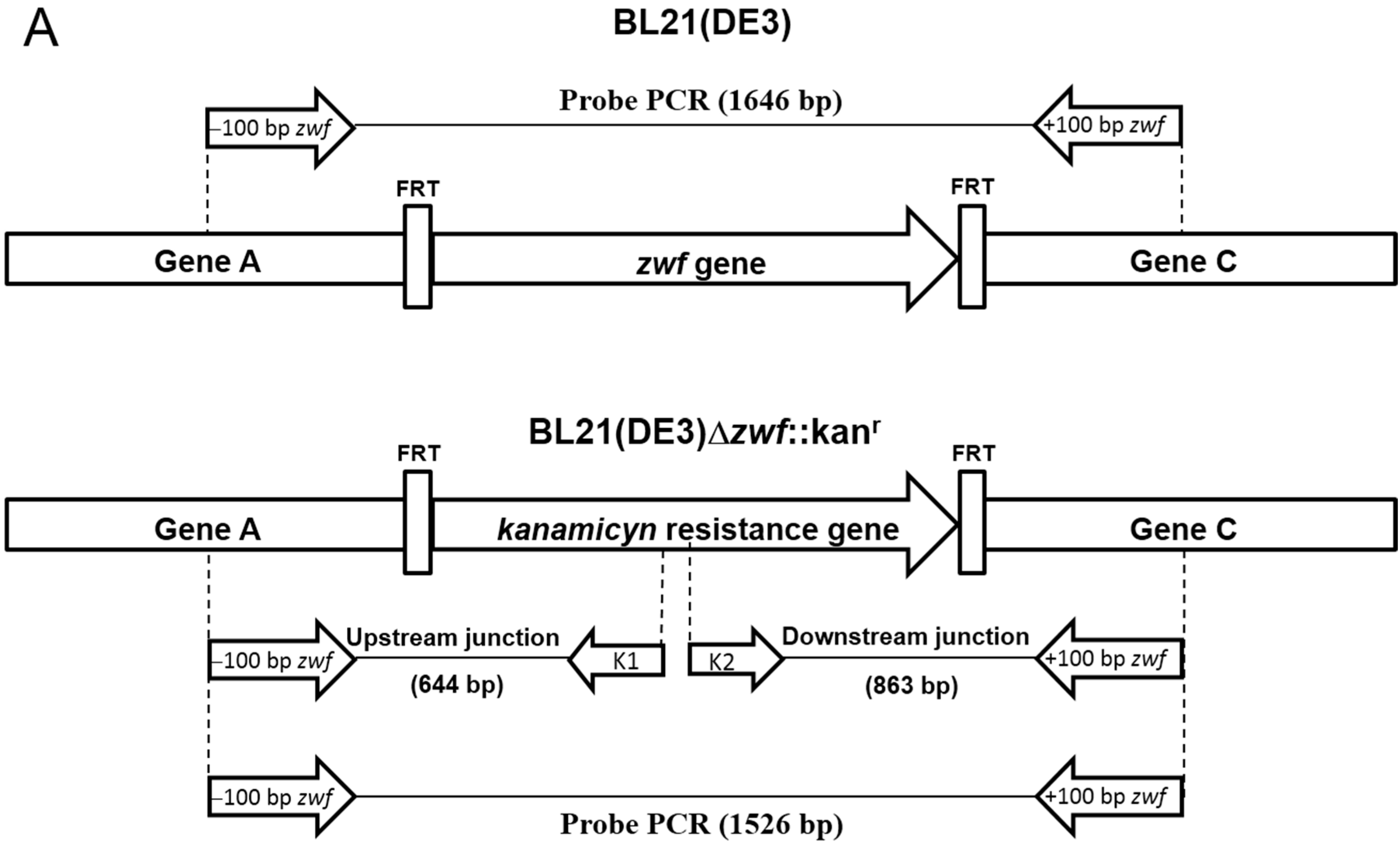
2.1. Construction of E. coli BL21(DE3)Δzwf::kanr Mutant

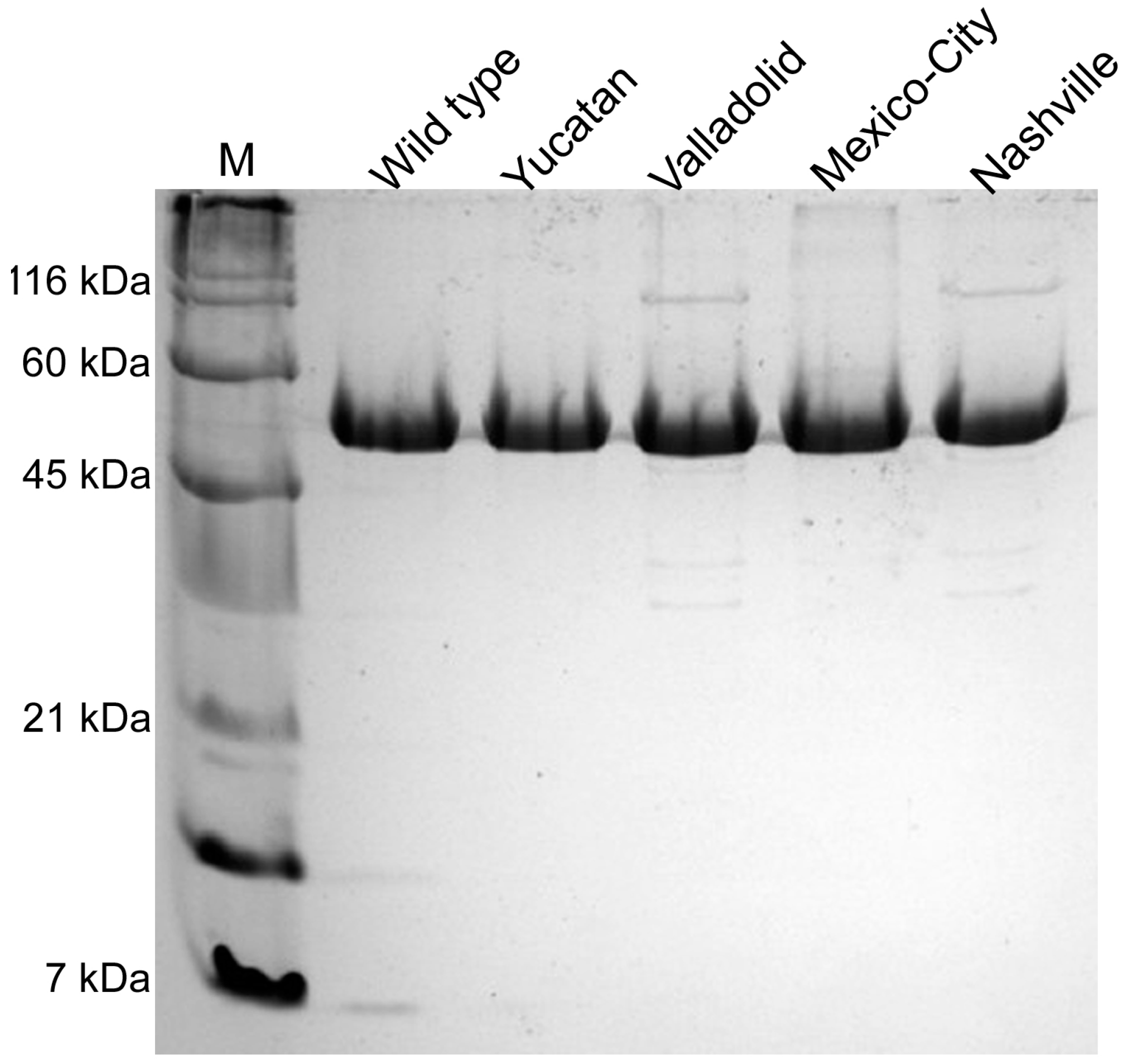
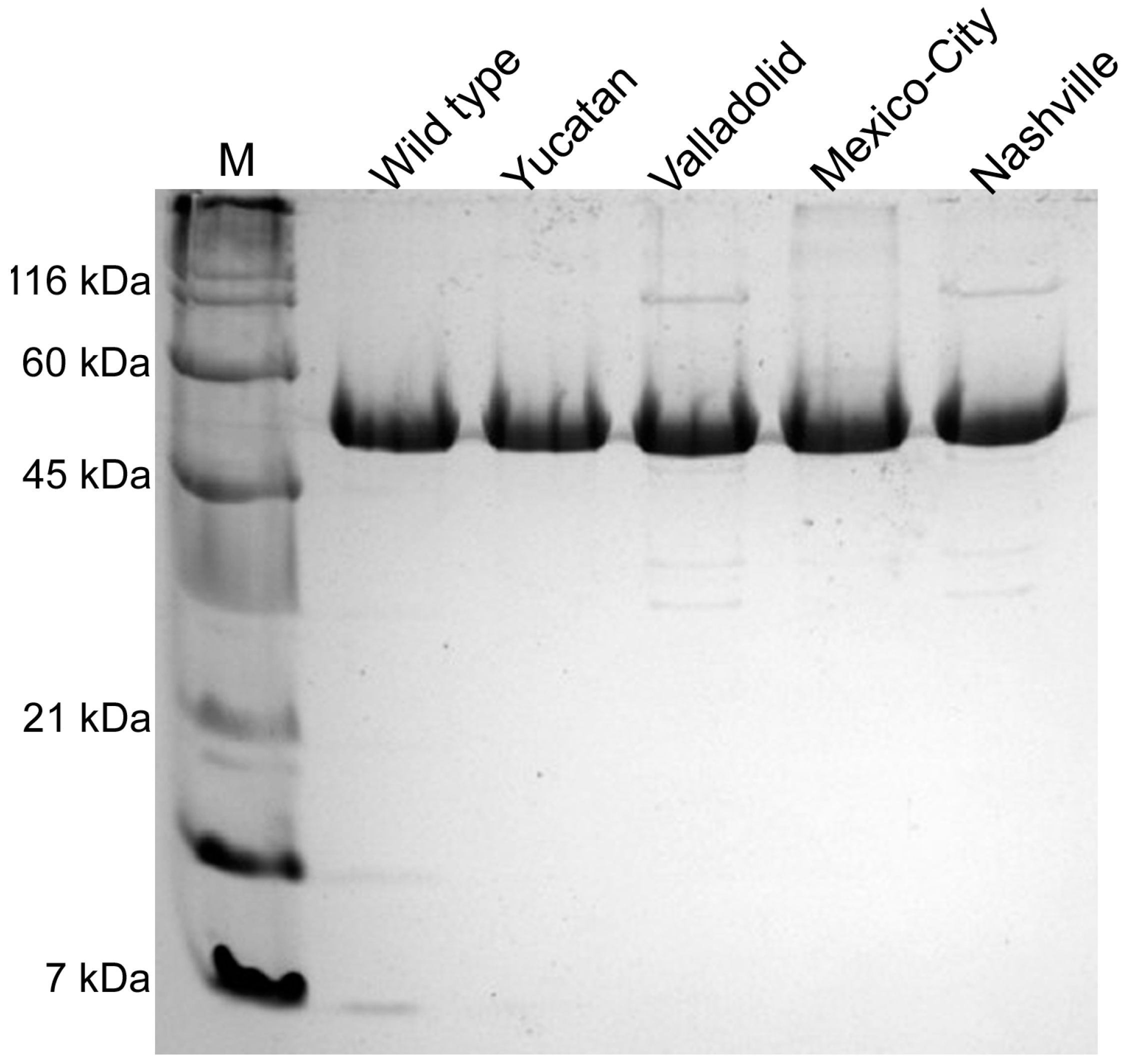
2.2. Construction, Expression and Purification of Recombinant Human G6PD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G6PD | Total Protein (mg) | Specific Activity (IU·mg−1) | Total Activity (IU) | Yield (%) |
|---|---|---|---|---|
| WT | 4.8 | 224 | 1075 | 61 |
| Yucatan (I) | 4.2 | 132 | 554 | 35 |
| Valladolid (II) | 4.3 | 92 | 395 | 29 |
| Mexico City (III) | 4.4 | 175 | 770 | 57 |
| * Nashville (I) | 1.7 | 103 | 175 | 13 |
2.3. Determination of Steady-State Kinetic Parameters
| G6PD | kcat (s−1) | KmG6P (µM) | KmNADP+ (µM) | kcat/KmG6P (s−1·M−1) | kcat/KmNADP+ (s−1·M−1) |
|---|---|---|---|---|---|
| Wild-type | 233 | 38.5 | 6.2 | 6.0 × 106 | 37.8 × 106 |
| Yucatan (I) | 138 | 39.9 | 6.4 | 3.5 × 106 | 21.7 × 106 |
| Valladolid (II) | 96 | 21.5 | 3.6 | 4.4 × 106 | 26.2 × 106 |
| Mexico City (III) | 182 | 24.9 | 9.1 | 7.3 × 106 | 19.1 × 106 |
| Nashville (I) | 119 | 90.6 | 31.2 | 1.3 × 106 | 3.8 × 106 |
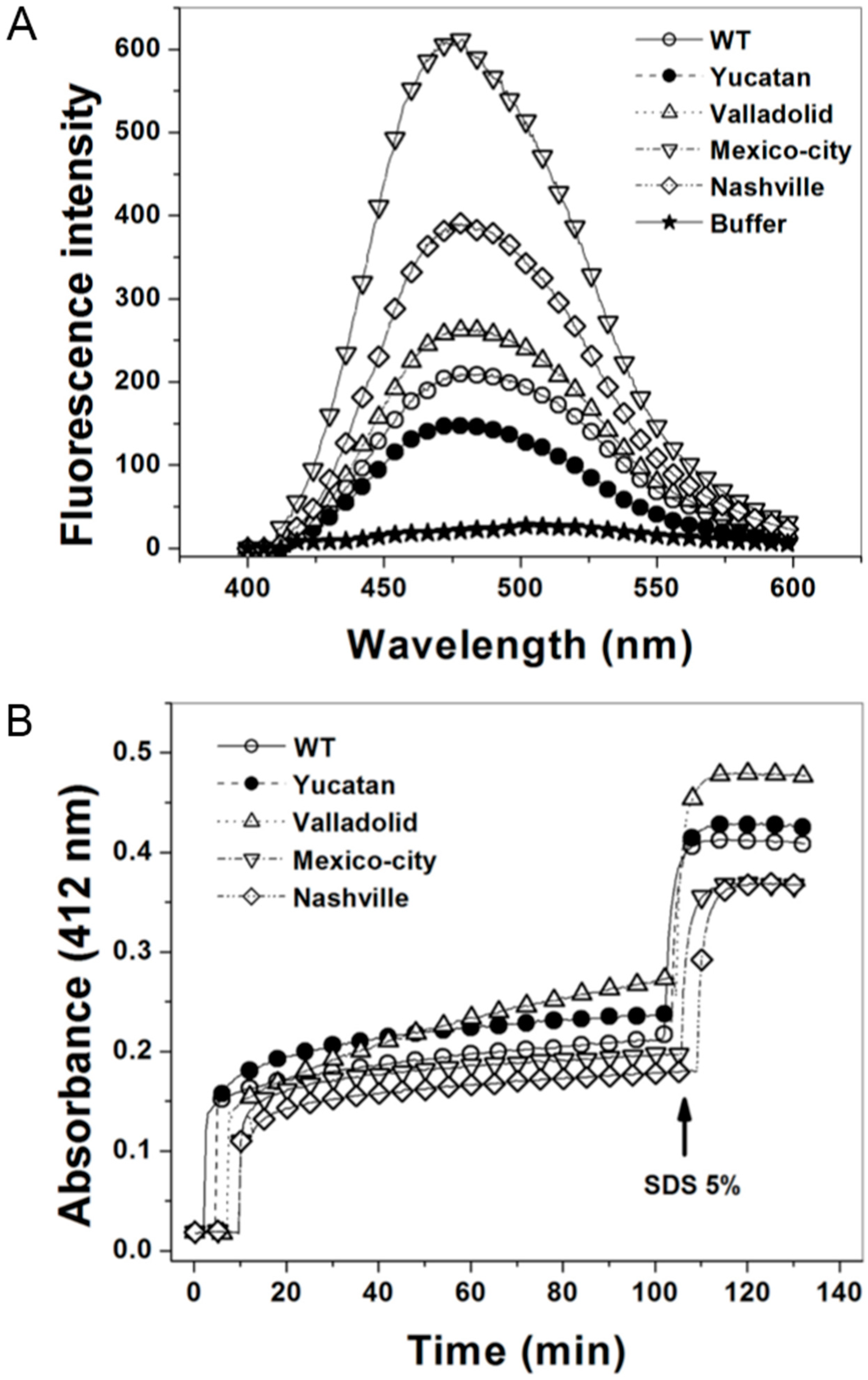
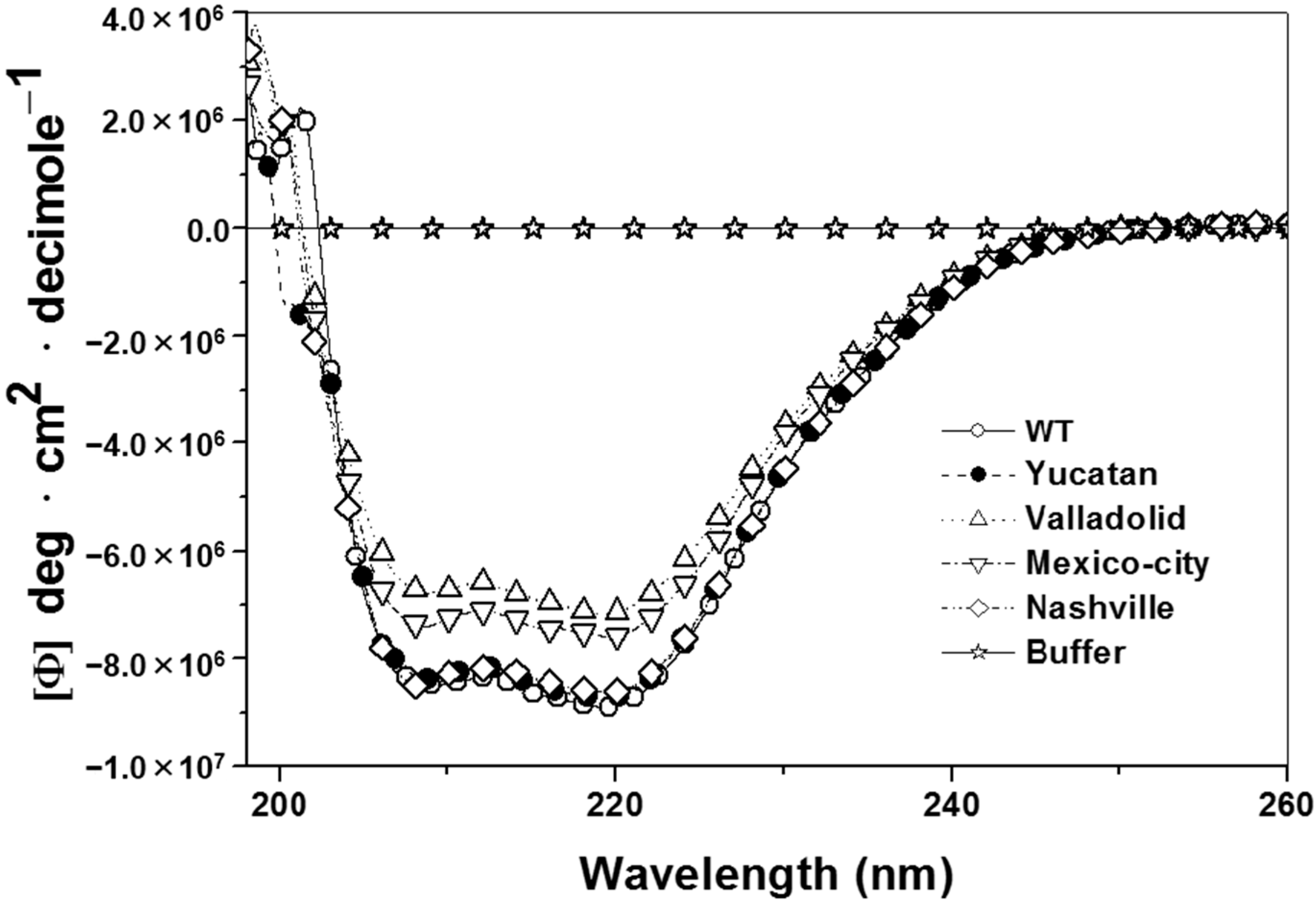
2.4. Structural Characterization of G6PD Enzymes
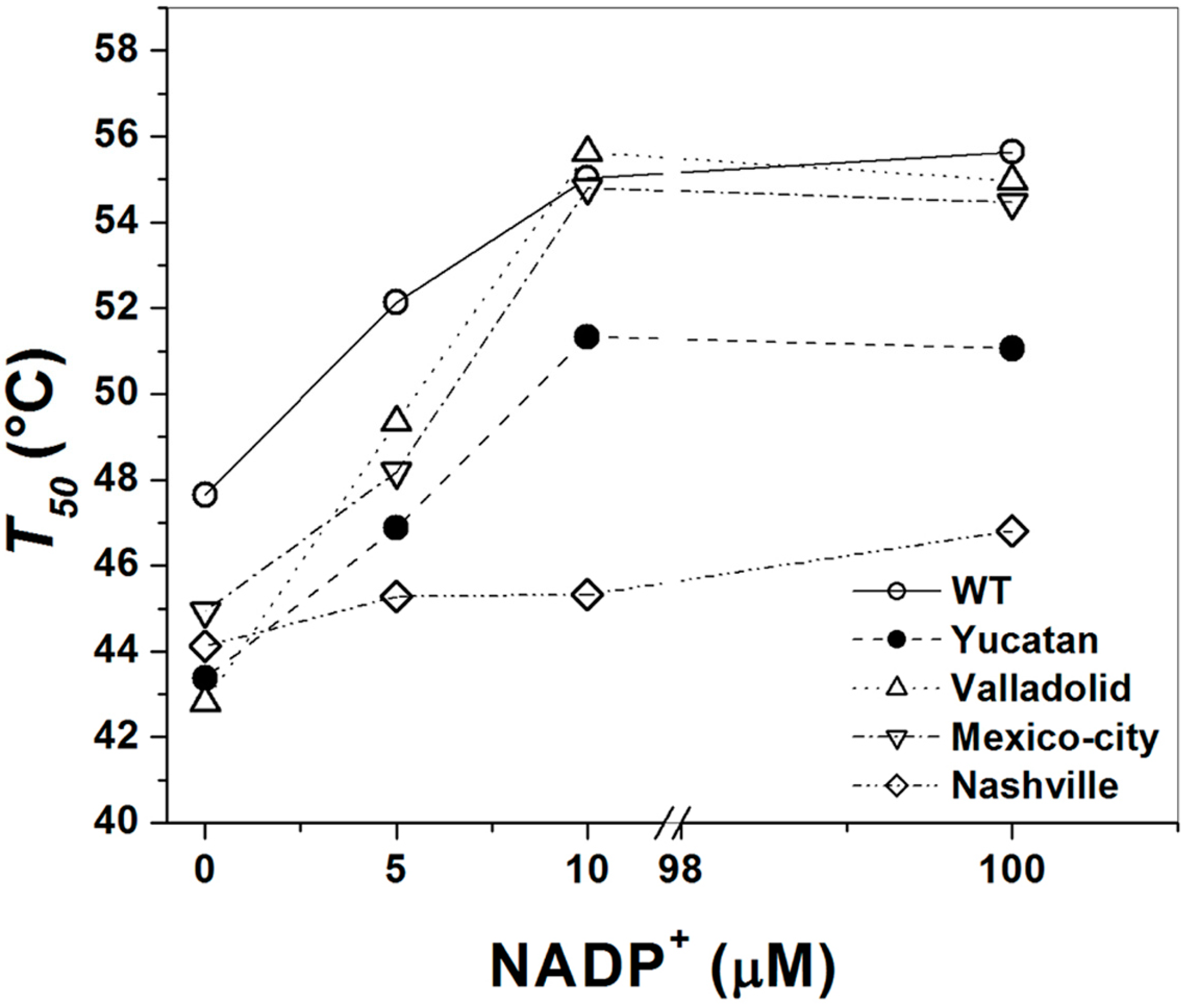
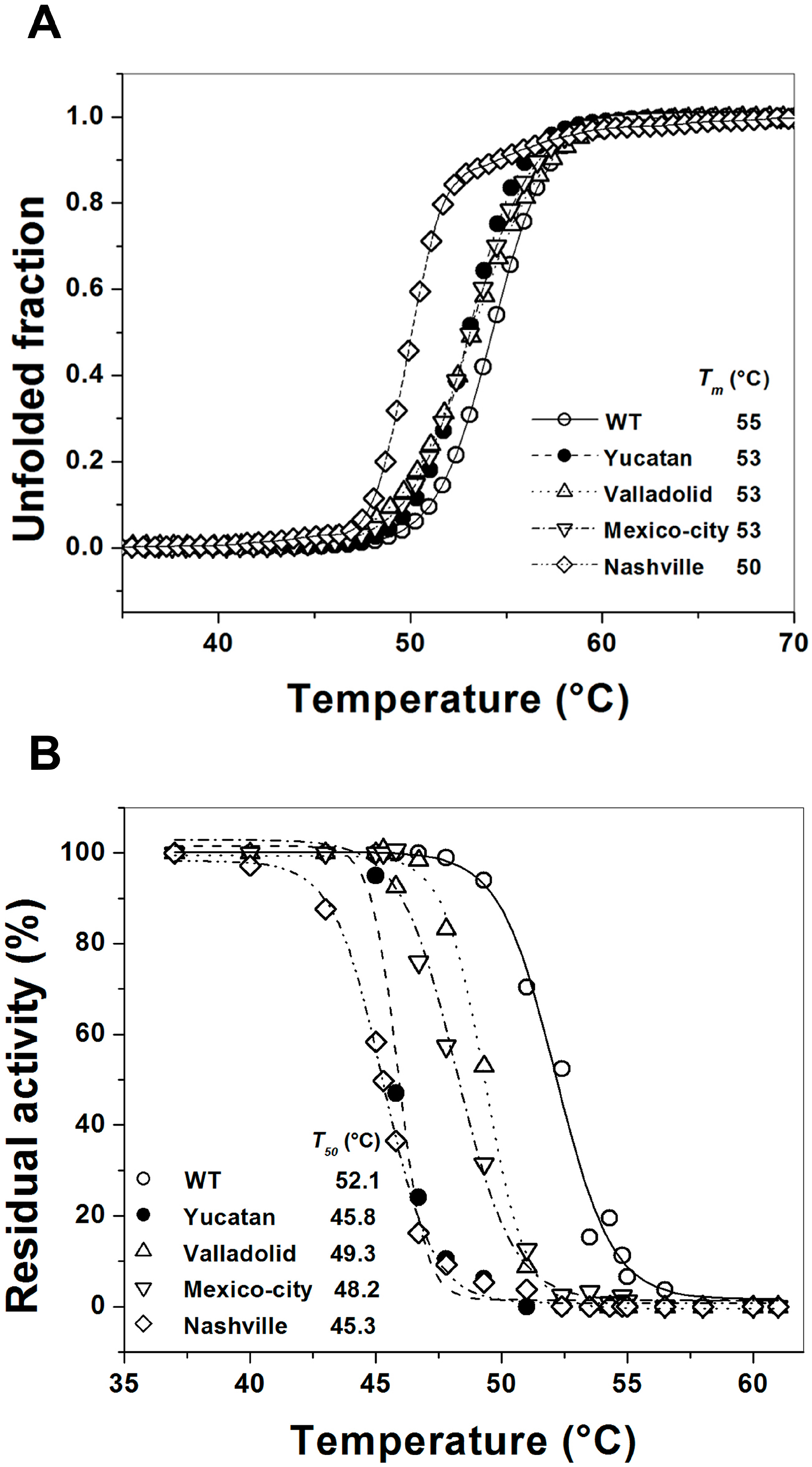
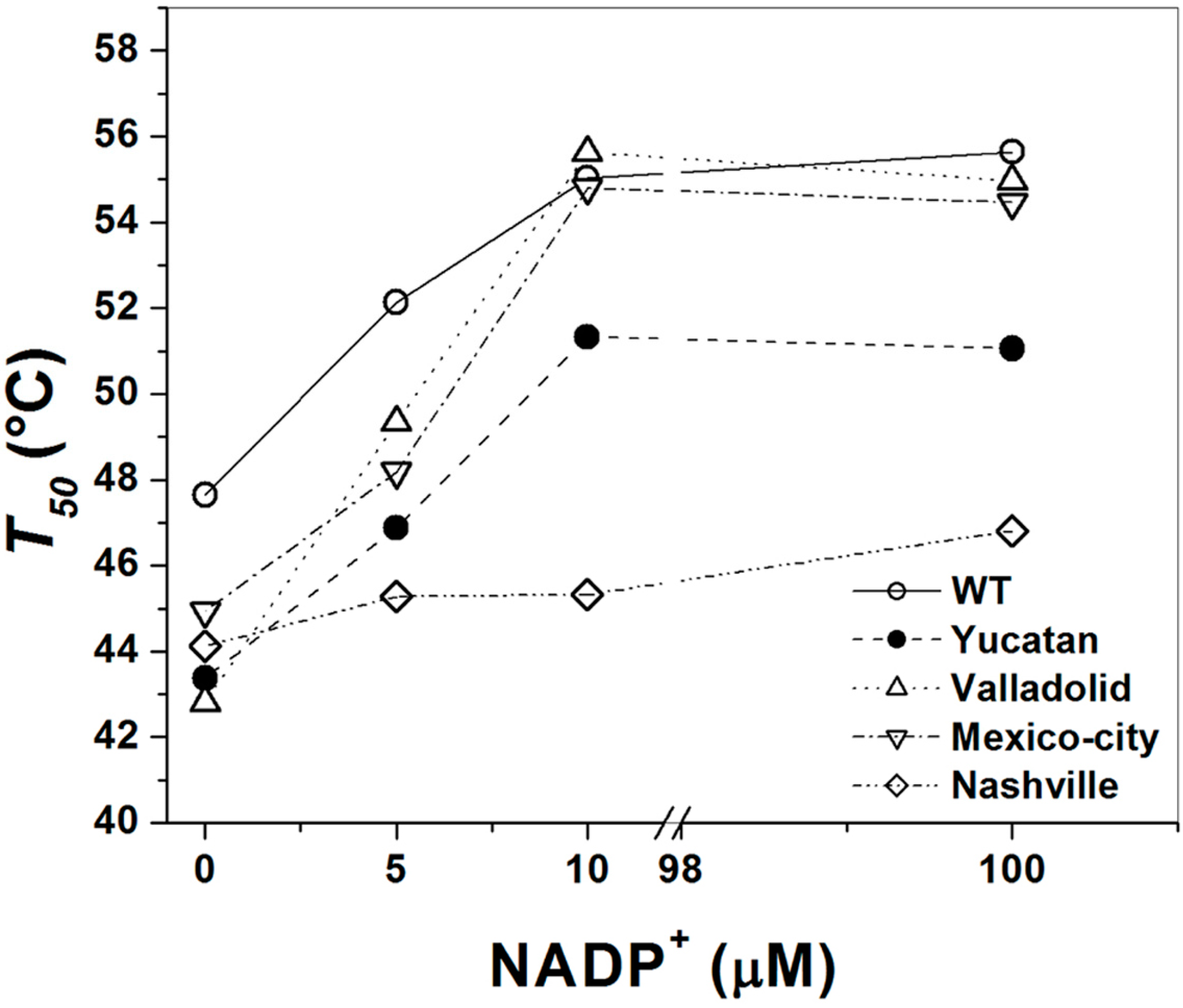
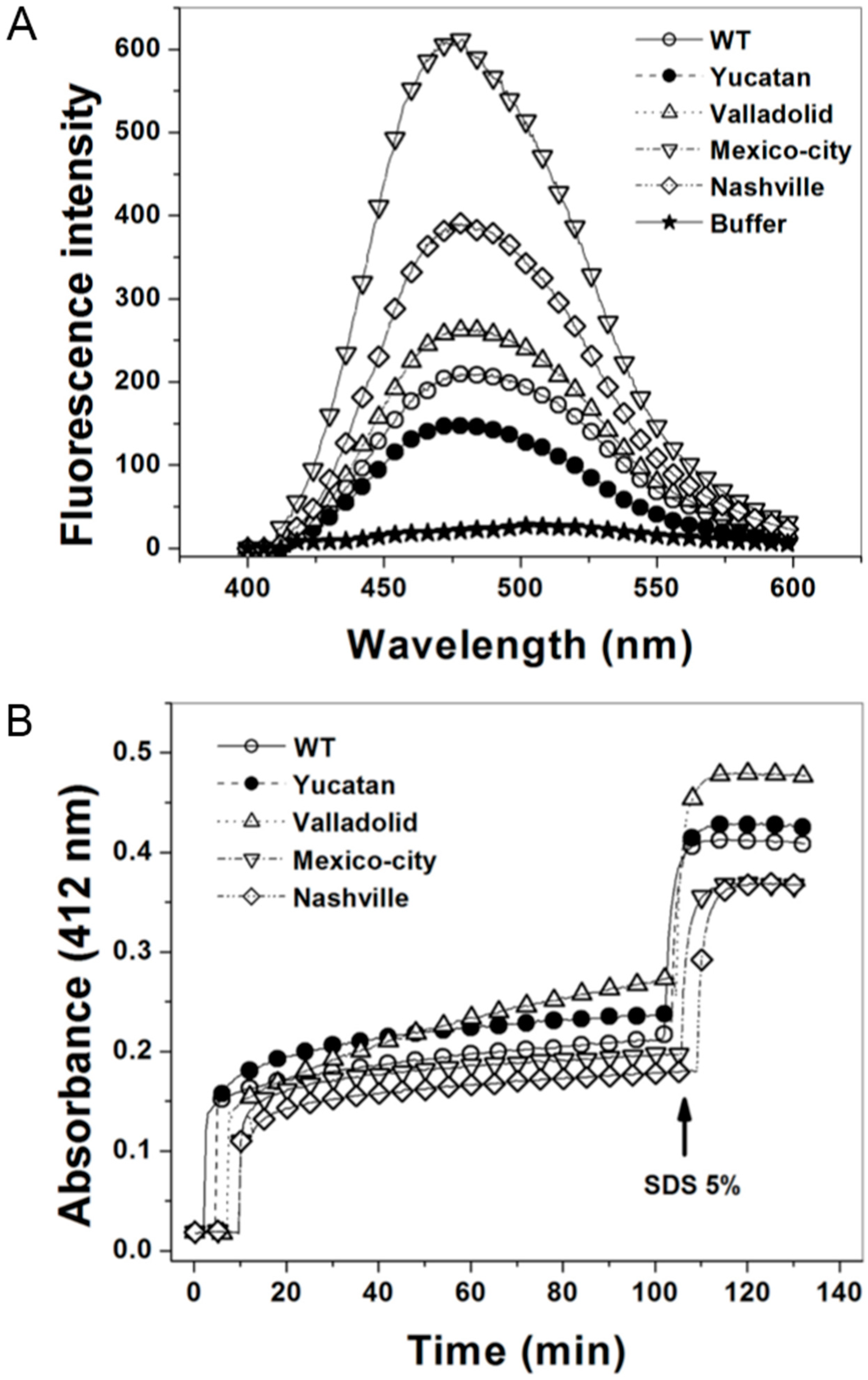
