1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone-Induced Cell Cycle Arrest in G1/G0 in HT-29 Cells Human Colon Adenocarcinoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
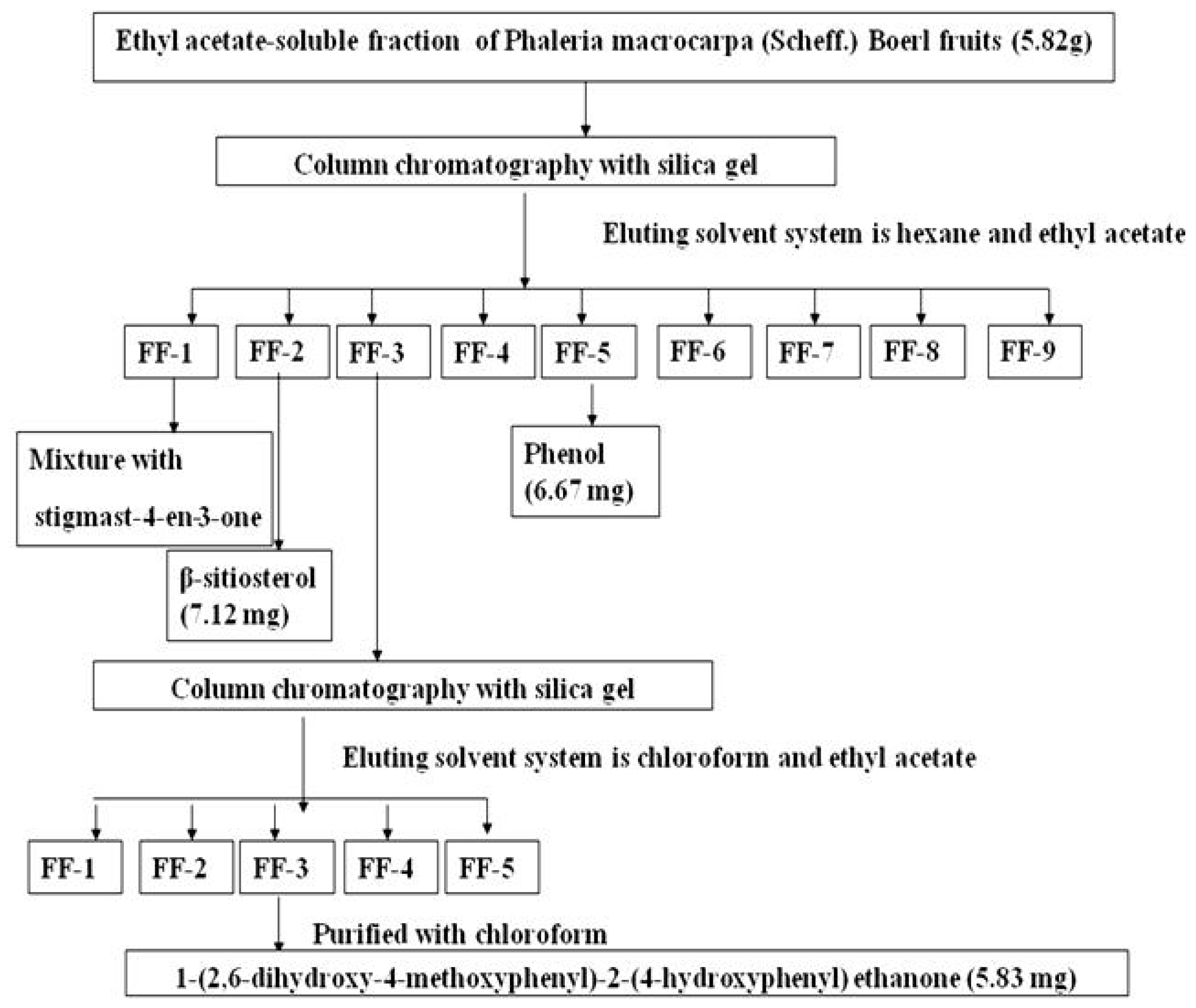
