Human Diseases Associated with Form and Function of the Golgi Complex


Abstract
:1. Introduction
2. When Traffic Comes to a Halt
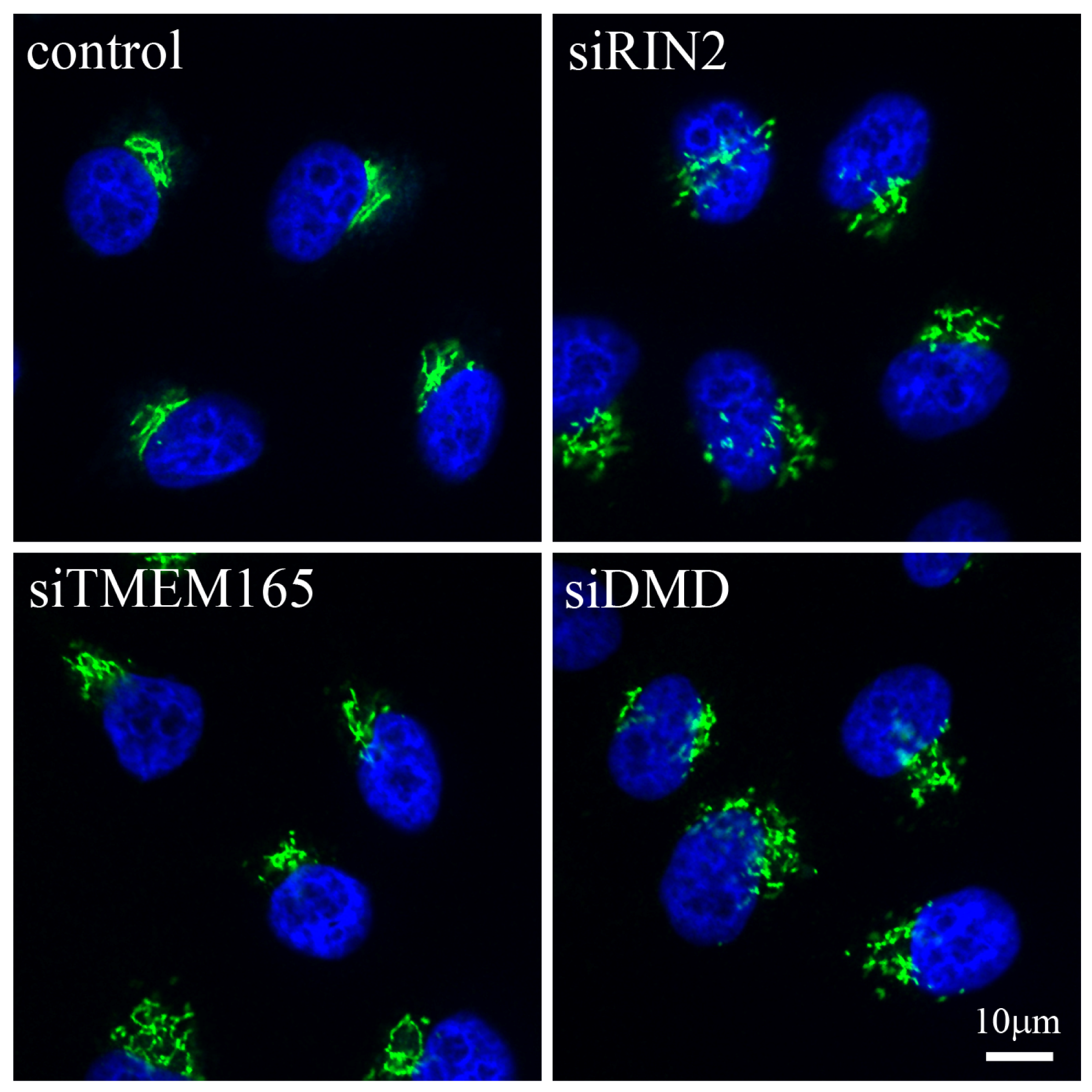
3. When Shape Changes
4. When Glycosylation Is Impaired
5. When Function Is Lost
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Aridor, M.; Hannan, L.A. Traffic jam: A compendium of human diseases that affect intracellular transport processes. Traffic 2000, 1, 836–851. [Google Scholar]
- Aridor, M.; Hannan, L.A. Traffic jams ii: An update of diseases of intracellular transport. Traffic 2002, 3, 781–790. [Google Scholar]
- Klumperman, J. Architecture of the mammalian golgi. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Lowe, M. Structural organization of the golgi apparatus. Curr. Opin. Cell Biol 2011, 23, 85–93. [Google Scholar]
- Inoue, K. Plp1-related inherited dysmyelinating disorders: Pelizaeus-merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005, 6, 1–16. [Google Scholar]
- Numata, Y.; Morimura, T.; Nakamura, S.; Hirano, E.; Kure, S.; Goto, Y.I.; Inoue, K. Depletion of molecular chaperones from the endoplasmic reticulum and fragmentation of the golgi apparatus associated with pathogenesis in pelizaeus-merzbacher disease. J. Biol. Chem 2013, 288, 7451–7466. [Google Scholar]
- Ting, C.H.; Wen, H.L.; Liu, H.C.; Hsieh-Li, H.M.; Li, H.; Lin-Chao, S. The spinal muscular atrophy disease protein smn is linked to the golgi network. PLoS One 2012, 7, e51826. [Google Scholar]
- Zhang, C.; Li, D.; Zhang, J.; Chen, X.; Huang, M.; Archacki, S.; Tian, Y.; Ren, W.; Mei, A.; Zhang, Q.; et al. Mutations in abcb6 cause dyschromatosis universalis hereditaria. J. Investig. Dermatol 2013, 133, 2221–2228. [Google Scholar]
- Presley, J.F.; Cole, N.B.; Schroer, T.A.; Hirschberg, K.; Zaal, K.J.; Lippincott-Schwartz, J. Er-to-golgi transport visualized in living cells. Nature 1997, 389, 81–85. [Google Scholar]
- Cole, N.B.; Sciaky, N.; Marotta, A.; Song, J.; Lippincott-Schwartz, J. Golgi dispersal during microtubule disruption: Regeneration of golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell 1996, 7, 631–650. [Google Scholar]
- Hirschberg, K.; Miller, C.M.; Ellenberg, J.; Presley, J.F.; Siggia, E.D.; Phair, R.D.; Lippincott-Schwartz, J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol 1998, 143, 1485–1503. [Google Scholar]
- Rendon, W.O.; Martinez-Alonso, E.; Tomas, M.; Martinez-Martinez, N.; Martinez-Menarguez, J.A. Golgi fragmentation is rab and snare dependent in cellular models of parkinson’s disease. Histochem. Cell Biol 2013, 139, 671–684. [Google Scholar]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks er-golgi traffic and rab1 rescues neuron loss in parkinson’s models. Science 2006, 313, 324–328. [Google Scholar]