2.5. Analysis of the Stability of the G6PD Enzymes
2.6. Structural Characterization of the G6PD Enzymes
3. Discussion
4. Experimental Section
4.1. Substrates and Reagents

4.2. Construction of the E. coli Mutant Strain BL21(DE3)Δzwf::kanr
4.3. Construction of G6PD Mutants and Cloning into an Expression Plasmid
4.4. Expression and Purification of Recombinant G6PD Enzymes
4.5. Kinetic Characterization of WT G6PD and Variants
4.6. Analysis of Secondary Structure, Thermal Stability, Thermoinactivation and Hydrophobic Surface Exposure
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luzzatto, L.; Mehta, A.; Vulliamy, T.J. Glucose-6-phosphate dehydrogenase deficiency. In Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., et al., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 179, pp. 4517–4553. [Google Scholar]
- Gaetani, G.F.; Galiano, S.; Canepa, L.; Ferraris, A.M.; Kirkman, H.N. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood 1989, 73, 334–339. [Google Scholar]
- Vulliamy, T.; Luzzatto, L.; Hirono, A.; Beutler, E. Hematologically important mutations: Glucose-6-phosphate dehydrogenase. Blood Cells Mol. Dis. 1997, 23, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.J.; Bautista, J.M.; Gilsanz, F. G6PD deficiency: The genotype-phenotype association. Blood Rev. 2007, 5, 267–283. [Google Scholar] [CrossRef]
- Minucci, B.; Giardina, C.; Zuppi, E. Glucose-6-phosphate dehydrogenase laboratory assay: How, when, and why? IUBMB Life 2009, 61, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Lam, V.M.S.; Engel, P.C. Functional properties of two mutants of human glucose 6-phosphate dehydrogenase, R393G and R393H, corresponding to the clinical variants G6PD Wisconsin and Nashville. Biochim. Biophys. Acta 2006, 1762, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Choi, M.Y.; Au, S.W.; Au, D.M.; Lam, V.M.S.; Engel, P.C. Purification and detailed study of two clinically different human glucose 6-phosphate dehydrogenase variants, G6PD (Plymouth) and G6PD (Mahidol): Evidence for defective protein folding as the basis of disease. Mol. Genet. Metab. 2008, 93, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Au, S.W.N.; Gover, S.; Lam, V.M.S.; Adams, M. Human glucose-6-phosphate dehydrogenase: The crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000, 8, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Vaca, G.; Arambula, E.; Esparza, A. Molecular heterogeneity of glucose-6-phosphate dehydrogenase deficiency in Mexico: Overall results of a 7-year project. Blood Cells Mol. Dis. 2002, 28, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Vaca, G.; Arámbula, E.; Monsalvo, A.; Medina, C.; Nuñez, C.; Sandoval, L.; López-Guido, B. Glucose-6-phosphate dehydrogenase (G6PD) mutations