Effect of GABA, a Bacterial Metabolite, on Pseudomonas fluorescens Surface Properties and Cytotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
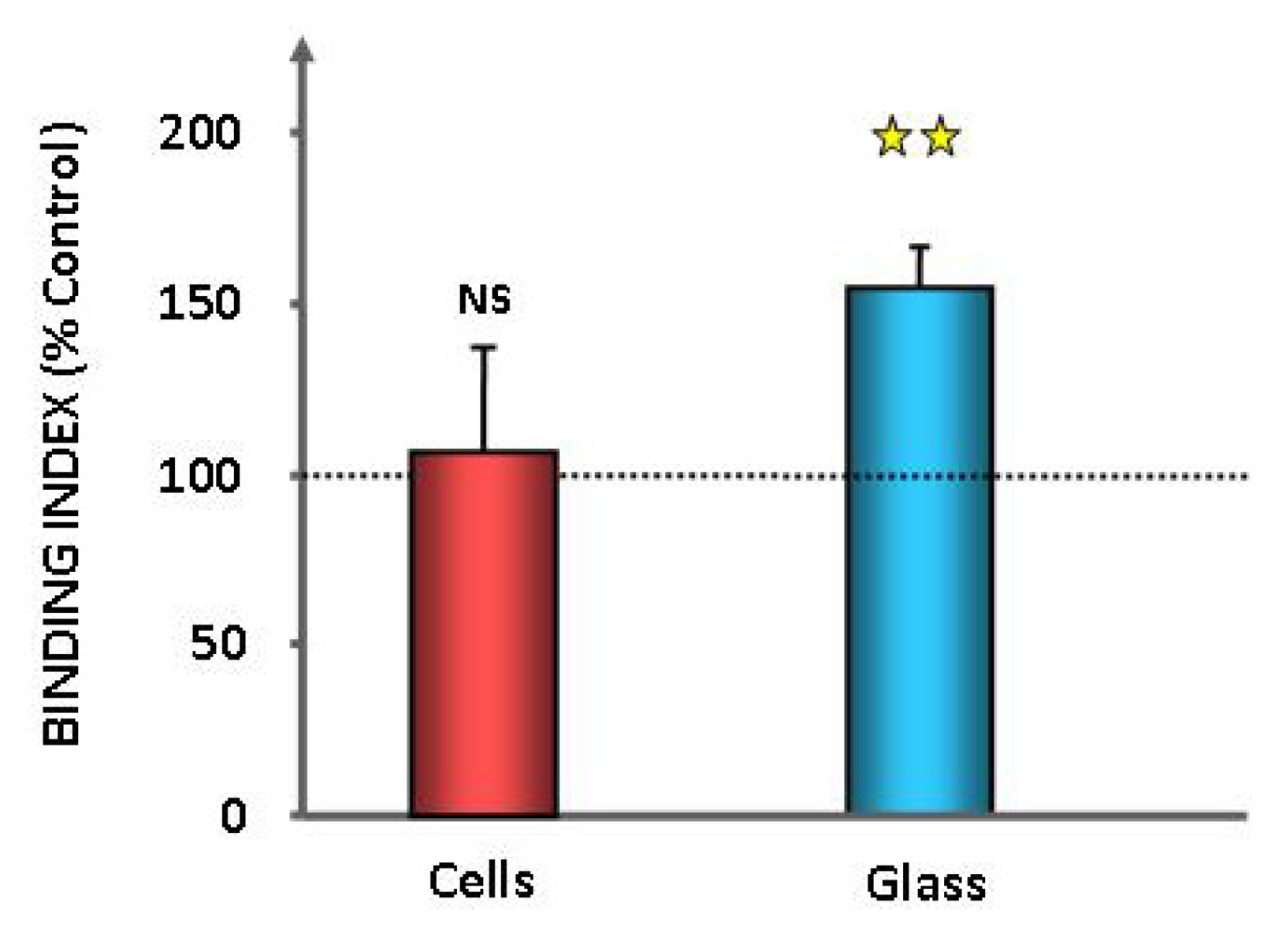
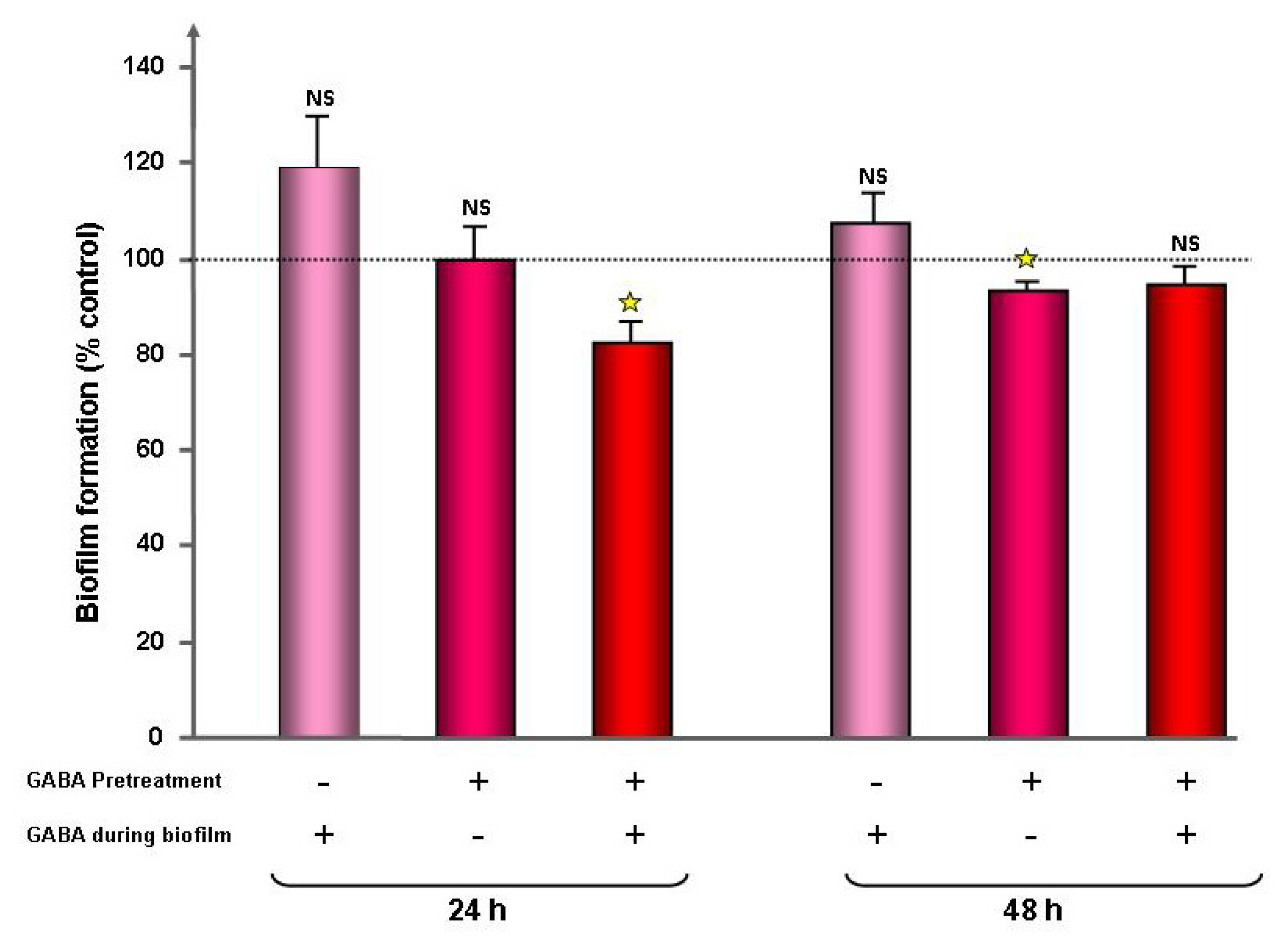
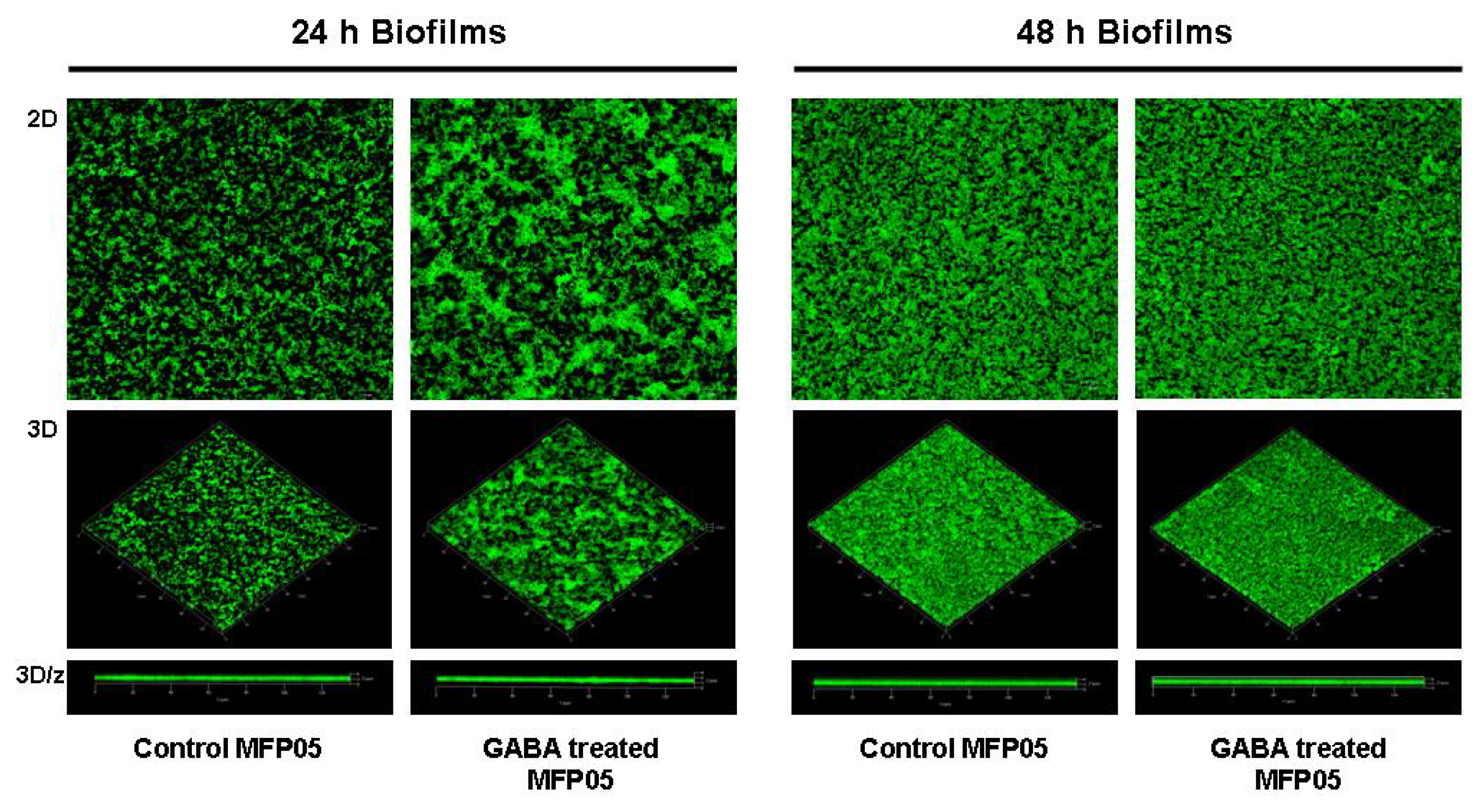
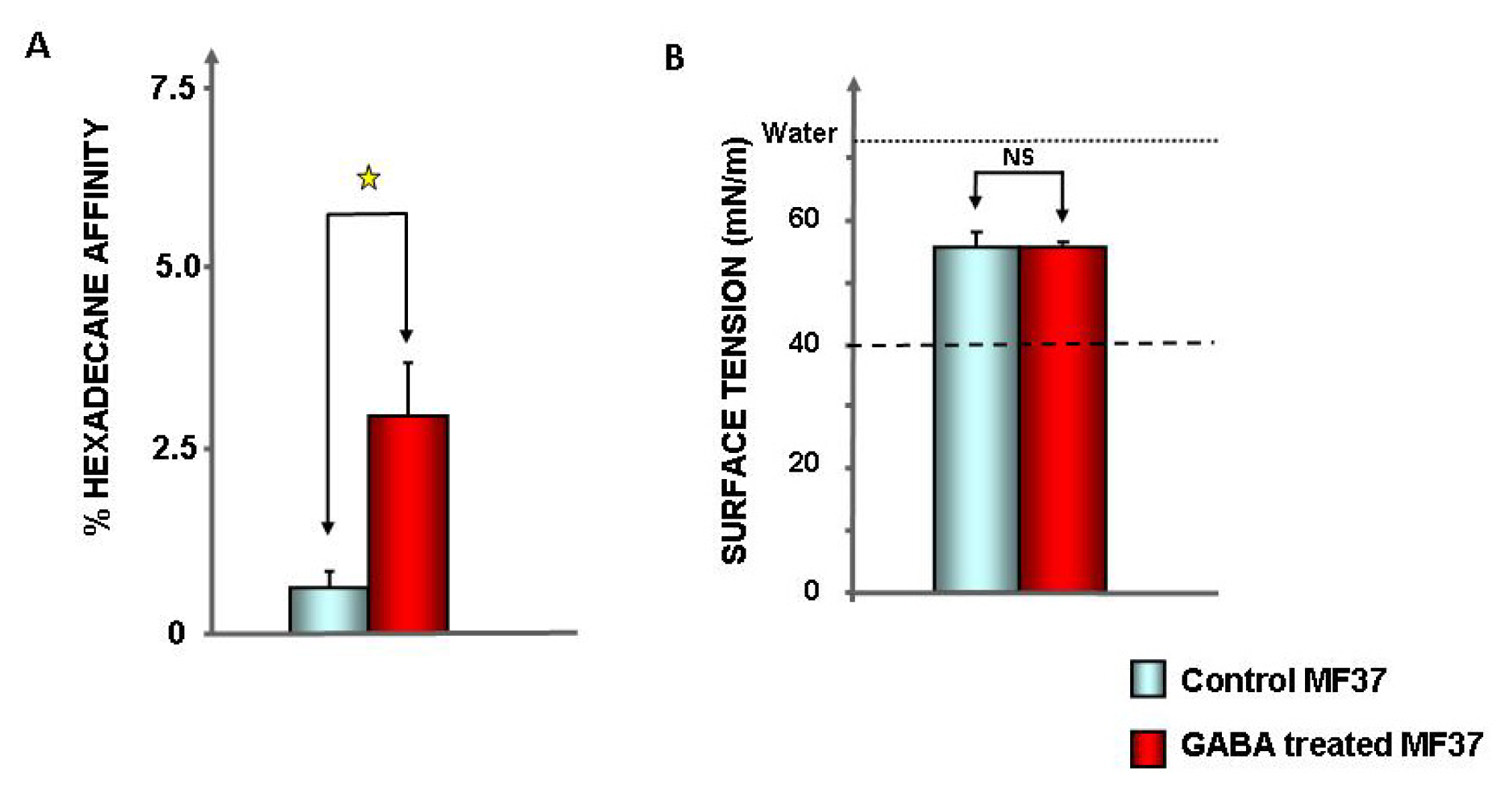
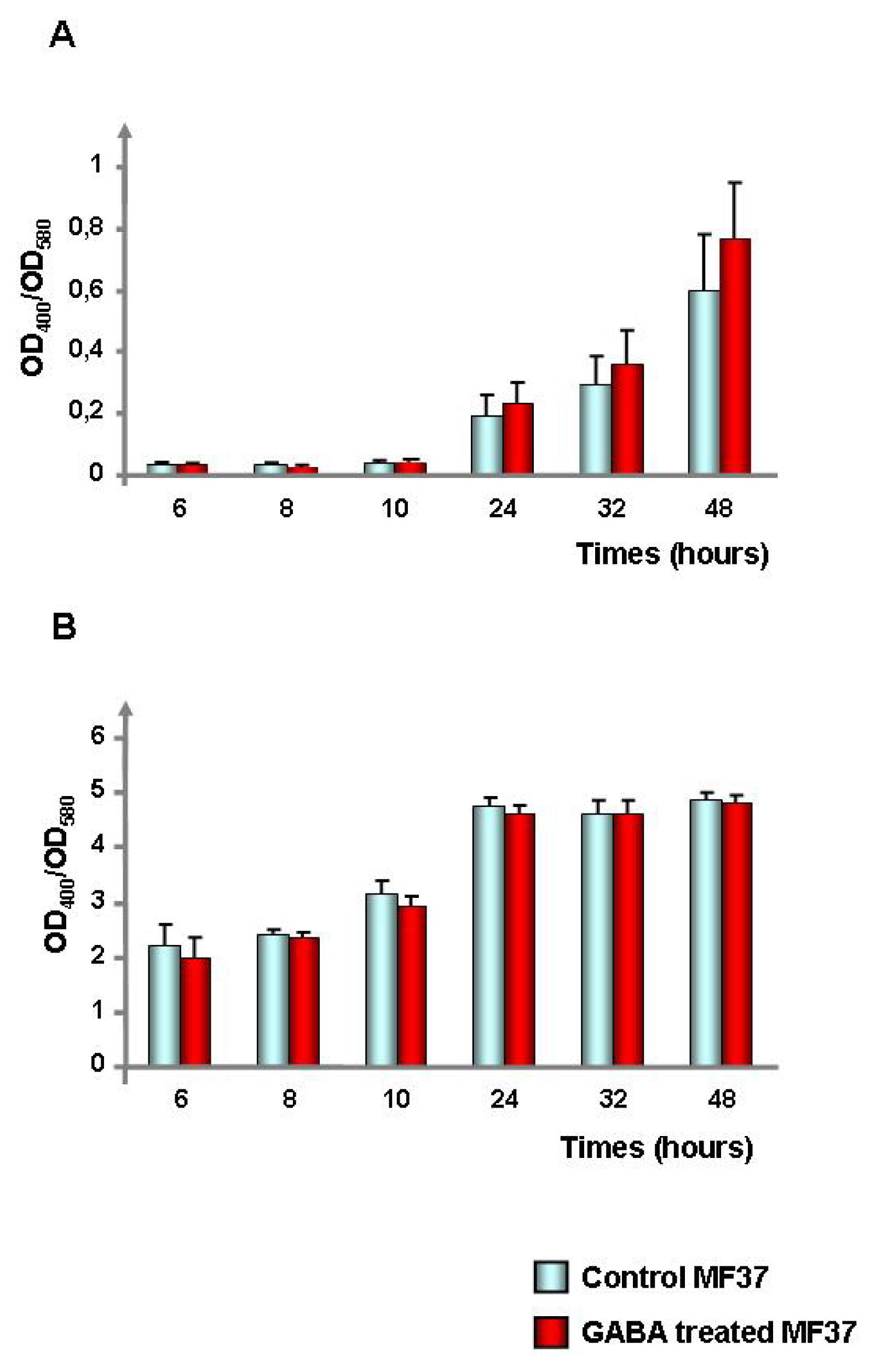
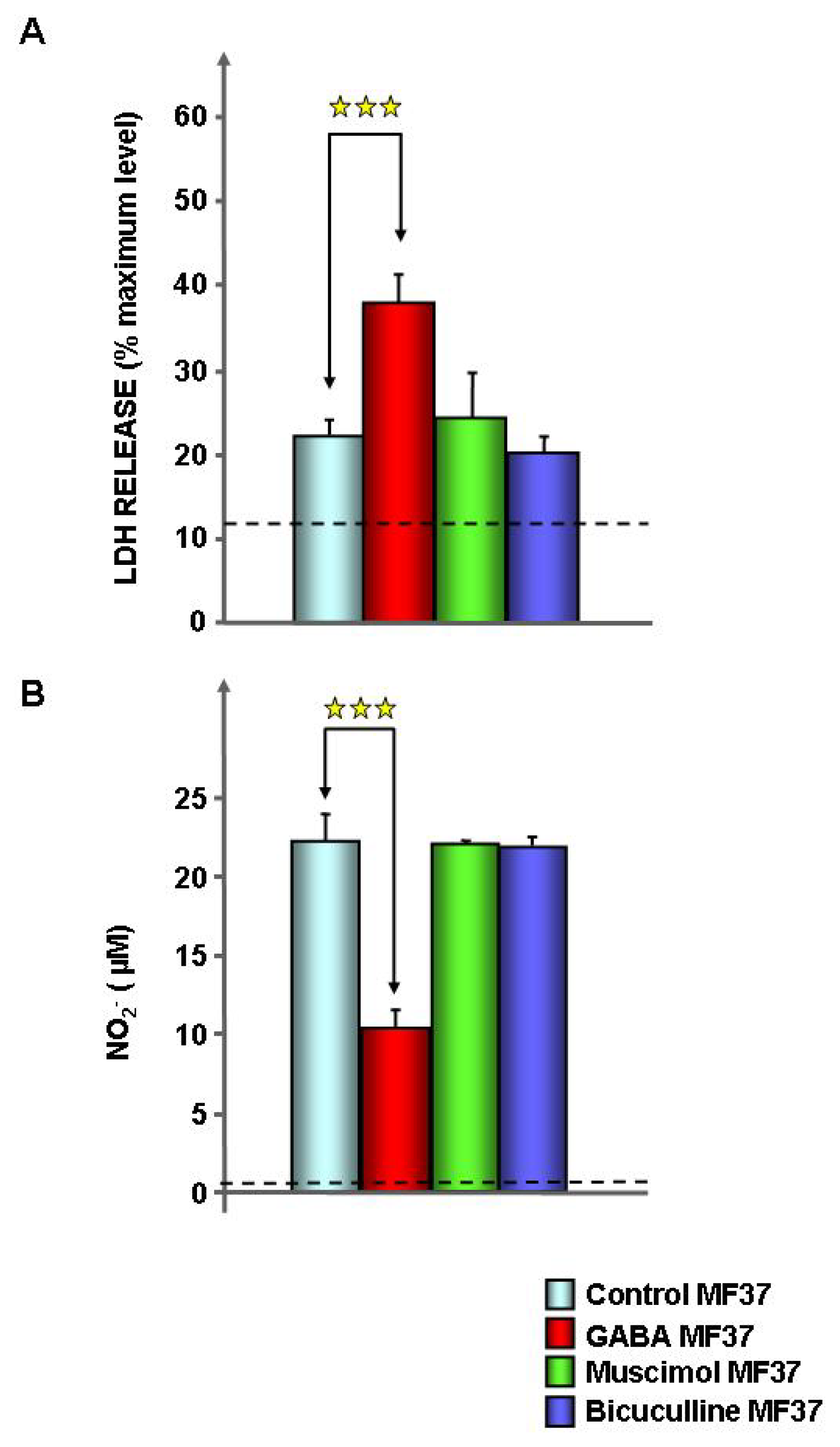
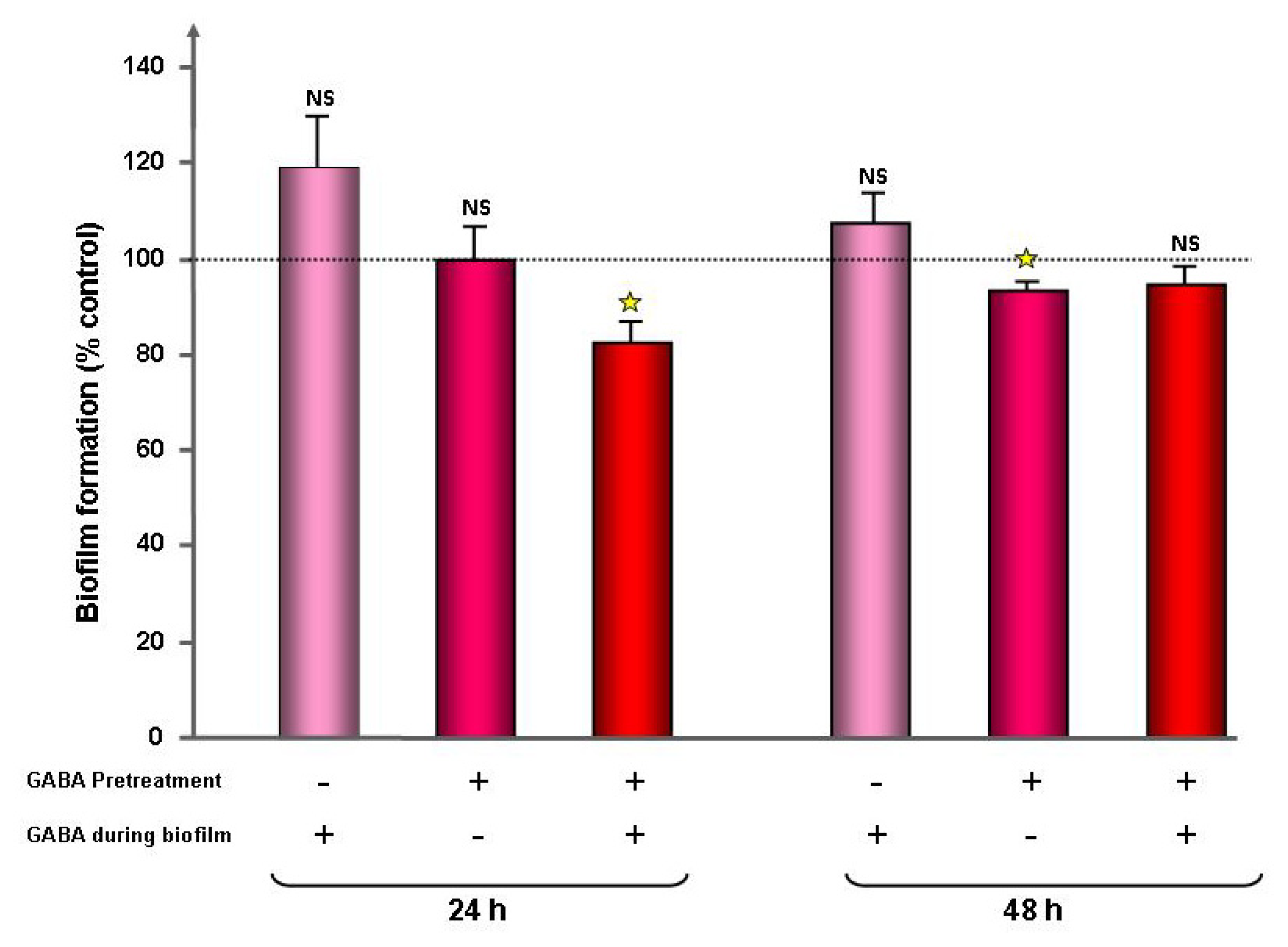
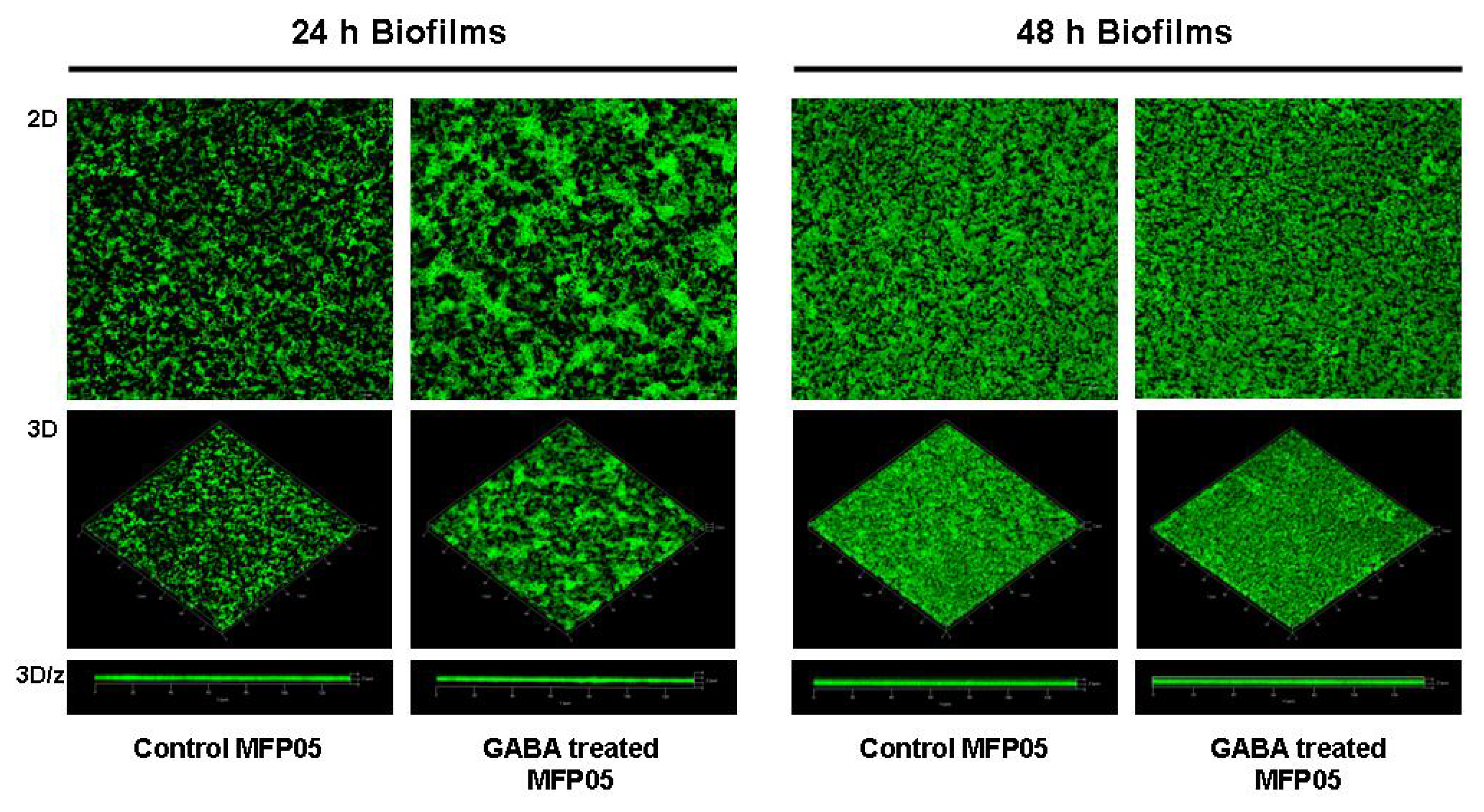
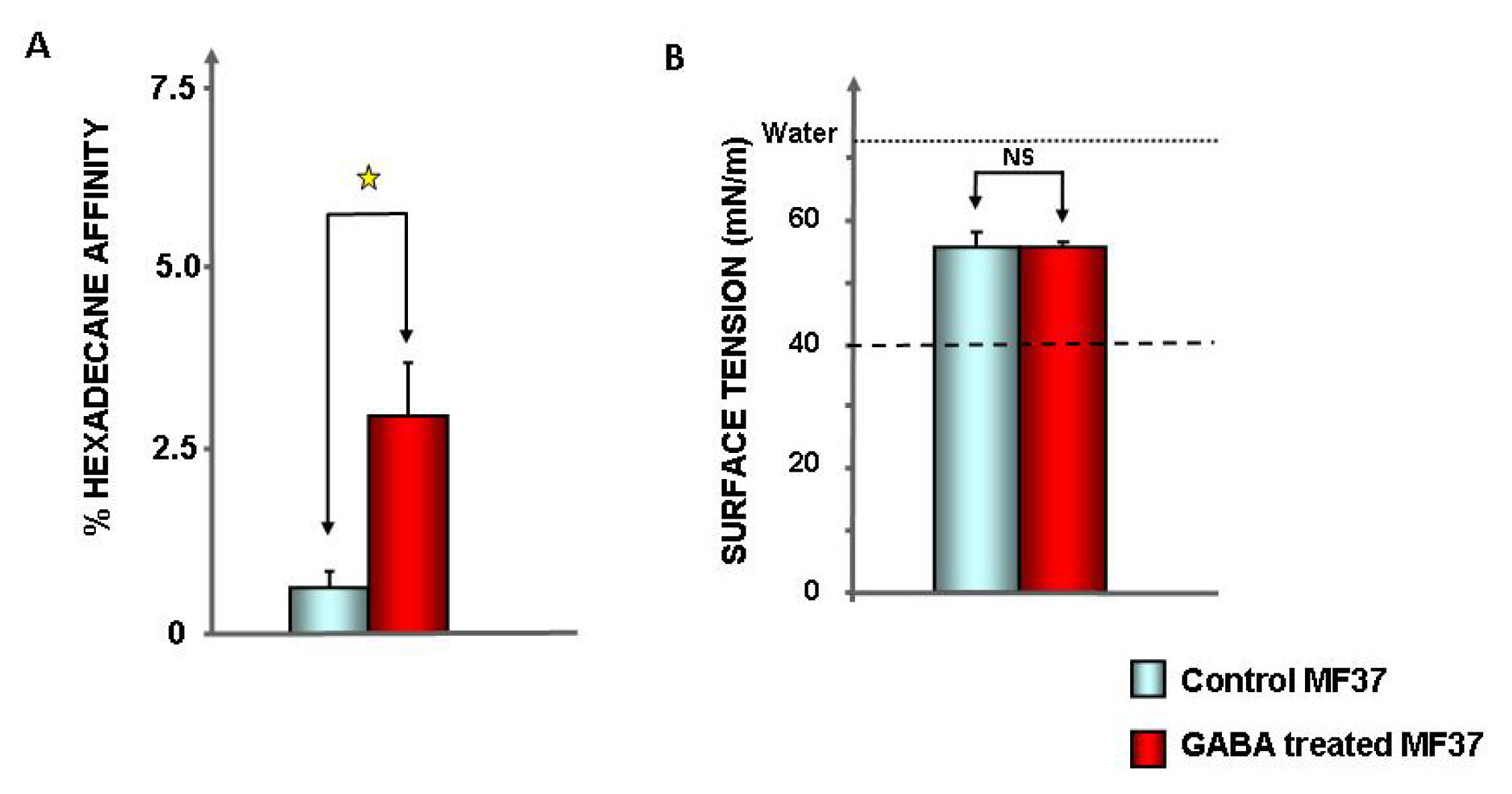
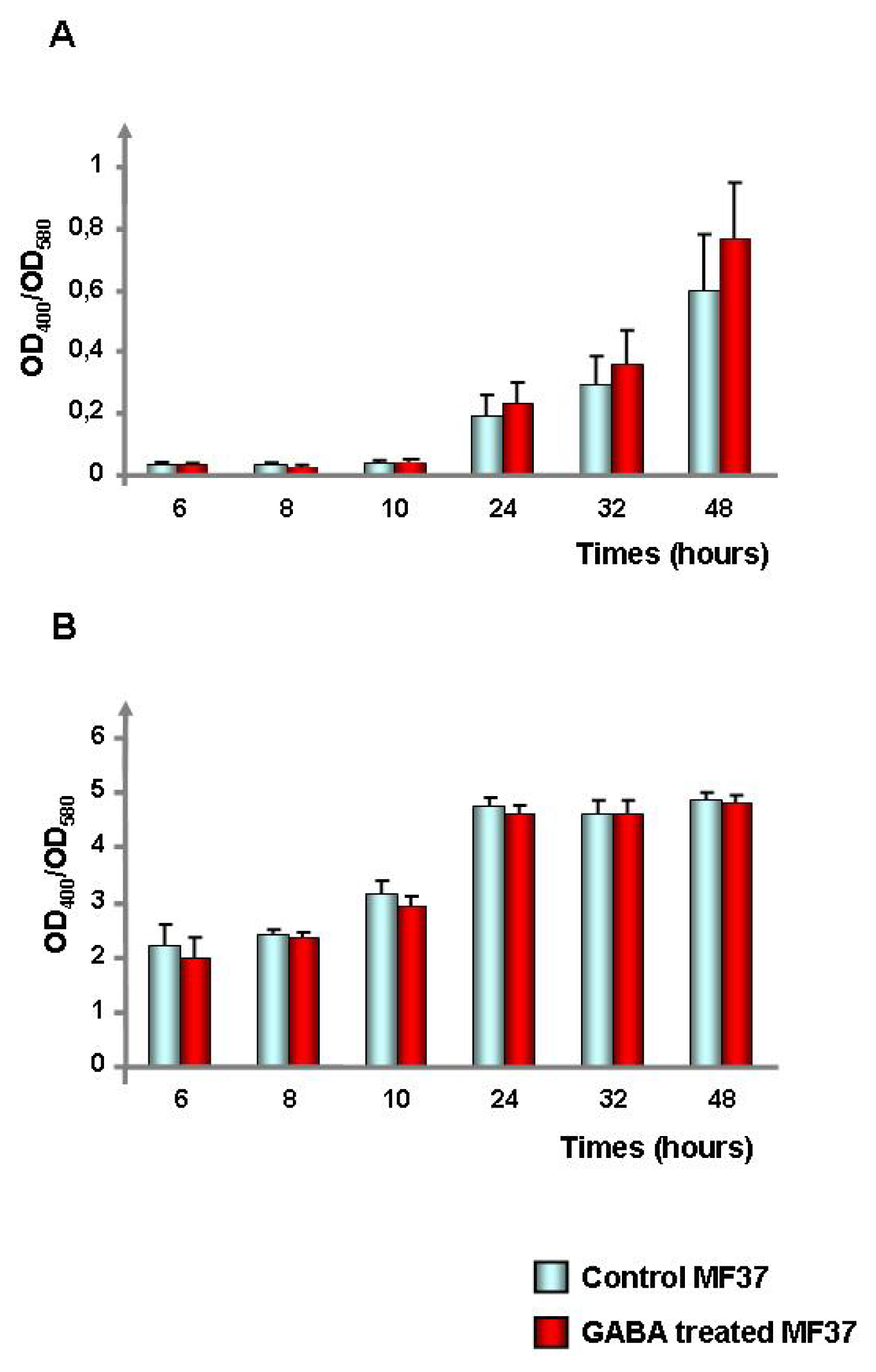
2.1. Effect of GABA on the Cytotoxic Activity, Adhesion Potential, Biofilm Formation Activity and Surface Properties of Pseudomonas fluorescens
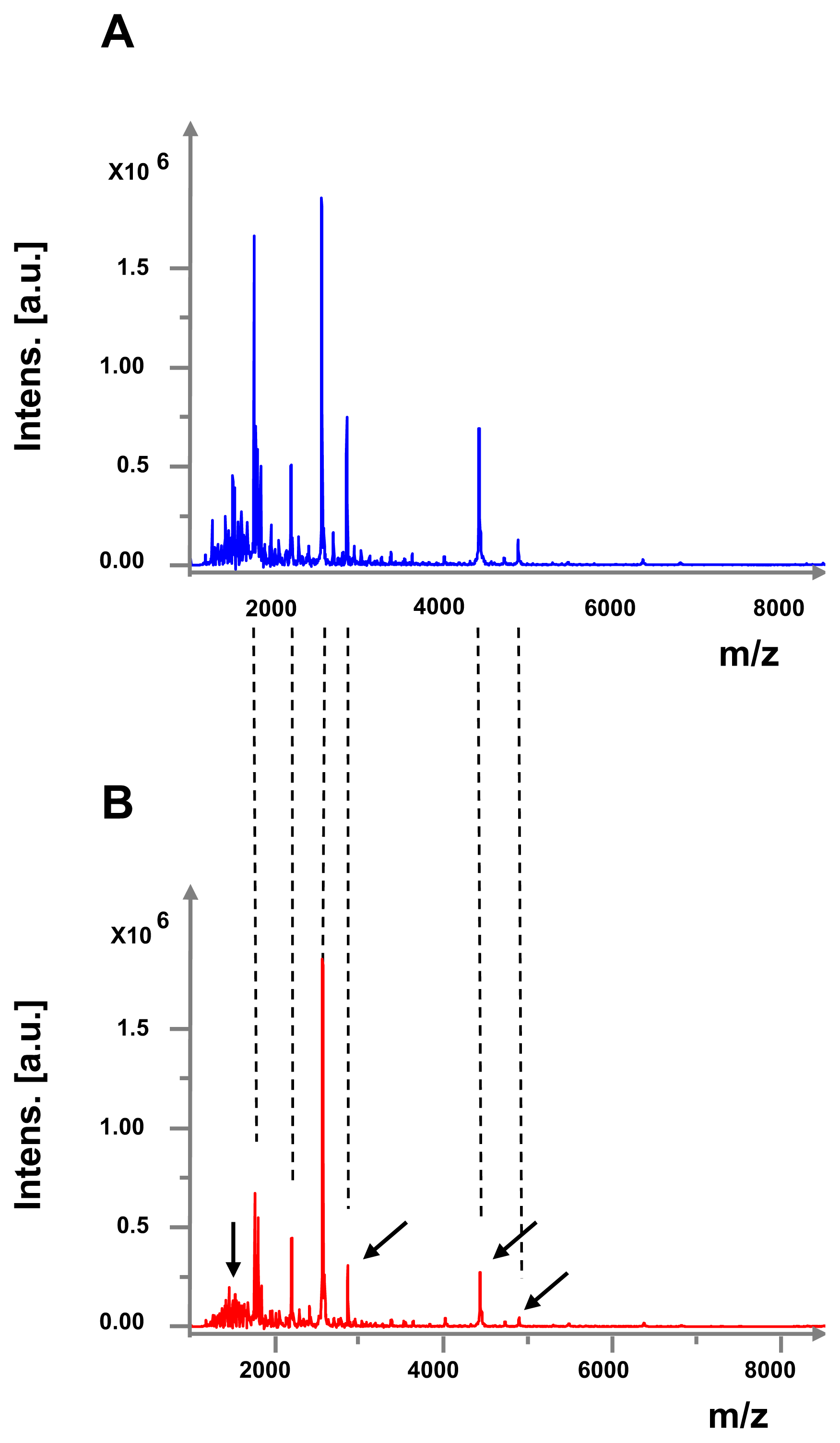
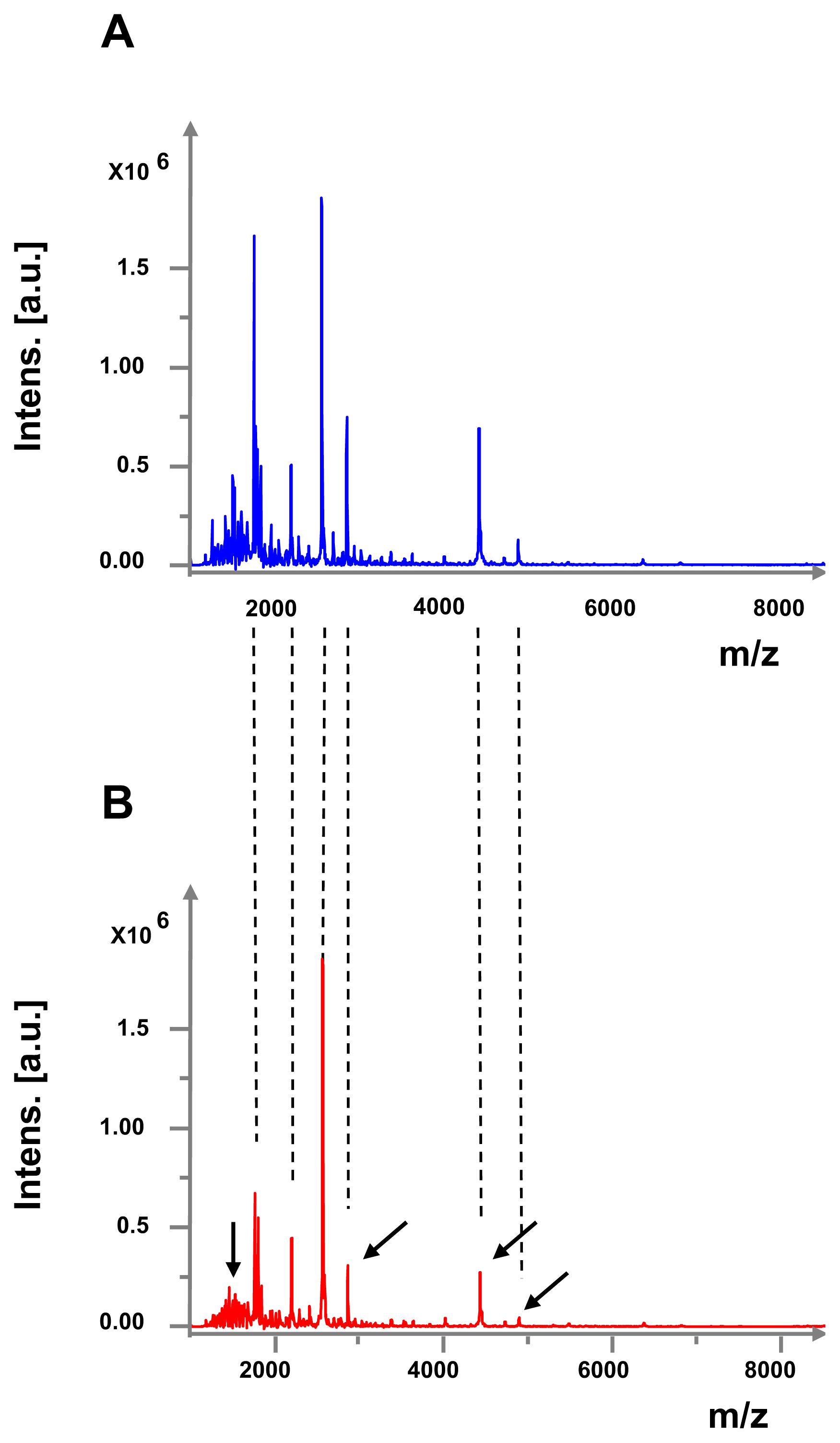
2.2. Effect of GABA on Secreted Diffusible Factors and Lipopolysaccharide Structure
3. Discussion
4. Experimental Section
4.1. Bacterial Strains and Culture Conditions
4.2. Swimming and Swarming Mobility Tests
4.3. Cytotoxic Activity Tests
4.4. Evaluation of the Bacterial Biofilm Formation Activity by the Crystal Violet Technique
4.5. GFP Transformation
4.6. Confocal Microscope Study of the Bacterial Biofilm Formation Activity on Glass Surfaces
4.7. Evaluation of Bacterial Surface Properties
4.8. Surface Tension and Biosurfactant Production in Bacterial Culture Medium
4.9. Study of the Lipopolysaccharide Structure
4.10. Detection of Diffusible Virulence Factors
4.11. Statistical Analysis
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Qi, Z.; Cort, A. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmos. Environ 2003, 37, 2247–2258. [Google Scholar]
- Noe, F.F.; Nickerson, W.J. Metabolism of 2-pyrrolidone and gamma aminobutyric acid by Pseudomonas aeruginosa. J. Bacteriol 1958, 75, 674–681. [Google Scholar]
- Chou, H.T.; Kwon, D.H.; Hegazy, M.; Lu, C.D. Transcriptome analysis of agmatine and putrescine catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol 2008, 190, 1966–1975. [Google Scholar]
- Tunnicliff, G. Inhibition of 4-aminobutyrate aminotransferase from Pseudomonas fluorescens by ATP. Biochem. Mol. Biol. Int 1993, 31, 41–47. [Google Scholar]
- Kaspar, H.F.; Mountfort, D.O.; Pybus, V. Degradation of gamma-aminobutyric acid (GABA) by marine microorganisms. FEMS Microbiol. Ecol 1991, 85, 313–318. [Google Scholar]
- Mountfort, D.O.; Pybus, V. Regulatory influences on the production of gamma-aminobutyric acid by a marine pseudomonad. Appl. Environ. Microbiol 1992, 58, 237–242. [Google Scholar]
- Siragusa, S.; de Angelis, M.; di Cagno, R.; Rizzello, C.G.; Coda, R.; Gobbetti, M. Synthesis of gamma-aminobutyric acid by lactic acid bacteria isolated from a variety of Italian cheeses. Appl. Environ. Microbiol 2007, 73, 7283–7290. [Google Scholar]
- Richard, H.T.; Foster, J.W. Acid resistance in Escherichia coli. Adv. Appl. Microbiol 2003, 52, 167–186. [Google Scholar]
- Brechtel, C.E.; King, S.C. 4-Aminobutyrate (GABA) transporters from the amine-polyamine-choline superfamily: Substrate specificity and ligand recognition profile of the 4-aminobutyrate permease from Bacillus subtilis. Biochem. J 1998, 333, 565–571. [Google Scholar]
- Hu, L.A.; King, S.C. Membrane topology of the Escherichia coli γ-aminobutyrate transporter: Implications on the topography and mechanism of prokaryotic and eukaryotic transporters from the APC superfamily. Biochem. J 1998, 336, 68–76. [Google Scholar]
- Zhao, Z.; Ding, J.Y.; Ma, W.H.; Zhou, N.Y.; Liu, S.J. Identification and characterization of gamma-aminobutyric acid uptake system GabPCg (NCgl0464) in Corynebacterium glutamicum. Appl. Environ. Microbiol 2012, 78, 2596–2601. [Google Scholar]
- Planamente, S.; Mondy, S.; Hommais, F.; Vigouroux, A.; Moréra, S.; Faure, D. Structural basis for selective GABA binding in bacterial pathogens. Mol. Microbiol 2012, 85, 1085–1099. [Google Scholar]
- Dagorn, A.; Hillion, M.; Chapalain, A.; Lesouhaitier, O.; Duclairoir-Poc, C.; Vieillard, J.; Chevalier, S.; Taupin, L.; Le Derf, F.; