Detecting and Comparing Non-Coding RNAs in the High-Throughput Era

Abstract
:1. Introduction
1.1. The Non-Coding RNA (New)-World
1.2. lncRNA Challenges
2. Comparing Non-Coding RNAs
2.1. RNA Structure Prediction
2.2. Structure Prediction and Alignment Strategies
3. Detecting ncRNA Homologues
4. High-Throughput Technologies and Genome-Wide Annotation of ncRNAs
4.1. Approaches for the High-Throughput Expression Detection
4.2. Datasets
4.3. NGS Challenges
4.4. Other Approaches
5. Discussion and Conclusions
Acknowledgments
Conflict of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafreniere, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar]
- Farazi, T.A.; Juranek, S.A.; Tuschl, T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development 2008, 135, 1201–1214. [Google Scholar]
- Bachellerie, J.P.; Cavaille, J.; Huttenhofer, A. The expanding snoRNA world. Biochimie 2002, 84, 775–790. [Google Scholar]
- Barrett, T.; Suzek, T.O.; Troup, D.B.; Wilhite, S.E.; Ngau, W.C.; Ledoux, P.; Rudnev, D.; Lash, A.E.; Fujibuchi, W.; Edgar, R. NCBI GEO: Mining millions of expression profiles— database and tools. Nucleic Acids Res 2005, 33, D562–D566. [Google Scholar]
- Parkinson, H.; Sarkans, U.; Shojatalab, M.; Abeygunawardena, N.; Contrino, S.; Coulson, R.; Farne, A.; Lara, G.G.; Holloway, E.; et al. ArrayExpress—A public repository for microarray gene expression data at the EBI. Nucleic Acids Res 2005, 33, D553–D555. [Google Scholar]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006, 34, D140–D144. [Google Scholar]
- Fraser, B.A.; Weadick, C.J.; Janowitz, I.; Rodd, F.H.; Hughes, K.A. Sequencing and characterization of the guppy (Poecilia reticulata) transcriptome. BMC Genomics 2011, 12, 202. [Google Scholar]
- Tuda, J.; Mongan, A.E.; Tolba, M.E.; Imada, M.; Yamagishi, J.; Xuan, X.; Wakaguri, H.; Sugano, S.; Sugimoto, C.; Suzuki, Y. Full-parasites: database of full-length cDNAs of apicomplexa parasites, 2010 update. Nucleic Acids Res 2011, 39, D625–D631. [Google Scholar]
- Mamidala, P.; Wijeratne, A.J.; Wijeratne, S.; Kornacker, K.; Sudhamalla, B.; Rivera-Vega, L.J.; Hoelmer, A.; Meulia, T.; Jones, S.C.; Mittapalli, O. RNA-Seq and molecular docking reveal multi-level pesticide resistance in the bed bug. BMC Genomics 2012, 13, 6. [Google Scholar]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Askarian-Amiri, M.E.; Ru, K.; Solda, G.; Simons, C.; et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res 2008, 18, 1433–1445. [Google Scholar]
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S.; Nikaido, I.; Osato, N.; Saito, R.; Suzuki, H.; et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res 2012, 22, 1760–1774. [Google Scholar]
- Managadze, D.; Lobkovsky, A.E.; Wolf, Y.I.; Shabalina, S.A.; Rogozin, I.B.; Koonin, E.V. The vast, conserved mammalian lincRNome. PLoS Comput. Biol 2013, 9, e1002917. [Google Scholar]
- Mattick, J.S.; Gagen, M.J. The evolution of controlled multitasked gene networks: The role of introns and other noncoding RNAs in the development of complex organisms. Mol. Biol. Evol 2001, 18, 1611–1630. [Google Scholar]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep 2001, 2, 986–991. [Google Scholar]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol 1958, 12, 138–163. [Google Scholar]
- Wang, J.; Zhang, J.; Zheng, H.; Li, J.; Liu, D.; Li, H.; Samudrala, R.; Yu, J.; Wong, G.K. Mouse transcriptome: Neutral evolution of “non-coding” complementary DNAs. Nature 2004, 431. [Google Scholar] [CrossRef]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Crowe, M.L.; Grimmond, S.M.; Mattick, J.S. NRED: A database of long noncoding RNA expression. Nucleic Acids Res 2009, 37, D122–D126. [Google Scholar]
- Mattick, J.S. The genetic signatures of noncoding RNAs. PLoS Genet 2009, 5, e1000459. [Google Scholar]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell. Biol 2011, 21, 354–361. [Google Scholar]
- Wang, X.; Song, X.; Glass, C.K.; Rosenfeld, M.G. The long arm of long noncoding RNAs: roles as sensors regulating gene transcriptional programs. Cold Spring Harb. Perspect. Biol 2011, 3, a003756. [Google Scholar]
- Satterlee, J.S.; Barbee, S.; Jin, P.; Krichevsky, A.; Salama, S.; Schratt, G.; Wu, D.Y. Noncoding RNAs in the brain. J. Neurosci 2007, 27, 11856–11859. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res 2011, 90, 430–440. [Google Scholar]
- Braidotti, G.; Baubec, T.; Pauler, F.; Seidl, C.; Smrzka, O.; Stricker, S.; Yotova, I.; Barlow, D.P. The Air noncoding RNA: An imprinted cis-silencing transcript. Cold Spring Harb. Symp. Quant. Biol 2004, 69, 55–66. [Google Scholar]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497–501. [Google Scholar]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem 2012, 81, 145–166. [Google Scholar]
- Amaral, P.P.; Clark, M.B.; Gascoigne, D.K.; Dinger, M.E.; Mattick, J.S. lncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res 2011, 39, D146–D151. [Google Scholar]
- Ravasi, T.; Suzuki, H.; Pang, K.C.; Katayama, S.; Furuno, M.; Okunishi, R.; Fukuda, S.; Ru, K.; Frith, M.C.; Gongora, M.M.; et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res 2006, 16, 11–19. [Google Scholar]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res 2004, 14, 1902–1910. [Google Scholar]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar]
- Ogawa, Y.; Sun, B.K.; Lee, J.T. Intersection of the RNA interference and X-inactivation pathways. Science 2008, 320, 1336–1341. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22, 1775–1789. [Google Scholar]
- Chen, Z.; Duan, X. Ribosomal RNA depletion for massively parallel bacterial RNA-sequencing applications. Methods Mol. Biol 2011, 733, 93–103. [Google Scholar]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab. initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol 2010, 28, 503–510. [Google Scholar]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011, 25, 1915–1927. [Google Scholar]
- Kutter, C.; Watt, S.; Stefflova, K.; Wilson, M.D.; Goncalves, A.; Ponting, C.P.; Odom, D.T.; Marques, A.C. Rapid turnover of long noncoding rnas and the evolution of gene expression. PLoS Genet 2012, 8, e1002841. [Google Scholar]
- ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816.
- Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.V.; Morillon, A.; et al. The reality of pervasive transcription. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [Green Version]
- Capel, B.; Swain, A.; Nicolis, S.; Hacker, A.; Walter, M.; Koopman, P.; Goodfellow, P.; Lovell-Badge, R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar]
- Cocquerelle, C.; Mascrez, B.; Hétuin, D.; Bailleul, B. Mis-splicing yields circular RNA molecules. FASEB J 1993, 7, 155–160. [Google Scholar]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar]
- Zaphiropoulos, P.G. Exon skipping and circular RNA formation in transcripts of the human cytochrome P-450 2C18 gene in epidermis and of the rat androgen binding protein gene in testis. Mol. Cell. Biol 1997, 17, 2985–2993. [Google Scholar]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar]
- Capriotti, E.; Marti-Renom, M.A. Quantifying the relationship between sequence and three-dimensional structure conservation in RNA. BMC Bioinforma 2010, 11, 322. [Google Scholar]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng 1999, 12, 85–94. [Google Scholar]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 2006, 22, 1–5. [Google Scholar]
- Bernhart, S.H.; Hofacker, I.L. From consensus structure prediction to RNA gene finding. Brief Funct. Genomic Proteomic 2009, 8, 461–471. [Google Scholar]
- Sun, Y.; Aljawad, O.; Lei, J.; Liu, A. Genome-scale NCRNA homology search using a Hamming distance-based filtration strategy. BMC Bioinforma 2012, 13, S12. [Google Scholar]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet 2005, 37, 766–770. [Google Scholar]
- Berezikov, E.; van Tetering, G.; Verheul, M.; van de Belt, J.; van Laake, L.; Vos, J.; Verloop, R.; van de Wetering, M.; Guryev, V.; Takada, S.; et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 2006, 16, 1289–1298. [Google Scholar]
- Guerra-Assuncao, J.A.; Enright, A.J. Large-scale analysis of microRNA evolution. BMC Genomics 2012, 13, 218. [Google Scholar]
- Missal, K.; Rose, D.; Stadler, P.F. Non-coding RNAs in Ciona intestinalis. Bioinformatics 2005, 21, ii77–ii78. [Google Scholar]
- Lindgreen, S.; Gardner, P.P.; Krogh, A. MASTR: Multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics 2007, 23, 3304–3311. [Google Scholar]
- Sperschneider, J.; Datta, A.; Wise, M.J. Predicting pseudoknotted structures across two RNA sequences. Bioinformatics 2012, 28, 3058–3065. [Google Scholar]
- Wong, T.K.F.; Wan, K.-L.; Hsu, B.-Y.; Cheung, B.W.Y.; Hon, W.-K.; Lam, T.-W.; Yiu, S.-M. RNASAlign: RNA structural alignment system. Bioinformatics 2011, 27, 2151–2152. [Google Scholar]
- Gardner, P.P.; Giegerich, R. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinforma 2004, 5, 140. [Google Scholar]
- Ravindran, P.P.; Heroux, A.; Ye, J.D. Improvement of the crystallizability and expression of an RNA crystallization chaperone. J. Biochem 2011, 150, 535–543. [Google Scholar]
- Furtig, B.; Richter, C.; Wohnert, J.; Schwalbe, H. NMR spectroscopy of RNA. Chembiochem 2003, 4, 936–962. [Google Scholar]
- Tzakos, A.G.; Grace, C.R.; Lukavsky, P.J.; Riek, R. NMR techniques for very large proteins and rnas in solution. Annu. Rev. Biophys. Biomol. Struct 2006, 35, 319–342. [Google Scholar]
- Zuker, M.; Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981, 9, 133–148. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31, 3406–3415. [Google Scholar]
- Mathews, D.H.; Turner, D.H.; Zuker, M. RNA secondary structure prediction. Curr. Protoc. Nucleic Acid Chem. 2007. [Google Scholar] [CrossRef]
- Dowell, R.D.; Eddy, S.R. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinforma 2004, 5, 71. [Google Scholar]
- Dima, R.I.; Hyeon, C.; Thirumalai, D. Extracting stacking interaction parameters for RNA from the data set of native structures. J. Mol. Biol 2005, 347, 53–69. [Google Scholar]
- Do, C.B.; Woods, D.A.; Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006, 22, e90–e98. [Google Scholar]
- Xia, T.; SantaLucia, J.J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol 1999, 288, 911–940. [Google Scholar]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar]
- Lu, Z.J.; Turner, D.H.; Mathews, D.H. A set of nearest neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res 2006, 34, 4912–4924. [Google Scholar]
- Lu, Z.J.; Gloor, J.W.; Mathews, D.H. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 2009, 15, 1805–1813. [Google Scholar]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, S.; Tacker, M.; Schuster, P. Fast folding and comparison of rna secondary structures. Monatshefte f. Chem. 1994, 125, 167–188. [Google Scholar]
- McCaskill, J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 1990, 29, 1105–1119. [Google Scholar]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis. Probabilistic Models of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK; p. 1998.
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. USA 2009, 106, 97–102. [Google Scholar]
- Doshi, K.J.; Cannone, J.J.; Cobaugh, C.W.; Gutell, R.R. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinforma 2004, 5, 105. [Google Scholar]
- Herschlag, D. RNA chaperones and the RNA folding problem. J. Biol. Chem 1995, 270, 20871–20874. [Google Scholar]
- Brennicke, A.; Marchfelder, A.; Binder, S. RNA editing. FEMS Microbiol. Rev 1999, 23, 297–316. [Google Scholar]
- Pan, T.; Sosnick, T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct 2006, 35, 161–175. [Google Scholar]
- Mandal, M.; Breaker, R.R. Gene regulation by riboswitches. Nat. Rev. Mol. Cell. Biol 2004, 5, 451–463. [Google Scholar]
- Soukup, J.K.; Soukup, G.A. Riboswitches exert genetic control through metabolite-induced conformational change. Curr. Opin. Struct. Biol 2004, 14, 344–349. [Google Scholar]
- Bengert, P.; Dandekar, T. Riboswitch finder—A tool for identification of riboswitch RNAs. Nucleic Acids Res 2004, 32, W154–W159. [Google Scholar]
- Voss, B.; Meyer, C.; Giegerich, R. Evaluating the predictability of conformational switching in RNA. Bioinformatics 2004, 20, 1573–1582. [Google Scholar]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res 2003, 31, 3429–3431. [Google Scholar]
- Sankoff, D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math 1985, 45, 810–825. [Google Scholar]
- Torarinsson, E.; Lindgreen, S. WAR: Webserver for aligning structural RNAs. Nucleic Acids Res 2008, 36, W79–W84. [Google Scholar]
- Bremges, A.; Schirmer, S.; Giegerich, R. Fine-tuning structural RNA alignments in the twilight zone. BMC Bioinforma 2010, 11, 222. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol 2000, 302, 205–217. [Google Scholar]
- Hofacker, I.L.; Fekete, M.; Stadler, P.F. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol 2002, 319, 1059–1066. [Google Scholar]
- Knudsen, B.; Hein, J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res 2003, 31, 3423–3428. [Google Scholar]
- Ruan, J.; Stormo, G.D.; Zhang, W. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics 2004, 20, 58–66. [Google Scholar]
- Luck, R.; Graf, S.; Steger, G. ConStruct: A tool for thermodynamic controlled prediction of conserved secondary structure. Nucleic Acids Res 1999, 27, 4208–4217. [Google Scholar]