in Mexico: Four new G6PD variants. Blood Cells Mol. Dis. 2003, 31, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Vulliamy, T.J. Hematologically important mutations: Glucose-6-phosphate dehydrogenase. Blood Cells. Mol. Dis. 2002, 28, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Zarza, R.; Pujades, A.; Rovira, A.; Saaevdra, R.; Fernánde, J.; Aymerich, M.; Vives-Corrons, J.L. Two new mutations of the glucose-6-hosphate dehydrogenase (G6PD) gene associated with haemolytic anemia: Clinical, biochemical and molecular relationships. Br. J. Haematol. 1997, 98, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Kuhl, W.; Fox, M.; Tabsh, K.; Crandall, B.F. Prenatal diagnosis of glucose-6-phosphate dehydrogenase deficiency. Acta Haematol. 1992, 87, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Engel, P.C. Clinical mutants of human glucose-6-phosphate dehydrogenase: Impairment of NADP+ binding affects both folding and stability. Biochim. Biophys. Acta 2009, 1792, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Thomason, L.C.; Constantino, N.; Court, D.L. E. coli manipulation by P1 transduction. Curr. Protoc. Mol. Biol. 2007. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Terrón-Hernández, J.; de la Mora-de la Mora, I.; García Torres, I.; López-Velázquez, G.; Reyes-Vivas, H.; Oria-Hernández, J. Cloning, expression, purification and characterization of His-tagged human glucose-6-phosphate dehydrogenase: A simplified method for protein yield. Protein J. 2013, 32, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Chan, T.F.; Lam, V.; Engel, P. What is the role of the second “structural” NADP-binding site in human glucose-6-phosphate dehydrogenase? Protein Sci. 2008, 17, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Scopes, D.A.; Bautista, J.M.; Naylor, C.E.; Adams, M.J.; Mason, P.J. Amino acid substitutions at the dimer interface of human glucose-6-phosphate dehydrogenase that increase thermostability and reduce the stabilising effect of NADP. Eur. J. Biochem. 1998, 251, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Lam, V.M.; Engel, P.C. Marked decrease in specific activity contributes to disease phenotype in two human glucose-6-phosphate dehydrogenase mutants, G6PDUnion and G6PDAndalus. Hum. Mutat. 2005, 26, 284–293. [Google Scholar] [CrossRef] [PubMed]
- De la Mora-de la Mora, I.; Torres-Larios, A.; Mendoza-Hernández, G.; Garcia-Torres, I.; Torres-Arroyo, A.; Gómez-Manzo, S.; Marcial-Quino, J.; Oria-Hernández, J.; López-Velázquez, G.; Reyes-Vivas, H. The E104D mutation increases the susceptibility of human triosephosphate isomerase to proteolysis. Asymmetric cleavage of the two monomers of the homodimeric enzyme. Biochim. Biophys. Acta 2013, 1834, 2702–2711. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Flores, S.; Rodríguez-Romero, A.; Hernández-Alcántara, G.; Oria-Hernéndez, J.; Gutiérrez-Castrellón, P.; Pérez-Hernández, G.; de la Mora-de la Mora, I.; Castillo-Villanueva, A.; García-Torres, I.; Méndez, S.T.; et al. Determining the molecular mechanism of inactivation by chemical modification of triosephosphate isomerase from the human parasite Giardia lamblia: A study for antiparasitic drug design. Proteins 2011, 79, 2711–2724. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.; Lysko, H. A precise method for the determination of whole blood and plasma sulfhydryl groups. Anal. Biochem. 1979, 93, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 9606, 64–74. [Google Scholar] [CrossRef]
- Bautista, J.M.; Mason, P.J.; Luzzatto, L. Purification and properties of human glucose-6-phosphate dehydrogenase made in E. coli. Biochim. Biophys. Acta 1992, 1119, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.K.; Yeh, C.H.; Huang, C.S.; Huang, M.J. Expression and biochemical characterization of human glucose-6-phosphate dehydrogenase in Escherichia coli: A system to analyze normal and mutant enzymes. Blood 1994, 83, 1436–1441. [Google Scholar] [PubMed]
- Wang, X.T.; Au, S.W.N.; Lam, V.M.S.; Engel, P.C. Recombinant human glucose-6-phosphate dehydrogenase: Evidence for a rapid-equilibrium random-order mechanism. Eur. J. Biochem. 2002, 269, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Allan, N.C. Different properties of glucose 6-phosphate dehydrogenase from human erythrocytes with normal and abnormal enzyme levels. Biochem. Biophys. Res. Commun. 1965, 21, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Town, M.; Bautista, J.M.; Mason, P.J.; Luzzatto, L. Both mutations in G6PD A− are necessary to produce the G6PD deficient phenotype. Hum. Mol. Genet. 1992, 1, 171–174. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System. DeLano Scientific: Palo Alto, CA, USA, 2002.
- Lennox, E.S. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1955, 1, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Budzik, J.M.; Rosche, W.A.; Rietsch, A.; O’Toole, G.A. Isolation and characterization of a generalized transducing phage for Pseudomonas aeruginosa strains PAO1 and PA14. J. Bacteriol. 2004, 186, 3270–3273. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Betke, K.; Beutler, E.; Brewer, G.J.; Kirkman, H.N.; Luzzatto, L.; Motulsky, A.G.; Ramot, B.; Siniscalco, M. Standardization of procedures for the study of glucose-6-phosphate dehydrogenase. World Health Organ Tech. Rep. Ser. 1967, 366, 1–53. [Google Scholar] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcántara, G.; Oria-Hernández, J.; et al. The Stability of G6PD Is Affected by Mutations with Different Clinical Phenotypes. Int. J. Mol. Sci. 2014, 15, 21179-21201. https://doi.org/10.3390/ijms151121179