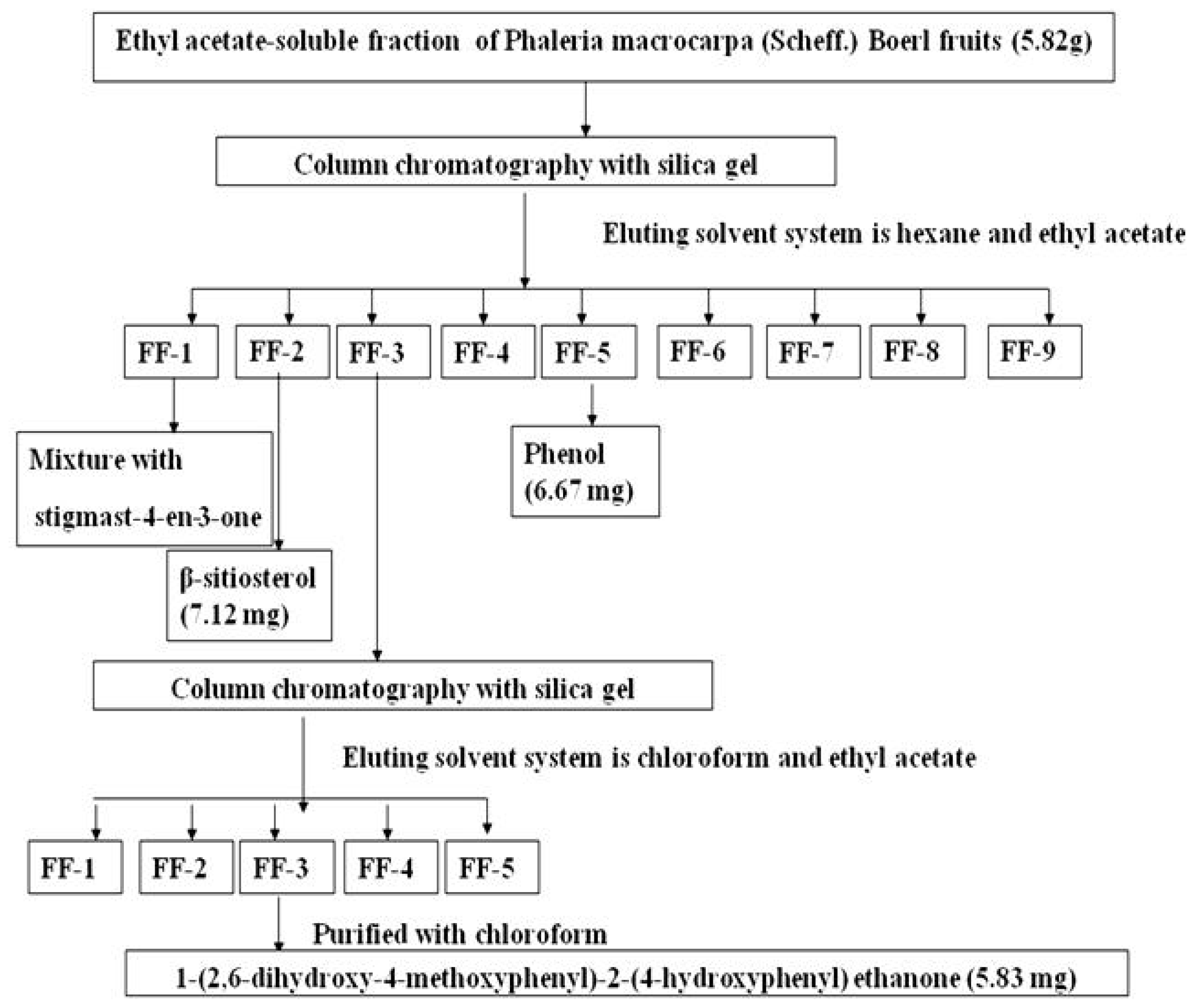
2.1. Isolation of 1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone (DMHE)
2.2. Identification of 1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone (DMHE)
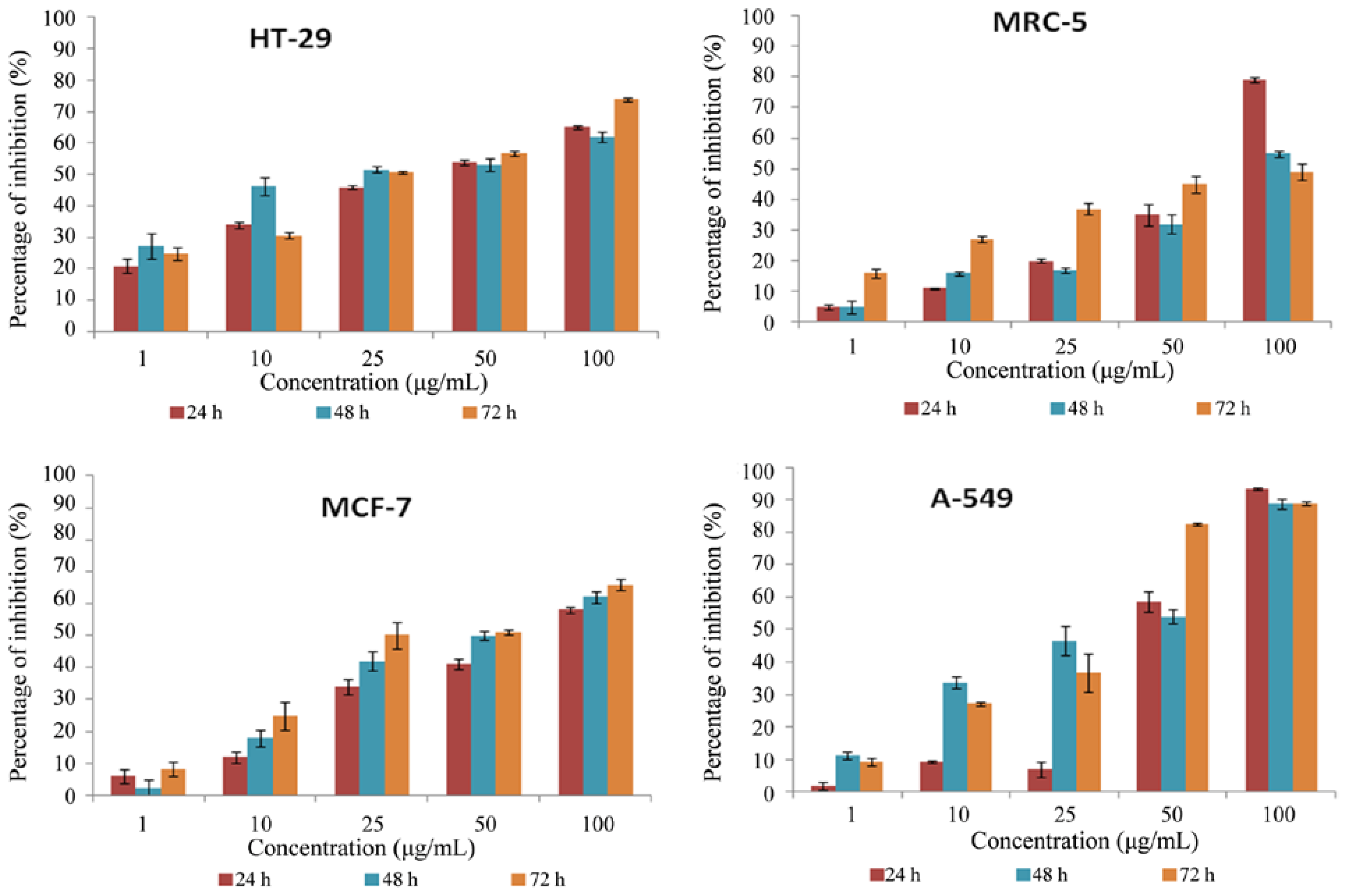
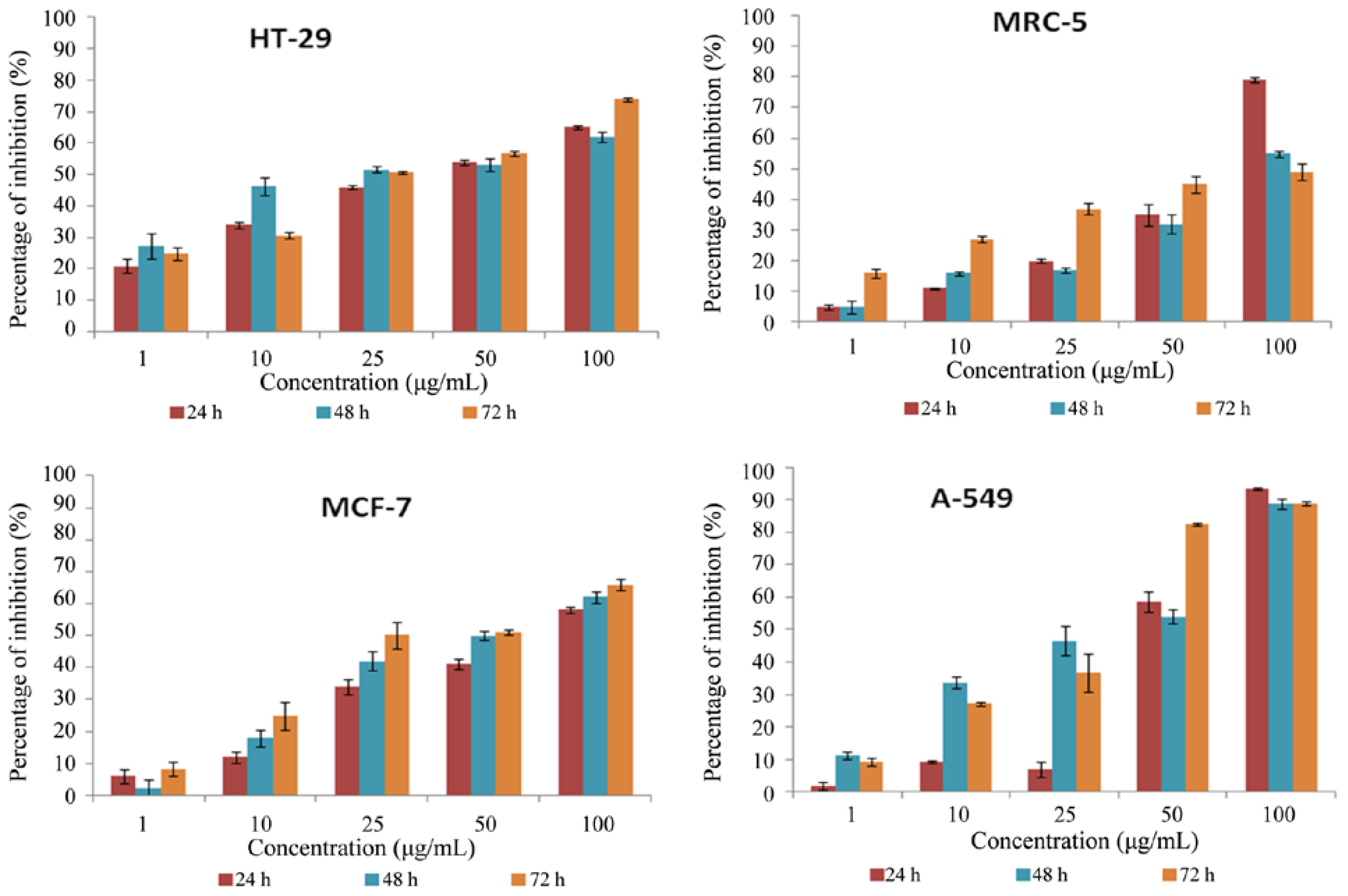
2.3. Cytotoxicity Screening Using MTT Cell Proliferation Assay
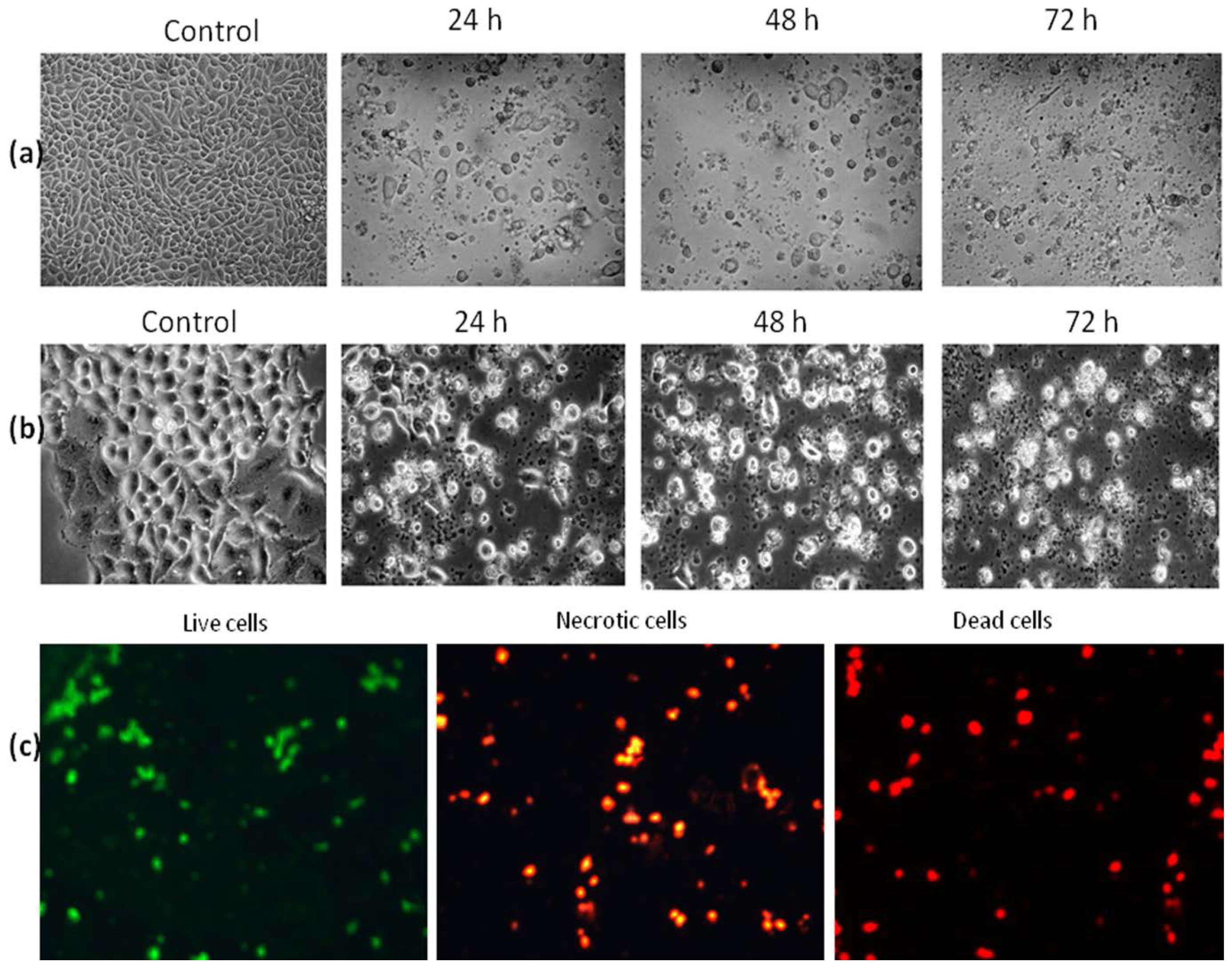
2.4. Morphological Examination of Apoptosis
2.4.1. Inverted and Phase Contrast Microscopic Examination