- Mathews, D.H.; Turner, D.H. Dynalign: An algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol 2002, 317, 191–203. [Google Scholar]
- Mathews, D.H. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics 2005, 21, 2246–2253. [Google Scholar]
- Gorodkin, J.; Heyer, L.J.; Stormo, G.D. Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res 1997, 25, 3724–3732. [Google Scholar]
- Havgaard, J.H.; Lyngso, R.B.; Stormo, G.D.; Gorodkin, J. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics 2005, 21, 1815–1824. [Google Scholar]
- Holmes, I. Accelerated probabilistic inference of RNA structure evolution. BMC Bioinforma 2005, 6, 73. [Google Scholar]
- Dowell, R.D.; Eddy, S.R. Efficient pairwise RNA structure prediction and alignment using sequence alignment constraints. BMC Bioinforma 2006, 7, 400. [Google Scholar]
- Hofacker, I.L.; Bernhart, S.H.; Stadler, P.F. Alignment of RNA base pairing probability matrices. Bioinformatics 2004, 20, 2222–2227. [Google Scholar]
- Wilm, A.; Higgins, D.G.; Notredame, C. R-Coffee: A method for multiple alignment of non-coding RNA. Nucleic Acids Res 2008, 36, e52. [Google Scholar]
- Reeder, J.; Giegerich, R. Consensus shapes: An alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics 2005, 21, 3516–3523. [Google Scholar]
- Bernhart, S.H.; Hofacker, I.L.; Stadler, P.F. Local RNA base pairing probabilities in large sequences. Bioinformatics 2006, 22, 614–615. [Google Scholar]
- Capriotti, E.; Marti-Renom, M.A. RNA structure alignment by a unit-vector approach. Bioinformatics 2008, 24, i112–i118. [Google Scholar]
- Ferre, F.; Ponty, Y.; Lorenz, W.A.; Clote, P. DIAL: A web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res 2007, 35, W659–W668. [Google Scholar]
- Wang, C.W.; Chen, K.T.; Lu, C.L. iPARTS: An improved tool of pairwise alignment of RNA tertiary structures. Nucleic Acids Res 2010, 38, W340–W347. [Google Scholar]
- Dror, O.; Nussinov, R.; Wolfson, H. ARTS: Alignment of RNA tertiary structures. Bioinformatics 2005, 21, ii47–ii53. [Google Scholar]
- Chang, Y.F.; Huang, Y.L.; Lu, C.L. SARSA: A web tool for structural alignment of RNA using a structural alphabet. Nucleic Acids Res 2008, 36, W19–W24. [Google Scholar]
- Bauer, R.P.; Rother, K.P.; Moor, P.P.; Reinert, K.P.; Steinke, T.P.; Bujnicki, J.P.; Preissner, R.P. Fast structural alignment of biomolecules using a hash table, N-grams and string descriptors. Algorithms 2009, 2, 692–709. [Google Scholar]
- Kirillova, S.; Tosatto, S.C.; Carugo, O. FRASS: The web-server for RNA structural comparison. BMC Bioinforma 2010, 11, 327. [Google Scholar]
- Freyhult, E.K.; Bollback, J.P.; Gardner, P.P. Exploring genomic dark matter: A critical assessment of the performance of homology search methods on noncoding RNA. Genome Res 2007, 17, 117–125. [Google Scholar]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol 1981, 147, 195–197. [Google Scholar]
- Rognes, T. Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation. BMC Bioinforma 2011, 12, 221. [Google Scholar]
- Lipman, D.J.; Pearson, W.R. Rapid and sensitive protein similarity searches. Science 1985, 227, 1435–1441. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar]
- Weinberg, Z.; Ruzzo, W.L. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics 2006, 22, 35–39. [Google Scholar]
- Holmes, I. A probabilistic model for the evolution of RNA structure. BMC Bioinforma 2004, 5, 166. [Google Scholar]
- Notredame, C.; O’Brien, E.A.; Higgins, D.G. RAGA: RNA sequence alignment by genetic algorithm. Nucleic Acids Res 1997, 25, 4570–4580. [Google Scholar]
- Eddy, S.R.; Durbin, R. RNA sequence analysis using covariance models. Nucleic Acids Res 1994, 22, 2079–2088. [Google Scholar]
- Eddy, S.R. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinforma 2002, 3, 18. [Google Scholar]
- Klein, R.J.; Eddy, S.R. RSEARCH: Finding homologs of single structured RNA sequences. BMC Bioinforma 2003, 4, 44. [Google Scholar]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res 2003, 31, 439–441. [Google Scholar]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res 2008, 36, D281–D288. [Google Scholar]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005, 33, D121–D124. [Google Scholar]
- Zhang, S.; Borovok, I.; Aharonowitz, Y.; Sharan, R.; Bafna, V. A sequence-based filtering method for ncRNA identification and its application to searching for riboswitch elements. Bioinformatics 2006, 22, e557–e565. [Google Scholar]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res 2009, 37, D136–D140. [Google Scholar]
- Roshan, U.; Chikkagoudar, S.; Livesay, D.R. Searching for evolutionary distant RNA homologs within genomic sequences using partition function posterior probabilities. BMC Bioinforma 2008, 9, 61. [Google Scholar]
- Bussotti, G.; Raineri, E.; Erb, I.; Zytnicki, M.; Wilm, A.; Beaudoing, E.; Bucher, P.; Notredame, C. BlastR—Fast and accurate database searches for non-coding RNAs. Nucleic Acids Res 2011, 39, 6886–6895. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar]
- Aparicio, S.; Chapman, J.; Stupka, E.; Putnam, N.; Chia, J.M.; Dehal, P.; Christoffels, A.; Rash, S.; Hoon, S.; Smit, A.; et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002, 297, 1301–1310. [Google Scholar]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar]
- Schwartz, S.; Kent, W.J.; Smit, A.; Zhang, Z.; Baertsch, R.; Hardison, R.C.; Haussler, D.; Miller, W. Human-mouse alignments with BLASTZ. Genome Res 2003, 13, 103–107. [Google Scholar]
- Boguski, M.S.; Tolstoshev, C.M.; Bassett, D.E.J. Gene discovery in dbEST. Science 1994, 265, 1993–1994. [Google Scholar]
- Dias Neto, E.; Correa, R.G.; Verjovski-Almeida, S.; Briones, M.R.; Nagai, M.A.; da Silva, W.J.; Zago, M.A.; Bordin, S.; Costa, F.F.; Goldman, G.H.; et al. Shotgun sequencing of the human transcriptome with ORF expressed sequence tags. Proc. Natl. Acad. Sci. USA 2000, 97, 3491–3496. [Google Scholar]
- Gerhard, D.S.; Wagner, L.; Feingold, E.A.; Shenmen, C.M.; Grouse, L.H.; Schuler, G.; Klein, S.L.; Old, S.; Rasooly, R.; Good, P.; et al. The status, quality, and expansion of the NIH full-length cDNA project: The Mammalian Gene Collection (MGC). Genome Res 2004, 14, 2121–2127. [Google Scholar]
- Boguski, M.S.; Lowe, T.M.; Tolstoshev, C.M. dbEST—Database for “expressed sequence tags.”. Nat Genet 1993, 4, 332–333. [Google Scholar]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F.; et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656. [Google Scholar]
- Bonaldo, M.F.; Lennon, G.; Soares, M.B. Normalization and subtraction: Two approaches to facilitate gene discovery. Genome Res 1996, 6, 791–806. [Google Scholar]
- Nagaraj, S.H.; Gasser, R.B.; Ranganathan, S. A hitchhiker’s guide to expressed sequence tag (EST) analysis. Brief. Bioinforma 2007, 8, 6–21. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Malone, J.H.; Oliver, B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol 2011, 9, 34. [Google Scholar]
- Eklund, A.C.; Turner, L.R.; Chen, P.; Jensen, R.V.; deFeo, G.; Kopf-Sill, A.R.; Szallasi, Z. Replacing cRNA targets with cDNA reduces microarray cross-hybridization. Nat. Biotechnol 2006, 24, 1071–1073. [Google Scholar]
- Okoniewski, M.J.; Miller, C.J. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinforma 2006, 7, 276. [Google Scholar]
- Casneuf, T.; van de Peer, Y.; Huber, W. In situ analysis of cross-hybridisation on microarrays and the inference of expression correlation. BMC Bioinforma 2007, 8, 461. [Google Scholar]
- Cox, W.G.; Beaudet, M.P.; Agnew, J.Y.; Ruth, J.L. Possible sources of dye-related signal correlation bias in two-color DNA microarray assays. Anal. Biochem 2004, 331, 243–254. [Google Scholar]
- Dombkowski, A.A.; Thibodeau, B.J.; Starcevic, S.L.; Novak, R.F. Gene-specific dye bias in microarray reference designs. FEBS Lett 2004, 560, 120–124. [Google Scholar]
- Rosenzweig, B.A.; Pine, P.S.; Domon, O.E.; Morris, S.M.; Chen, J.J.; Sistare, F.D. Dye bias correction in dual-labeled cDNA microarray gene expression measurements. Environ. Health Perspect 2004, 112, 480–487. [Google Scholar]
- Dobbin, K.K.; Kawasaki, E.S.; Petersen, D.W.; Simon, R.M. Characterizing dye bias in microarray experiments. Bioinformatics 2005, 21, 2430–2437. [Google Scholar]
- Martin-Magniette, M.L.; Aubert, J.; Cabannes, E.; Daudin, J.J. Evaluation of the gene-specific dye bias in cDNA microarray experiments. Bioinformatics 2005, 21, 1995–2000. [Google Scholar]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar]
- Royce, T.E.; Rozowsky, J.S.; Bertone, P.; Samanta, M.; Stolc, V.; Weissman, S.; Snyder, M.; Gerstein, M. Issues in the analysis of oligonucleotide tiling microarrays for transcript mapping. Trends Genet 2005, 21, 466–475. [Google Scholar]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet 2007, 8, 413–423. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet 2009, 10, 57–63. [Google Scholar]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008, 18, 1509–1517. [Google Scholar]
- Fu, X.; Fu, N.; Guo, S.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Chen, W.; Li, Y.; Zeng, R.; et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 2009, 10, 161. [Google Scholar]
- Ozsolak, F.; Milos, P.M. RNA Sequencing: Advances, challenges and opportunities. Nat. Rev. Genet 2011, 12, 87–98. [Google Scholar]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar]
- Harbers, M.; Carninci, P. Tag-based approaches for transcriptome research and genome annotation. Nat. Methods 2005, 2, 495–502. [Google Scholar]
- Saha, S.; Sparks, A. B.; Rago, C.; Akmaev, V.; Wang, C.J.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Using the transcriptome to annotate the genome. Nat. Biotechnol 2002, 20, 508–512. [Google Scholar]
- Gowda, M.; Jantasuriyarat, C.; Dean, R.A.; Wang, G.L. Robust-LongSAGE (RL-SAGE): A substantially improved LongSAGE method for gene discovery and transcriptome analysis. Plant. Physiol 2004, 134, 890–897. [Google Scholar]
- Matsumura, H.; Ito, A.; Saitoh, H.; Winter, P.; Kahl, G.; Reuter, M.; Kruger, D.H.; Terauchi, R. SuperSAGE. Cell. Microbiol 2005, 7, 11–18. [Google Scholar]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Luo, S.; McCurdy, S.; Foy, M.; Ewan, M.; et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol 2000, 18, 630–634. [Google Scholar]
- Ng, P.; Wei, C.L.; Sung, W.K.; Chiu, K.P.; Lipovich, L.; Ang, C.C.; Gupta, S.; Shahab, A.; Ridwan, A.; Wong, C.H.; et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat. Methods 2005, 2, 105–111. [Google Scholar]
- Schaefer, B.C. Revolutions in rapid amplification of cDNA ends: New strategies for polymerase chain reaction cloning of full-length cDNA ends. Anal. Biochem 1995, 227, 255–273. [Google Scholar]
- Kapranov, P.; Drenkow, J.; Cheng, J.; Long, J.; Helt, G.; Dike, S.; Gingeras, T.R. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res 2005, 15, 987–997. [Google Scholar]
- Olivarius, S.; Plessy, C.; Carninci, P. High-throughput verification of transcriptional starting sites by Deep-RACE. BioTechniques 2009, 46, 130–132. [Google Scholar]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M.; et al. CAGE: Cap analysis of gene expression. Nat. Methods 2006, 3, 211–222. [Google Scholar]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar]
- Mercer, T.R.; Gerhardt, D.J.; Dinger, M.E.; Crawford, J.; Trapnell, C.; Jeddeloh, J.A.; Mattick, J.S.; Rinn, J.L. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol 2011, 30, 99–104. [Google Scholar]
- Mathavan, S.; Lee, S.G.; Mak, A.; Miller, L.D.; Murthy, K.R.; Govindarajan, K.R.; Tong, Y.; Wu, Y.L.; Lam, S.H.; Yang, H.; et al. Transcriptome analysis of zebrafish embryogenesis using microarrays. PLoS Genet 2005, 1, 260–276. [Google Scholar]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar]
- Graveley, B.R.; Brooks, A.N.; Carlson, J.W.; Duff, M.O.; Landolin, J.M.; Yang, L.; Artieri, C.G.; van Baren, M.J.; Boley, N.; Booth, B.W.; et al. The developmental transcriptome of Drosophila melanogaster. Nature 2011, 471, 473–479. [Google Scholar]
- Pang, K.C.; Stephen, S.; Dinger, M.E.; Engström, P.G.; Lenhard, B.; Mattick, J.S. RNAdb 2.0— An expanded database of mammalian non-coding RNAs. Nucleic Acids Res 2007, 35, D178–D182. [Google Scholar]
- Kin, T.; Yamada, K.; Terai, G.; Okida, H.; Yoshinari, Y.; Ono, Y.; Kojima, A.; Kimura, Y.; Komori, T.; Asai, K. fRNAdb: A platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res 2007, 35, D145–D148. [Google Scholar]
- Liu, C.; Bai, B.; Skogerbø, G.; Cai, L.; Deng, W.; Zhang, Y.; Bu, D.; Zhao, Y.; Chen, R. NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res 2005, 33, D112–D125. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res 2009, 37, D32–D36. [Google Scholar]
- Harrow, J.; Denoeud, F.; Frankish, A.; Reymond, A.; Chen, C.K.; Chrast, J.; Lagarde, J.; Gilbert, J.G.; Storey, R.; Swarbreck, D.; et al. GENCODE: Producing a reference annotation for ENCODE. Genome Biol. 2006, 7, S41–S49. [Google Scholar]
- Havana team. Available online: http://www.sanger.ac.uk/research/projects/vertebrategenome/havana/ (accessed 6 March 2013).
- Loveland, J.E.; Gilbert, J.G.; Griffiths, E.; Harrow, J.L. Community gene annotation in practice. Database (Oxford) 2012, 2012. [Google Scholar] [CrossRef]
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucleic Acids Res 2012, 40, D84–D90. [Google Scholar]
- Kawai, J.; Shinagawa, A.; Shibata, K.; Yoshino, M.; Itoh, M.; Ishii, Y.; Arakawa, T.; Hara, A.; Fukunishi, Y.; Konno, H.; et al. Functional annotation of a full-length mouse cDNA collection. Nature 2001, 409, 685–690. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI reference sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res 2012, 40, D130–D135. [Google Scholar]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res 2002, 12, 996–1006. [Google Scholar]
- Stalker, J.; Gibbins, B.; Meidl, P.; Smith, J.; Spooner, W.; Hotz, H.R.; Cox, A.V. The Ensembl web site: Mechanics of a genome browser. Genome Res 2004, 14, 951–955. [Google Scholar]
- Wilming, L.G.; Gilbert, J.G.; Howe, K.; Trevanion, S.; Hubbard, T.; Harrow, J.L. The vertebrate genome annotation (Vega) database. Nucleic Acids Res 2008, 36, D753–D760. [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74.
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet 2011, 12, 671–682. [Google Scholar]
- Rogers, M.F.; Thomas, J.; Reddy, A.S.; Ben-Hur, A. SpliceGrapher: Detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol 2012, 13, R4. [Google Scholar]
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet 2010, 11, 31–46. [Google Scholar]
- Butler, J.; MacCallum, I.; Kleber, M.; Shlyakhter, I.A.; Belmonte, M.K.; Lander, E.S.; Nusbaum, C.; Jaffe, D.B. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res 2008, 18, 810–820. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008, 18, 821–829. [Google Scholar]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res 2009, 19, 1117–1123. [Google Scholar]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar]
- Lin, H.; Zhang, Z.; Zhang, M.Q.; Ma, B.; Li, M. ZOOM! Zillions of oligos mapped. Bioinformatics 2008, 24, 2431–2437. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009, 10, R25. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar]
- Schatz, M.C. CloudBurst: Highly sensitive read mapping with MapReduce. Bioinformatics 2009, 25, 1363–1369. [Google Scholar]
- Ahmadi, A.; Behm, A.; Honnalli, N.; Li, C.; Weng, L.; Xie, X. Hobbes: Optimized gram-based methods for efficient read alignment. Nucleic Acids Res 2012, 40, e41. [Google Scholar]
- Derrien, T.; Estelle, J.; Marco Sola, S.; Knowles, D.G.; Raineri, E.; Guigo, R.; Ribeca, P. Fast computation and applications of genome mappability. PLoS One 2012, 7, e30377. [Google Scholar]
- Cloonan, N.; Xu, Q.; Faulkner, G.J.; Taylor, D.F.; Tang, D.T.; Kolle, G.; Grimmond, S.M. RNA-MATE: A recursive mapping strategy for high-throughput RNA-sequencing data. Bioinformatics 2009, 25, 2615–2616. [Google Scholar]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008, 18, 1851–1858. [Google Scholar]
- Smith, A.D.; Xuan, Z.; Zhang, M.Q. Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinforma 2008, 9, 128. [Google Scholar]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998, 8, 186–194. [Google Scholar]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998, 8, 175–185. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 2010, 28, 511–515. [Google Scholar]
- Li, W.; Feng, J.; Jiang, T. IsoLasso: A LASSO regression approach to RNA-Seq based transcriptome assembly. J. Comput. Biol 2011, 18, 1693–1707. [Google Scholar]
- Palmieri, N.; Nolte, V.; Suvorov, A.; Kosiol, C.; Schlötterer, C. Evaluation of different reference based annotation strategies using RNA-Seq—A case study in Drososphila pseudoobscura. PLoS One 2012. [Google Scholar] [CrossRef]
- Garg, R.; Patel, R.K.; Tyagi, A.K.; Jain, M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 2011, 18, 53–63. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 2011, 29, 644–652. [Google Scholar]
- Jager, M.; Ott, C.E.; Grunhagen, J.; Hecht, J.; Schell, H.; Mundlos, S.; Duda, G.N.; Robinson, P.N.; Lienau, J. Composite transcriptome assembly of RNA-seq data in a sheep model for delayed bone healing. BMC Genomics 2011, 12, 158. [Google Scholar]
- Keiler, K.C. Biology of trans-translation. Annu. Rev. Microbiol 2008, 62, 133–151. [Google Scholar]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res 2012, 40, 5034–5051. [Google Scholar]
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433. [Google Scholar]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar]
- Lin, M.F.; Carlson, J.W.; Crosby, M.A.; Matthews, B.B.; Yu, C.; Park, S.; Wan, K.H.; Schroeder, A.J.; Gramates, L.S.; St Pierre, S.E.; et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Res 2007, 17, 1823–1836. [Google Scholar]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res 2011, 39, 3864–3878. [Google Scholar]
- Marchler-Bauer, A.; Panchenko, A.R.; Shoemaker, B.A.; Thiessen, P.A.; Geer, L.Y.; Bryant, S.H. CDD: A database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res 2002, 30, 281–283. [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40, D71–D75.
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 2007, 35, W345–W349. [Google Scholar]
- Arrial, R.T.; Togawa, R.C.; Brigido, M.M. Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: Case study of the pathogenic fungus Paracoccidioides brasiliensis. BMC Bioinforma 2009, 10, 239. [Google Scholar]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res 2013, 41, e74. [Google Scholar]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar]
- Lu, Z.J.; Yip, K.Y.; Wang, G.; Shou, C.; Hillier, L.W.; Khurana, E.; Agarwal, A.; Auerbach, R.; Rozowsky, J.; Cheng, C.; et al. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res 2011, 21, 276–285. [Google Scholar]
- Esteve-Codina, A.; Kofler, R.; Palmieri, N.; Bussotti, G.; Notredame, C.; Perez-Enciso, M. Exploring the gonad transcriptome of two extreme male pigs with RNA-seq. BMC Genomics 2011, 12, 552. [Google Scholar]
- Nam, J.W.; Bartel, D. Long non-coding RNAs in C. elegans. Genome Res 2012, 22, 2529–2540. [Google Scholar]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar]
- Eveland, A.L.; McCarty, D.R.; Koch, K.E. Transcript profiling by 3′-untranslated region sequencing resolves expression of gene families. Plant Physiol 2008, 146, 32–44. [Google Scholar]
- Hillier, L.W.; Reinke, V.; Green, P.; Hirst, M.; Marra, M.A.; Waterston, R.H. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 2009, 19, 657–666. [Google Scholar]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar]
- Shendure, J. The beginning of the end for microarrays? Nat. Methods 2008, 5, 585–587. [Google Scholar]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet 2009, 10, 135–151. [Google Scholar]
- Auer, P.L.; Doerge, R.W. Statistical design and analysis of RNA sequencing data. Genetics 2010, 185, 405–416. [Google Scholar]