- Dupuis, N.; Lebon, S.; Kumar, M.; Drunat, S.; Graul-Neumann, L.M.; Gressens, P.; El Ghouzzi, V. A novel rab33b mutation in smith-mccort dysplasia. Hum. Mutat 2013, 34, 283–286. [Google Scholar]
- Singan, V.R.; Handzic, K.; Simpson, J.C. Quantitative image analysis approaches for probing rab gtpase localization and function in mammalian cells. Biochem. Soc. Trans 2012, 40, 1389–1393. [Google Scholar]
- Saito, K.; Murai, J.; Kajiho, H.; Kontani, K.; Kurosu, H.; Katada, T. A novel binding protein composed of homophilic tetramer exhibits unique properties for the small gtpase rab5. J. Biol. Chem 2002, 277, 3412–3418. [Google Scholar]
- Basel-Vanagaite, L.; Sarig, O.; Hershkovitz, D.; Fuchs-Telem, D.; Rapaport, D.; Gat, A.; Isman, G.; Shirazi, I.; Shohat, M.; Enk, C.D.; et al. Rin2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: Macs syndrome. Am. J. Hum. Genet 2009, 85, 254–263. [Google Scholar]
- Syx, D.; Malfait, F.; van Laer, L.; Hellemans, J.; Hermanns-Le, T.; Willaert, A.; Benmansour, A.; de Paepe, A.; Verloes, A. The rin2 syndrome: A new autosomal recessive connective tissue disorder caused by deficiency of ras and rab interactor 2 (rin2). Hum. Genet 2010, 128, 79–88. [Google Scholar]
- Simpson, J.C.; Joggerst, B.; Laketa, V.; Verissimo, F.; Cetin, C.; Erfle, H.; Bexiga, M.G.; Singan, V.R.; Heriche, J.K.; Neumann, B.; et al. Genome-wide rnai screening identifies human proteins with a regulatory function in the early secretory pathway. Nat. Cell Biol 2012, 14, 764–774. [Google Scholar]
- Chia, J.; Goh, G.; Racine, V.; Ng, S.; Kumar, P.; Bard, F. Rnai screening reveals a large signaling network controlling the golgi apparatus in human cells. Mol. Syst. Biol 2012, 8, 629. [Google Scholar]
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; Bammens, R.; Morelle, W.; Rosnoblet, C.; Legrand, D.; et al. Tmem165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet 2012, 91, 15–26. [Google Scholar]
- Zeevaert, R.; de Zegher, F.; Sturiale, L.; Garozzo, D.; Smet, M.; Moens, M.; Matthijs, G.; Jaeken, J. Bone dysplasia as a key feature in three patients with a novel congenital disorder of glycosylation (cdg) type ii due to a deep intronic splice mutation in tmem165. JIMD Rep 2013, 8, 145–152. [Google Scholar]
- Rosnoblet, C.; Legrand, D.; Demaegd, D.; Hacine-Gherbi, H.; de Bettignies, G.; Bammens, R.; Borrego, C.; Duvet, S.; Morsomme, P.; Matthijs, G.; et al. Impact of disease-causing mutations on tmem165 subcellular localization, a recently identified protein involved in cdg-ii. Hum. Mol. Genet 2013, 22, 2914–2928. [Google Scholar]
- Ungar, D. Golgi linked protein glycosylation and associated diseases. Semin. Cell Dev. Biol 2009, 20, 762–769. [Google Scholar]