Gómez-Manzo S, Terrón-Hernández J, De la Mora-De la Mora I, González-Valdez A, Marcial-Quino J, García-Torres I, Vanoye-Carlo A, López-Velázquez G, Hernández-Alcántara G, Oria-Hernández J, et al. The Stability of G6PD Is Affected by Mutations with Different Clinical Phenotypes. International Journal of Molecular Sciences. 2014; 15(11):21179-21201. https://doi.org/10.3390/ijms151121179
Chicago/Turabian StyleGómez-Manzo, Saúl, Jessica Terrón-Hernández, Ignacio De la Mora-De la Mora, Abigail González-Valdez, Jaime Marcial-Quino, Itzhel García-Torres, America Vanoye-Carlo, Gabriel López-Velázquez, Gloria Hernández-Alcántara, Jesús Oria-Hernández, and et al. 2014. "The Stability of G6PD Is Affected by Mutations with Different Clinical Phenotypes" International Journal of Molecular Sciences 15, no. 11: 21179-21201. https://doi.org/10.3390/ijms151121179
APA StyleGómez-Manzo, S., Terrón-Hernández, J., De la Mora-De la Mora, I., González-Valdez, A., Marcial-Quino, J., García-Torres, I., Vanoye-Carlo, A., López-Velázquez, G., Hernández-Alcántara, G., Oria-Hernández, J., Reyes-Vivas, H., & Enríquez-Flores, S. (2014). The Stability of G6PD Is Affected by Mutations with Different Clinical Phenotypes. International Journal of Molecular Sciences, 15(11), 21179-21201. https://doi.org/10.3390/ijms151121179