- Hiller, M.; Pudimat, R.; Busch, A.; Backofen, R. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res 2006, 34, e117. [Google Scholar]
- Chang, T.-H.; Huang, H.-D.; Chuang, T.-N.; Shien, D.-M.; Horng, J.-T. RNAMST: Efficient and flexible approach for identifying RNA structural homologs. Nucleic Acids Res 2006, 34, W423–W428. [Google Scholar]
- Yao, Z.; Weinberg, Z.; Ruzzo, W.L. CMfinder—A covariance model based RNA motif finding algorithm. Bioinformatics 2006, 22, 445–452. [Google Scholar]
- Ji, Y.; Xu, X.; Stormo, G.D. A graph theoretical approach for predicting common RNA secondary structure motifs including pseudoknots in unaligned sequences. Bioinformatics 2004, 20, 1591–1602. [Google Scholar]
- Riccitelli, N.J.; Lupták, A. Computational discovery of folded RNA domains in genomes and in vitro selected libraries. Methods 2010, 52, 133–140. [Google Scholar]
- Gautheret, D.; Major, F.; Cedergren, R. Pattern searching/alignment with RNA primary and secondary structures: An effective descriptor for tRNA. Comput. Appl. Biosci 1990, 6, 325–331. [Google Scholar]
- Gautheret, D.; Lambert, A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol 2001, 313, 1003–1011. [Google Scholar]
- König, J.; Zarnack, K.; Luscombe, N.M.; Ule, J. Protein-RNA interactions: New genomic technologies and perspectives. Nat. Rev. Genet 2011, 13, 77–83. [Google Scholar]
- Rivas, E.; Eddy, S.R. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics 2000, 16, 583–605. [Google Scholar]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma 2005, 6, 31. [Google Scholar]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and genomewise. Genome Res 2004, 14, 988–995. [Google Scholar]
- Eyras, E.; Reymond, A.; Castelo, R.; Bye, J.M.; Camara, F.; Flicek, P.; Huckle, E.J.; Parra, G.; Shteynberg, D.D.; Wyss, C.; et al. Gene finding in the chicken genome. BMC Bioinforma 2005, 6, 131. [Google Scholar]
- Mariotti, M.; Guigo, R. Selenoprofiles: Profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics 2010, 26, 2656–2663. [Google Scholar]
- Vieira, F.G.; Rozas, J. Comparative genomics of the odorant-binding and chemosensory protein gene families across the Arthropoda: Origin and evolutionary history of the chemosensory system. Genome Biol. Evol 2011, 3, 476–490. [Google Scholar]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V.; Şenergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012, 338, 1469–1472. [Google Scholar]
- Santoro, F.; Pauler, F.M. Silencing by the imprinted Airn macro lncRNA: Transcription is the answer. Cell. Cycle 2013, 12, 711–712. [Google Scholar]

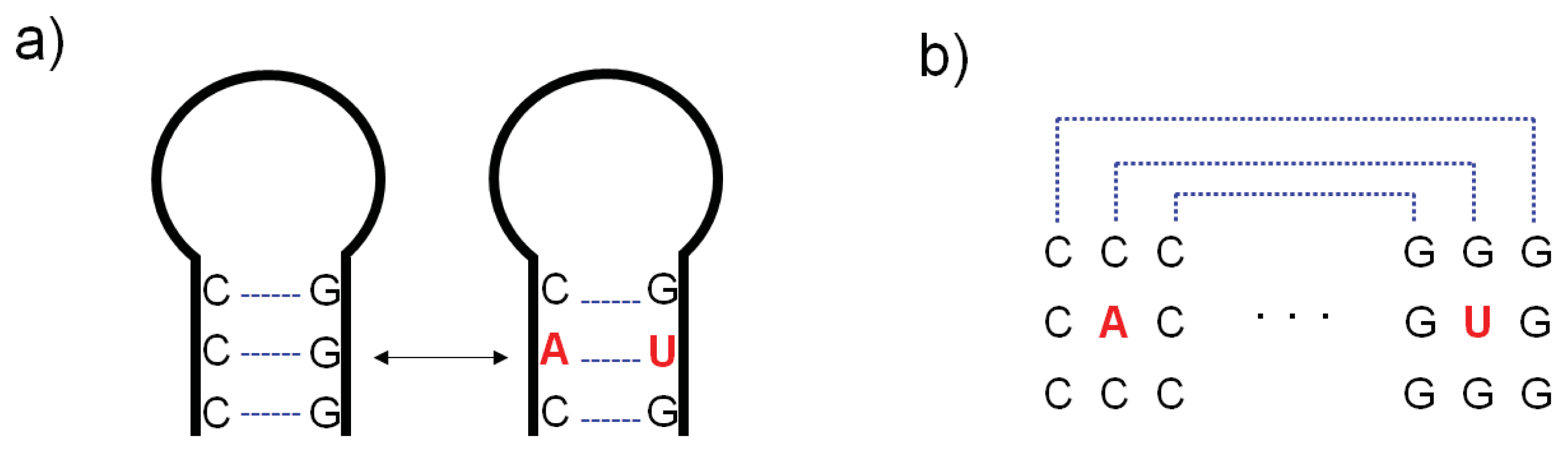


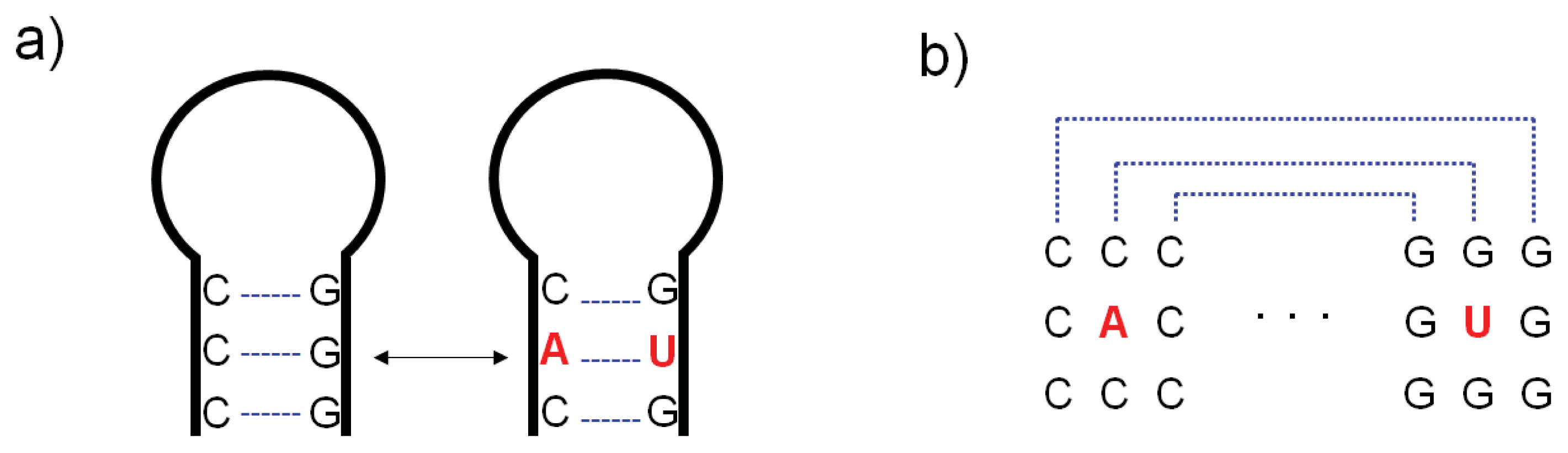
{kind=link}
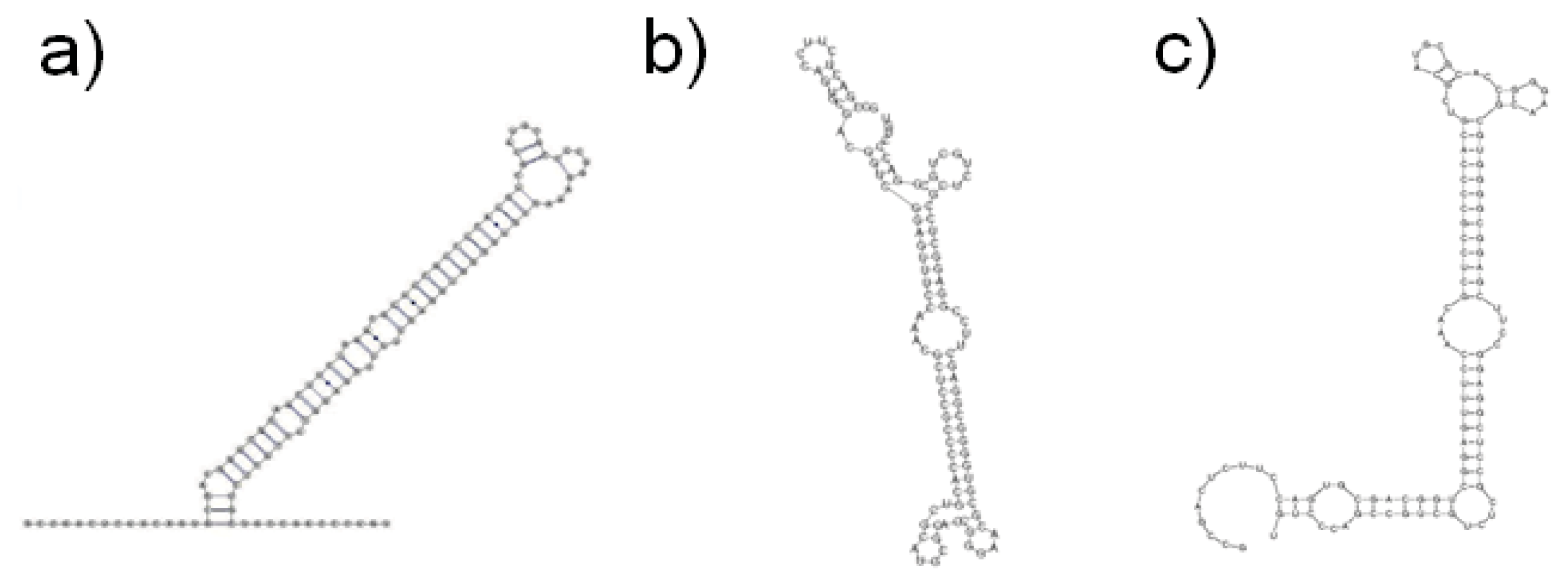
{kind=link}
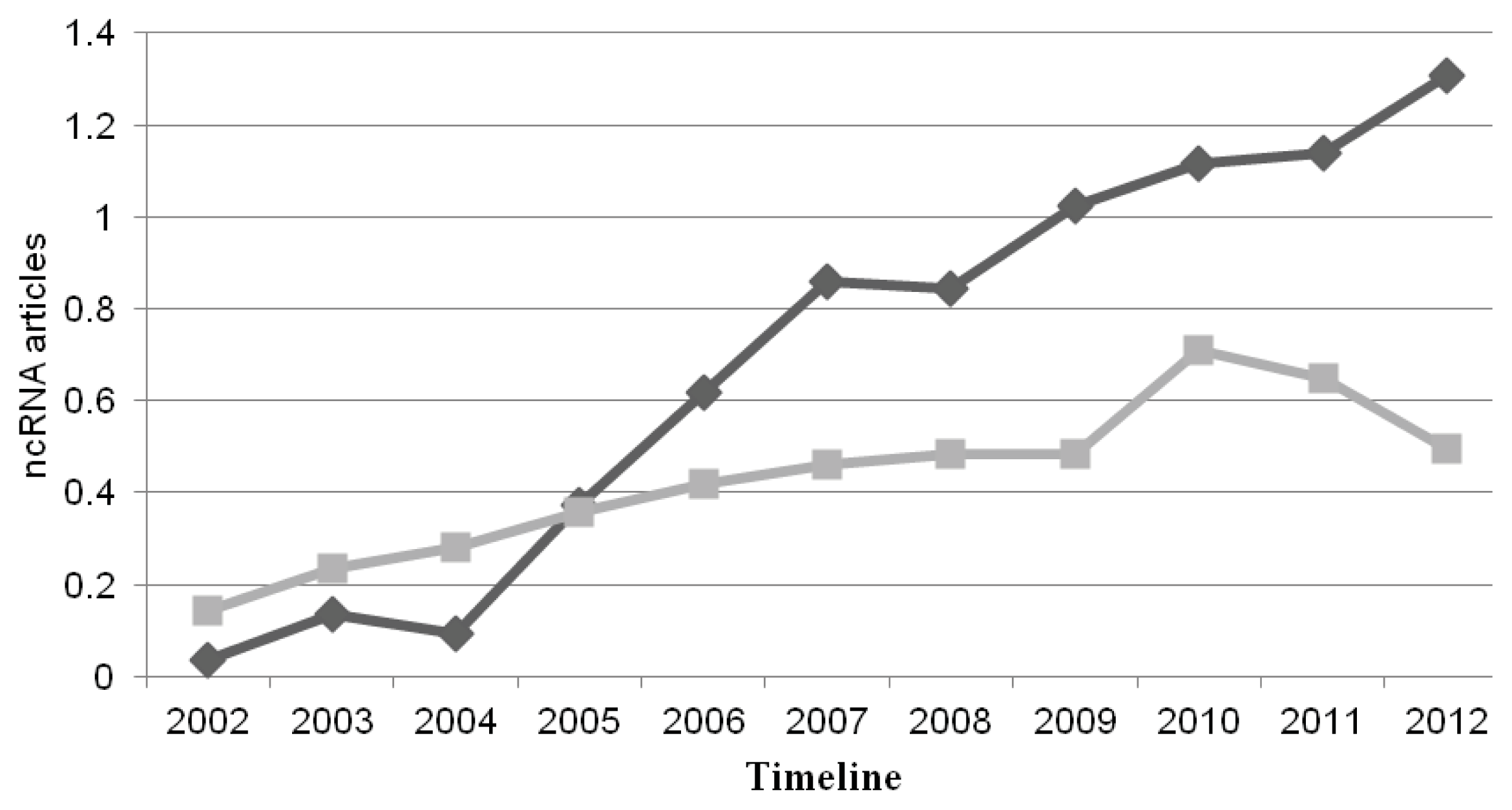
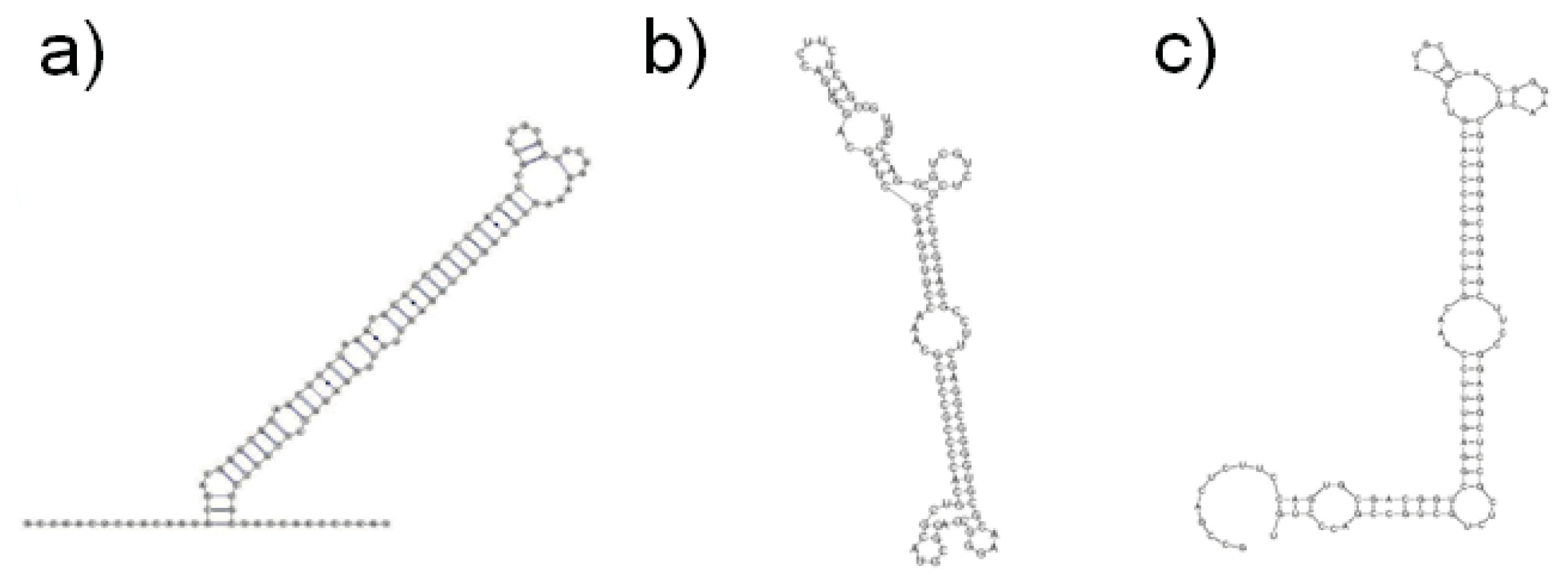{kind=link}