- Rosnoblet, C.; Peanne, R.; Legrand, D.; Foulquier, F. Glycosylation disorders of membrane trafficking. Glycoconj. J 2013, 30, 23–31. [Google Scholar]
- Miller, V.J.; Ungar, D. Re’cog’nition at the golgi. Traffic 2012, 13, 891–897. [Google Scholar]
- Freeze, H.H.; Ng, B.G. Golgi glycosylation and human inherited diseases. Cold Spring Harb. Perspect. Biol 2011, 3, a005371. [Google Scholar]
- Percival, J.M.; Froehner, S.C. Golgi complex organization in skeletal muscle: A role for golgi-mediated glycosylation in muscular dystrophies? Traffic 2007, 8, 184–194. [Google Scholar]
- Taupenot, L.; Harper, K.L.; O’Connor, D.T. Role of h+-atpase-mediated acidification in sorting and release of the regulated secretory protein chromogranin a: Evidence for a vesiculogenic function. J. Biol. Chem 2005, 280, 3885–3897. [Google Scholar]
- Condon, K.H.; Ho, J.; Robinson, C.G.; Hanus, C.; Ehlers, M.D. The angelman syndrome protein ube3a/e6ap is required for golgi acidification and surface protein sialylation. J. Neurosci 2013, 33, 3799–3814. [Google Scholar]
- Kornak, U.; Reynders, E.; Dimopoulou, A.; van Reeuwijk, J.; Fischer, B.; Rajab, A.; Budde, B.; Nurnberg, P.; Foulquier, F.; Lefeber, D.; et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular h+-atpase subunit atp6v0a2. Nat. Genet 2008, 40, 32–34. [Google Scholar]
- Fischer, B.; Dimopoulou, A.; Egerer, J.; Gardeitchik, T.; Kidd, A.; Jost, D.; Kayserili, H.; Alanay, Y.; Tantcheva-Poor, I.; Mangold, E.; et al. Further characterization of atp6v0a2-related autosomal recessive cutis laxa. Hum. Genet 2012, 131, 1761–1773. [Google Scholar]
- Hucthagowder, V.; Morava, E.; Kornak, U.; Lefeber, D.J.; Fischer, B.; Dimopoulou, A.; Aldinger, A.; Choi, J.; Davis, E.C.; Abuelo, D.N.; et al. Loss-of-function mutations in atp6v0a2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet 2009, 18, 2149–2165. [Google Scholar]
- Coman, D.; Irving, M.; Kannu, P.; Jaeken, J.; Savarirayan, R. The skeletal manifestations of the congenital disorders of glycosylation. Clin. Genet 2008, 73, 507–515. [Google Scholar]
- Percival, J.M.; Gregorevic, P.; Odom, G.L.; Banks, G.B.; Chamberlain, J.S.; Froehner, S.C. Raav6-microdystrophin rescues aberrant golgi complex organization in mdx skeletal muscles. Traffic 2007, 8, 1424–1439. [Google Scholar]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet 2013, 14, 373–378. [Google Scholar]
- Starr, T.; Sun, Y.; Wilkins, N.; Storrie, B. Rab33b and rab6 are functionally overlapping regulators of golgi homeostasis and trafficking. Traffic 2010, 11, 626–636. [Google Scholar]
- Valsdottir, R.; Hashimoto, H.; Ashman, K.; Koda, T.; Storrie, B.; Nilsson, T. Identification of rabaptin-5, rabex-5, and gm130 as putative effectors of rab33b, a regulator of retrograde traffic between the golgi apparatus and er. FEBS Lett 2001, 508, 201–209. [Google Scholar]