2.4.2. Fluorescence Microscopic Examination
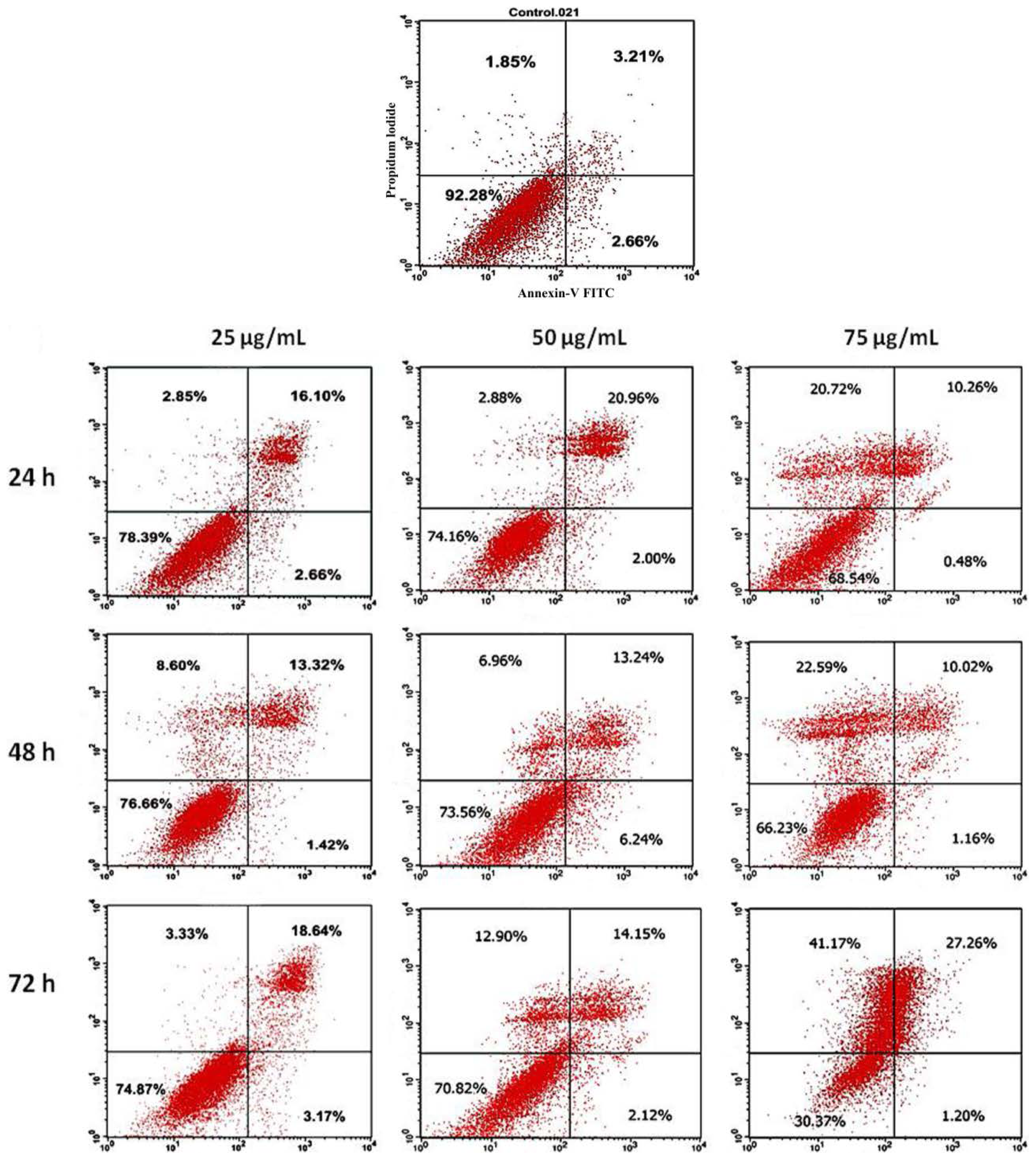
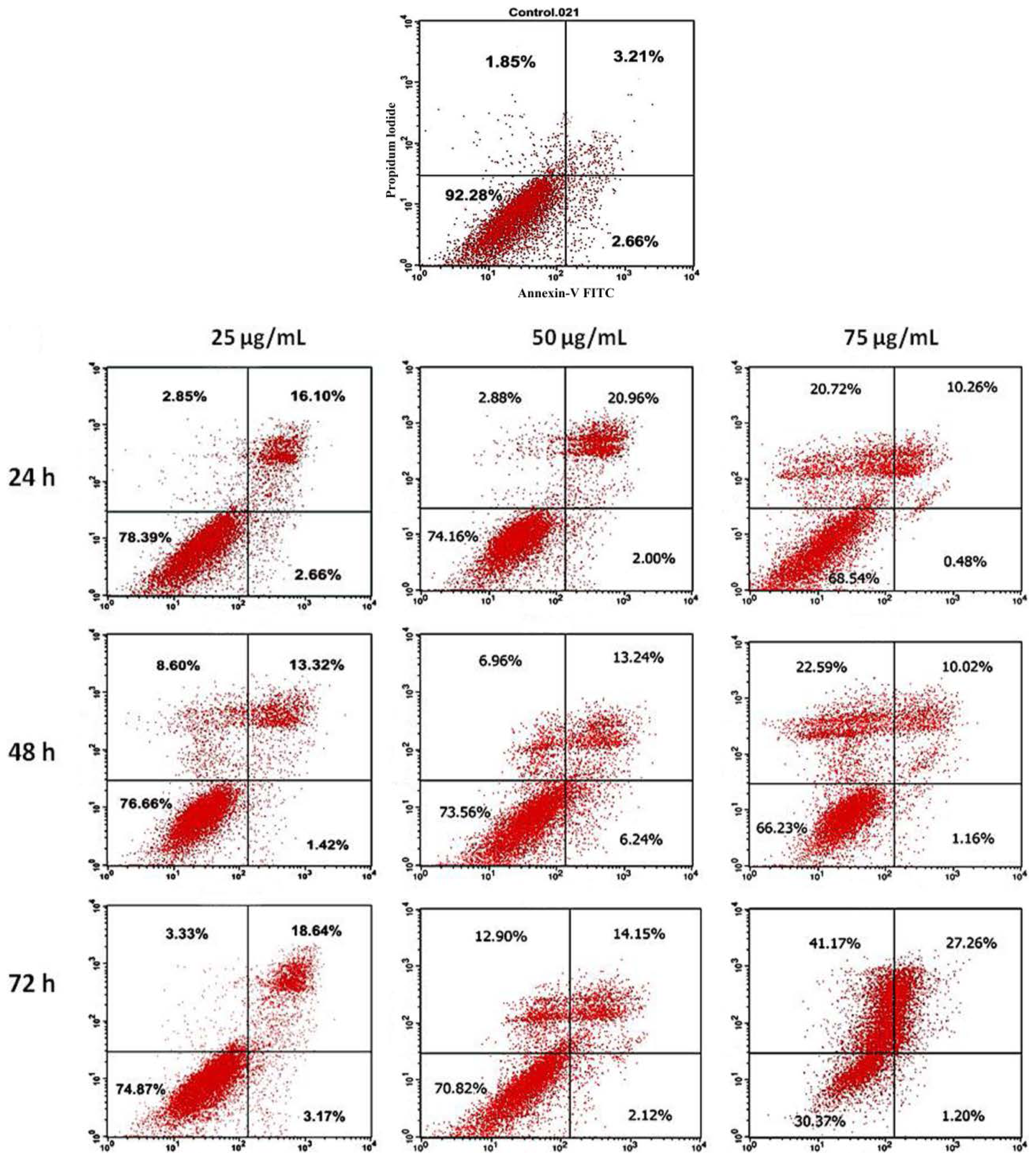
2.5. Annexin V Staining Assay
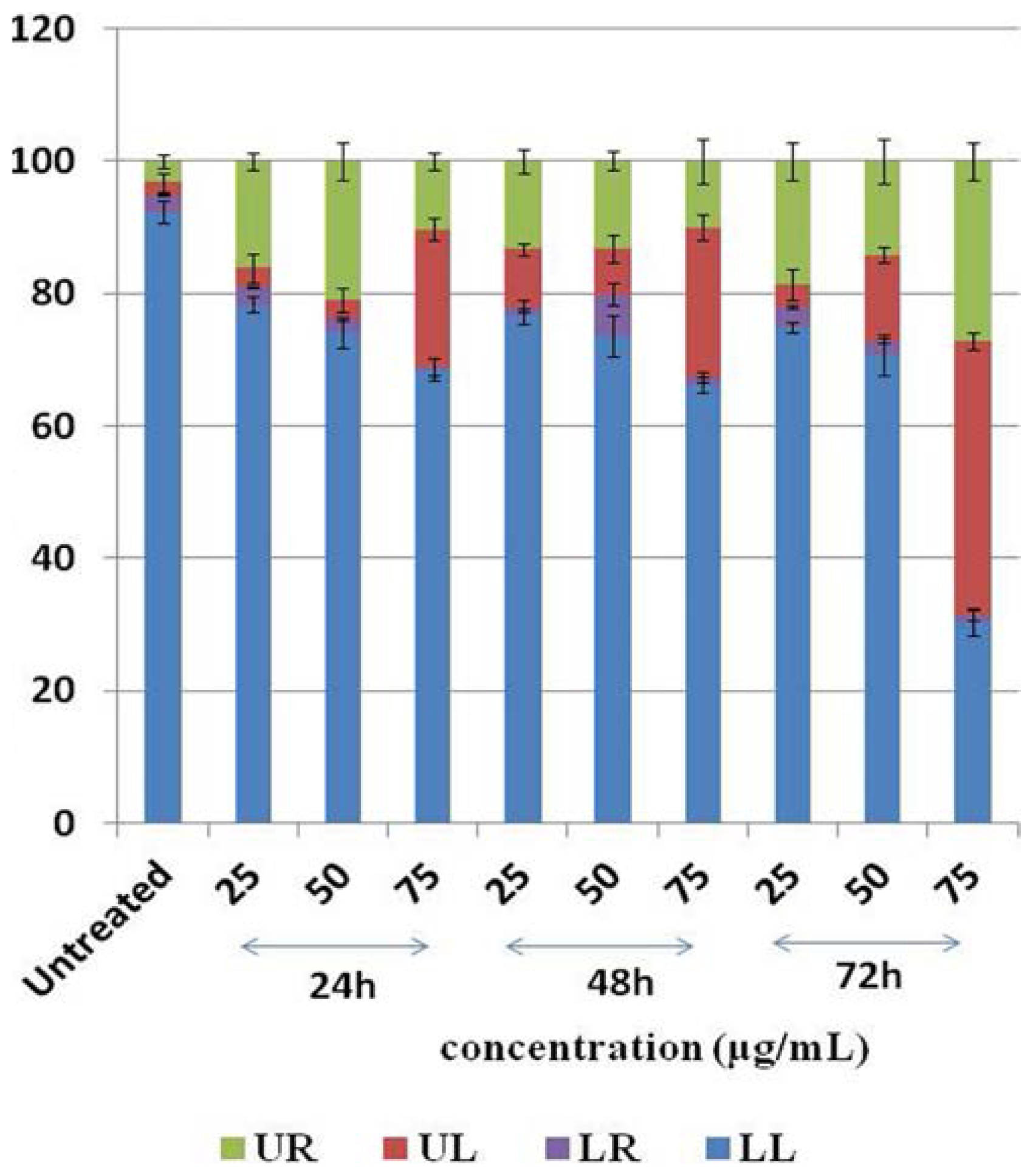
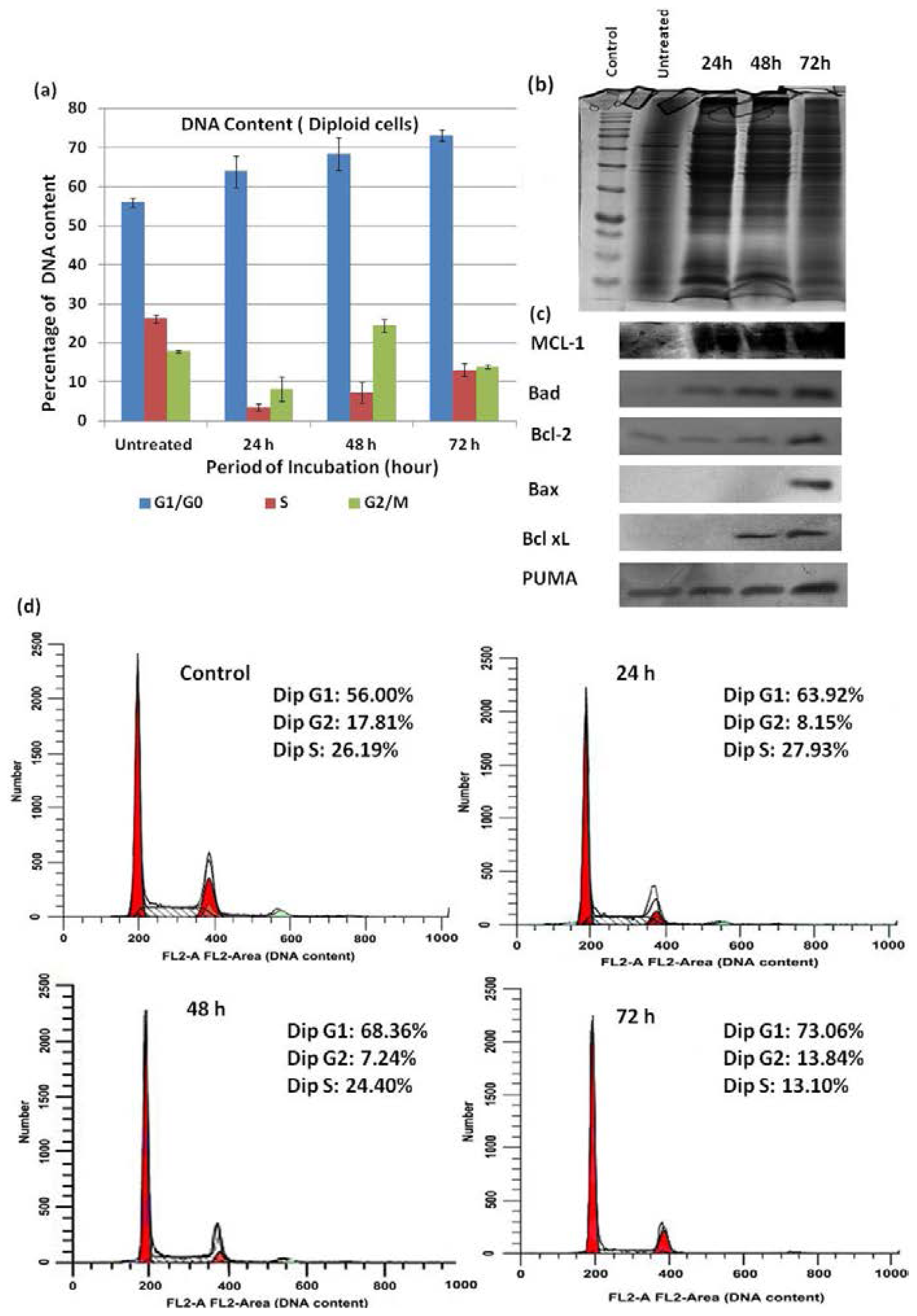
2.6. Cell Cycle Analysis
2.7. Western Blot Analysis
3. Experimental Section
3.1. Isolation of Bioactive Compound
3.2. GC-MS (Gas Chromatography-Mass Spectrophotometry) and NMR (Nuclear Magnetic Resonance) Analysis of Bioactive Compound
3.3. Cytotoxicity Screening
3.3.1. Cell Culture
3.3.2. MTT Cell Proliferation Assay
3.4. Morphological Examination of Apoptosis
3.4.1. Inverted and Phase-Contrast Microscopy
3.4.2. Fluorescence Microscopy
3.5. Annexin V Staining Assay
3.6. Cell Cycle Analysis
3.7. Western Blot Analysis
3.8. Data Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kinghorn, A.D.; Balandrin, N.F. (Eds.) American Chemical Society: Washington, DC, USA, 1993; p. 534.
- Farnsworth, N.R.; Akerele, O.; Bingel, A.S.; Soejarto, D.D.; Guo, Z. Medicinal plants in therapy. Bull. World Health Organ 1985, 63, 965–981. [Google Scholar]
- Triastuti, A.; Hee-Juhn, P.; Jong, W.C. Phaleria macrocarpa suppresses oxidative stress in alloxan-induced diabetic rats by enhancing hepatic antioxidant enzyme activity. Nat. Prod. Sci 2009, 15, 37–43. [Google Scholar]
- Rudi, H.; Ahmad, S.; Oskoueian, E.; Sukari, A.; Shukor, M.Y. Antioxidant, anti-inflammatory and cytotoxicity of Phaleria macrocarpa (Boerl.) Scheff Fruit. BMC Complement. Altern. Med 2011. [Google Scholar] [CrossRef]
- Susanthy, E. N-Hexane Extract Cytotoxicity Beans and Meat Mahkota Dewa Fruit (Phaleria macrocarpa [Scheff.] Boerl.) Against HeLa and Myeloma Cell Line; Jurusan Kimia FMIPA UGM: Yogyakarta, Indonesia, 2005; pp. 47–50. [Google Scholar]
- Faried, A.; Kurnia, D.; Faried, L.S.; Usman, N.; Miyazaki, T.; Kato, H.; Kuwano, H. Anticancer effects of gallic acid isolated from Indonesian herbal medicine, Phaleria macrocarpa (Scheff.) Boerl, on human cancer cell lines. Int. J. Oncol 2007, 30, 605–613. [Google Scholar]
- Aripin, A.; Arifin, P.F.; Tjandrawinata, R.R. Isolated Compounds from Phaleria macrocarpa as Anti-Cancer Agents. U.S. Patent US8569382 B2, 29 October 2013. [Google Scholar]
- Raymond, R.T.; Dwi, N.; Liana, W.S.; Prihatini, H.; Audrey, C. Symptomatic treatment of premenstrual syndrome and/or primary dysmenorrhea with DLBS1442, a bioactive extract of Phaleria macrocarpa. Int. J. Gen. Med 2011, 4, 465–476. [Google Scholar]
- Arunporn, I.; Peter, J.; Houghton, E.; Amooquaye, E. In vitro cytotoxic activity of Thai medicinal plants used traditionally to treat cancer. J. Ethnopharmacol 2004, 90, 33–38. [Google Scholar]
- Potten, C.S.; James, W.W. Apoptosis: The Life and Death of Cells; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Call, J.A.; Eckhardt, S.G.; Camidge, D.R. Targeted manipulation of apoptosis in cancer treatment. Lancet Oncol 2008, 9, 1002–1011. [Google Scholar]
- Dickson, M.A.; Schwartz, G.K. Development of cell cycle inhibitors for cancer therapy. Curr. Opin. Oncol 2009, 16, 120–127. [Google Scholar]
- Reed, J.; Pellecchia, M. Apoptosis-based therapies for hematological malignancies. Blood 2005, 106, 408–418. [Google Scholar]
- Malek, S.N.A.; Phang, C.W.; Ibrahim, H.; Norhanom, A.W.; Sim, K.S. Phytochemical and cytotoxic investigations of Alpinia mutica rhizomes. Molecules 2011, 16, 583–589. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival, modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar]
- Malek, S.N.A.; Shin, S.K.; Wahab, N.A.; Yaacob, H. Cytotoxic components of Pereskia bleo (Kunth) DC. (Cactaceae) leaves. Molecules 2009, 14, 1713–1724. [Google Scholar]
- Malek, S.N.A.; Lee, G.S.; Hong, S.L.; Yaacob, H.; Wahab, N.A.; Faizal Weber, J.-F.; Shah, S.A.A. Phytochemical and cytotoxic investigations of Curcuma mangga rhizomes. Molecules 2011, 16, 4539–4548. [Google Scholar]
- Scherliess, R. The MTT assay as tool to evaluate and compare excipient toxicity in vitro on respiratory epithelial cells. Int. J. Pharm 2011, 411, 98–105. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lay, M.M.; Karsani, S.A.; Malek, S.N.A. 1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone-Induced Cell Cycle Arrest in G1/G0 in HT-29 Cells Human Colon Adenocarcinoma Cells. Int. J. Mol. Sci. 2014, 15, 468-483. https://doi.org/10.3390/ijms15010468
Lay MM, Karsani SA, Malek SNA. 1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone-Induced Cell Cycle Arrest in G1/G0 in HT-29 Cells Human Colon Adenocarcinoma Cells. International Journal of Molecular Sciences. 2014; 15(1):468-483. https://doi.org/10.3390/ijms15010468
Chicago/Turabian StyleLay, Ma Ma, Saiful Anuar Karsani, and Sri Nurestri Abd Malek. 2014. "1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone-Induced Cell Cycle Arrest in G1/G0 in HT-29 Cells Human Colon Adenocarcinoma Cells" International Journal of Molecular Sciences 15, no. 1: 468-483. https://doi.org/10.3390/ijms15010468
APA StyleLay, M. M., Karsani, S. A., & Malek, S. N. A. (2014). 1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl) Ethanone-Induced Cell Cycle Arrest in G1/G0 in HT-29 Cells Human Colon Adenocarcinoma Cells. International Journal of Molecular Sciences, 15(1), 468-483. https://doi.org/10.3390/ijms15010468