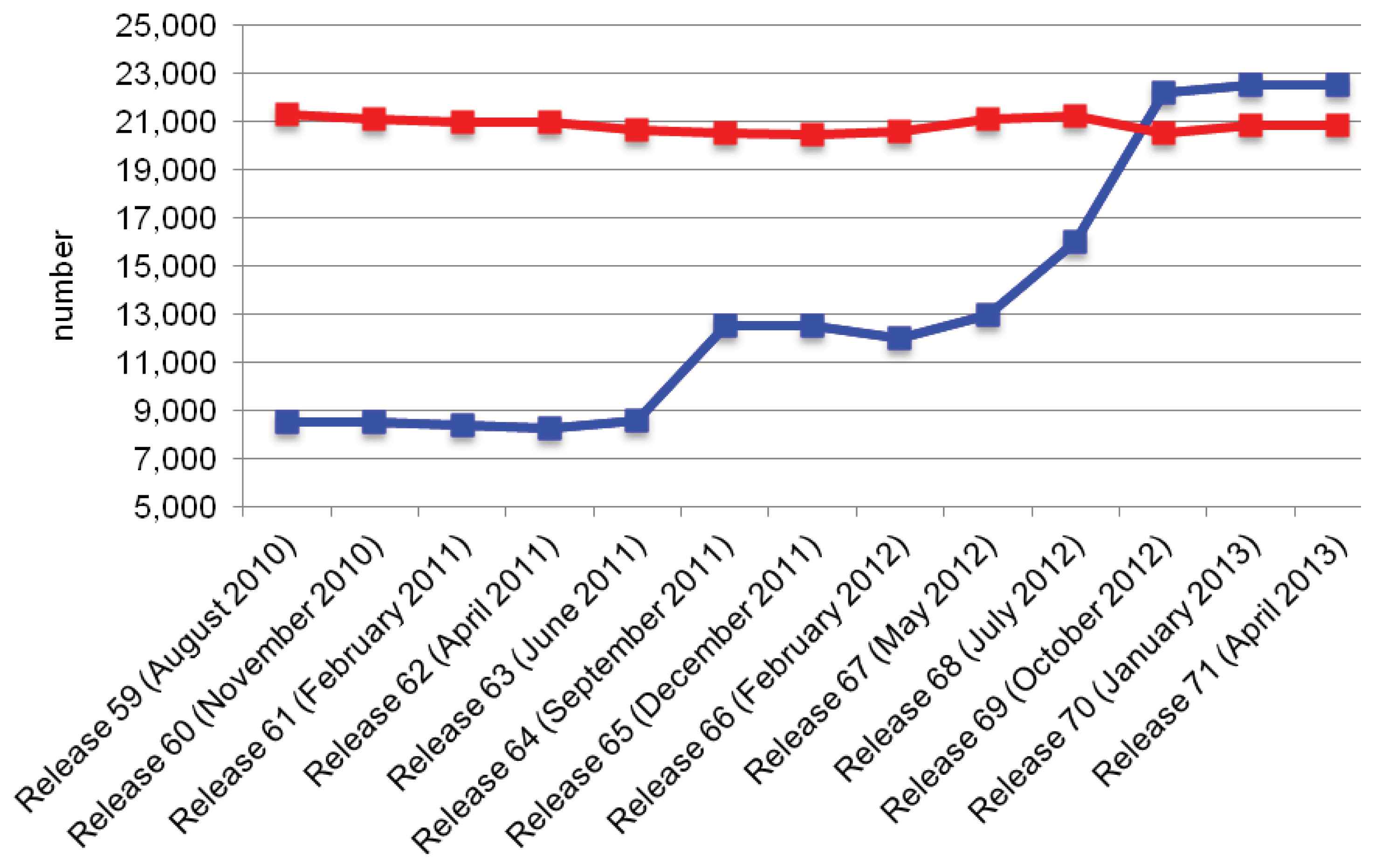{kind=link}
| Resource | Pubmed ID | Description | |
|---|---|---|---|
| Comparing ncRNAs (Section 2) | Mfold | 6163133 | Single sequence RNA secondary structure prediction. |
| RNAfold | 12824340 | ||
| WAR | 18492721 | WEB server allowing the execution of different alignment methods | |
| RNAalifold | 12079347 | Folding previously aligned RNAs (Plan A) | |
| PFOLD | 12824339 | ||
| ILM | 14693809 | ||
| Construct | 10518612 | ||
| Dynalign | 11902836 | Sankoff derived algorithm for the simultaneous alignment and secondary structure prediction (Plan B) | |
| Foldalign | 9278497 | ||
| Stemloc | 15790387 | ||
| Consan | 16952317 | ||
| pmmulti | 15073017 | ||
| R-Coffee | 18420654 | Aligners taking into account previously estimated secondary structure (Plan C) | |
| RNAcast | 16020472 | ||
| SARA | 18689811 | 3D structure alignment method | |
| DIAL | 17567620 | ||
| iPARTS | 20507908 | ||
| ARTS | 16204124 | ||
| SARSA | 18502774 | ||
| LaJolla | https://www.mdpi.com/1999-4893/2/2/692 | ||
| FRASS | 20553602 | ||
| Detecting ncRNAs (section 3) | ML-heuristic | 16267089 | Profile HMM |
| RAGA | 9358168 | Genetic algorithm | |
| RSEARCH | 14499004 | Covariance model | |
| Infernal | 12095421 | ||
| BlastR | 21624887 | BLAST-based dinucleotide homology search | |
| Datasets and browsers (section 4) | ENCODE | 22955616 | Consortium |
| Ensembl | 22086963 | ||
| FANTOM | 11217851 | ||
| HAVANA | http://www.sanger.ac.uk/research/projects/vertebrategenome/havana/ | Annotation team | |
| GENCODE | 22955987 | Project for the annotation of all human gene features | |
| UCSC | 12045153 | Genome browser | |
| VEGA | 18003653 | ||
| RefSeq | 18927115 | Collection of DNA, transcripts, and proteins | |
| Rfam | 12520045 | ncRNA database | |
| NRED | 18829717 | ||
| lncRNAdb | 21112873 | ||
| RNAdb | 17145715 | ||
| fRNAdb | 17099231 | ||
| NONCODE | 15608158 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bussotti, G.; Notredame, C.; Enright, A.J. Detecting and Comparing Non-Coding RNAs in the High-Throughput Era. Int. J. Mol. Sci. 2013, 14, 15423-15458. https://doi.org/10.3390/ijms140815423
Bussotti G, Notredame C, Enright AJ. Detecting and Comparing Non-Coding RNAs in the High-Throughput Era. International Journal of Molecular Sciences. 2013; 14(8):15423-15458. https://doi.org/10.3390/ijms140815423
Chicago/Turabian StyleBussotti, Giovanni, Cedric Notredame, and Anton J. Enright. 2013. "Detecting and Comparing Non-Coding RNAs in the High-Throughput Era" International Journal of Molecular Sciences 14, no. 8: 15423-15458. https://doi.org/10.3390/ijms140815423
APA StyleBussotti, G., Notredame, C., & Enright, A. J. (2013). Detecting and Comparing Non-Coding RNAs in the High-Throughput Era. International Journal of Molecular Sciences, 14(8), 15423-15458. https://doi.org/10.3390/ijms140815423