- Itoh, T.; Fujita, N.; Kanno, E.; Yamamoto, A.; Yoshimori, T.; Fukuda, M. Golgi-resident small gtpase rab33b interacts with atg16l and modulates autophagosome formation. Molecular Biol. Cell 2008, 19, 2916–2925. [Google Scholar]
- Fukuda, M.; Itoh, T. Direct link between atg protein and small gtpase rab: Atg16l functions as a potential rab33 effector in mammals. Autophagy 2008, 4, 824–826. [Google Scholar]
- Alshammari, M.J.; Al-Otaibi, L.; Alkuraya, F.S. Mutation in rab33b, which encodes a regulator of retrograde golgi transport, defines a second dyggve-melchior-clausen locus. J. Med. Genet 2012, 49, 455–461. [Google Scholar]
- Paupe, V.; Gilbert, T.; Le Merrer, M.; Munnich, A.; Cormier-Daire, V.; El Ghouzzi, V. Recent advances in dyggve-melchior-clausen syndrome. Mol. Genet. Metab 2004, 83, 51–59. [Google Scholar]
- Cheng, H.; Ma, Y.; Ni, X.; Jiang, M.; Guo, L.; Ying, K.; Xie, Y.; Mao, Y. Isolation and characterization of a human novel rab (rab39b) gene. Cytogenet. Genome Res 2002, 97, 72–75. [Google Scholar]
- Giannandrea, M.; Bianchi, V.; Mignogna, M.L.; Sirri, A.; Carrabino, S.; D’Elia, E.; Vecellio, M.; Russo, S.; Cogliati, F.; Larizza, L.; et al. Mutations in the small gtpase gene rab39b are responsible for x-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am. J. Hum. Genet 2010, 86, 185–195. [Google Scholar]
- Hennies, H.C.; Kornak, U.; Zhang, H.; Egerer, J.; Zhang, X.; Seifert, W.; Kuhnisch, J.; Budde, B.; Natebus, M.; Brancati, F.; et al. Gerodermia osteodysplastica is caused by mutations in scyl1bp1, a rab-6 interacting golgin. Nat. Genet 2008, 40, 1410–1412. [Google Scholar]
- Al-Dosari, M.; Alkuraya, F.S. A novel missense mutation in scyl1bp1 produces geroderma osteodysplastica phenotype indistinguishable from that caused by nullimorphic mutations. Am. J. Med. Genet. Part A 2009, 149A, 2093–2098. [Google Scholar]
- Corbett, M.A.; Schwake, M.; Bahlo, M.; Dibbens, L.M.; Lin, M.; Gandolfo, L.C.; Vears, D.F.; O’Sullivan, J.D.; Robertson, T.; Bayly, M.A.; et al. A mutation in the golgi qb-snare gene gosr2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet 2011, 88, 657–663. [Google Scholar]
- Boisse Lomax, L.; Bayly, M.A.; Hjalgrim, H.; Moller, R.S.; Vlaar, A.M.; Aaberg, K.M.; Marquardt, I.; Gandolfo, L.C.; Willemsen, M.; Kamsteeg, E.J.; et al. ‘North sea’ progressive myoclonus epilepsy: Phenotype of subjects with gosr2 mutation. Brain 2013, 136, 1146–1154. [Google Scholar]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 2013, 12, 105–118. [Google Scholar]
- Millott, R.; Dudek, E.; Michalak, M. The endoplasmic reticulum in cardiovascular health and disease. Can. J. Physiol. Pharmacol 2012, 90, 1209–1217. [Google Scholar]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol 2012, 199, 723–734. [Google Scholar]
- Tumer, Z. An overview and update of atp7a mutations leading to menkes disease and occipital horn syndrome. Hum. Mutat 2013, 34, 417–429. [Google Scholar]
- Huster, D.; Hoppert, M.; Lutsenko, S.; Zinke, J.; Lehmann, C.; Mossner, J.; Berr, F.; Caca, K. Defective cellular localization of mutant atp7b in wilson’s disease patients and hepatoma cell lines. Gastroenterology 2003, 124, 335–345. [Google Scholar]
- Egorov, M.V.; Capestrano, M.; Vorontsova, O.A.; Di Pentima, A.; Egorova, A.V.; Mariggio, S.; Ayala, M.I.; Tete, S.; Gorski, J.L.; Luini, A.; et al. Faciogenital dysplasia protein (fgd1) regulates export of cargo proteins from the golgi complex via cdc42 activation. Mol. Biol. Cell 2009, 20, 2413–2427. [Google Scholar]


{kind=link}
| Affected gene | Disease | Primary clinical manifestation | Comments/Cellular effect/Reference |
|---|---|---|---|
| ABCB6 | Dyschromatosis universalis hereditaria | Skin disorder | Mutation leads to retention of the protein in the Golgi [8] |
| ATP6V0A2 | Cutis laxa | Connective tissue disorder | Mutations lead to abnormal glycosylation of serum proteins (CDG-II) and impairment of Golgi trafficking [31–33] |
| ATP7A | Menkes disease, occipital horn syndrome | Neurodegeneration and connective tissue disorder | Protein is localized to the TGN and is essential for copper metabolism. A wide variety of reported mutations affect its localization and trafficking pathways through the Golgi [52] |
| ATP7B | Wilson disease | Hepatic and neurological disorders | Protein is localized to the TGN and is essential for copper metabolism. A wide variety of reported mutations affect its localization and trafficking pathways through the Golgi [53] |
| COG1, COG4, COG5, COG6, COG6, COG7, COG8 | Congenital disorders of glycosylation | Multi-system disorders | Typically reduced levels of the COG member occur, leading to defects in glycosylation [26] |
| DMD | Duchenne muscular dystrophy | Muscular disease | Absence of DMD leads to aberrant organization of the Golgi [35] |
| FGD1 | Aarskog-Scott syndrome/faciogenital dysplasia | Skeletal and genital abnormalities | Localized to the TGN with mutants causing a reduction in trafficking from the Golgi [54] |
| GOSR2 | “North Sea” progressive myoclonus epilepsy | Neurological disease | Mutant protein fails to localize to the cis-Golgi [47,48] |
| PLP1 | Pelizaeus-Merzbacher disease | Neurological disease | Mutation in PLP1 leads to depletion of ER Chaperones with a KDEL motif and Golgi fragmentation [6] |
| RAB1, RAB2, RAB8, STX5 | Parkinson’s disease | Neurological disease | Altered expression of the proteins leads to Golgi fragmentation [12] |
| RAB33B | Dyggve-Melchior-Clausen disease | Skeletal dysplasia | Missense mutation leads to decreased protein expression [41] |
| RAB33B | Smith-McCort Dysplasia | Skeletal dysplasia | Missense mutation leads to lower protein expression and swollen and fragmented Golgi in many cells [14] |
| RAB39B | X-linked mental retardation associated with autism, epilepsy and macrocephaly | Neurological disease | Loss of function mutations in RAB39B lead to altered number and morphology of neurite growth cones and reduction of presynaptic buttons [44] |
| RIN2 | Macrocephaly, alopecia, cutis laxa and scoliosis (MACS) syndrome | Connective tissue disorder | Loss of function mutation leads to presence of vacuoles in the Golgi [17] |
| RIN2 | RIN2 syndrome | Connective tissue disorder | Loss of function mutation leads to dilation of ER, and rarified and dilated Golgi cisternae [18] |
| SCYL1BP1/GORAB | Gerodermia osteodysplastica | Connective tissue disorder | Loss of function mutation [45,46] |
| SMN | Proximal spinal muscular atrophy | Neurological disease | Decreased expression of SMN leads to accumulation of SMN granules in the trans-Golgi network and a global blockage of granule secretion [7] |
| TMEM165 | Congenital disorder of glycosylation type II (CGD-II) | Psychomotor retardation and bone dysplasia | Mutation leads to lower protein expression and altered subcellular localization with overall Golgi swelling and fragmented trans-Golgi network. Decreased protein glycosylation is also observed [21–23] |
| UBE3A | Angelman syndrome | Neurodevelopmental Disorder | Loss of protein expression leads to an altered Golgi morphology and pH, which is associated with a reduction in protein sialylation [30] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bexiga, M.G.; Simpson, J.C. Human Diseases Associated with Form and Function of the Golgi Complex. Int. J. Mol. Sci. 2013, 14, 18670-18681. https://doi.org/10.3390/ijms140918670
Bexiga MG, Simpson JC. Human Diseases Associated with Form and Function of the Golgi Complex. International Journal of Molecular Sciences. 2013; 14(9):18670-18681. https://doi.org/10.3390/ijms140918670
Chicago/Turabian StyleBexiga, Mariana G., and Jeremy C. Simpson. 2013. "Human Diseases Associated with Form and Function of the Golgi Complex" International Journal of Molecular Sciences 14, no. 9: 18670-18681. https://doi.org/10.3390/ijms140918670
APA StyleBexiga, M. G., & Simpson, J. C. (2013). Human Diseases Associated with Form and Function of the Golgi Complex. International Journal of Molecular Sciences, 14(9), 18670-18681. https://doi.org/10.3390/ijms140918670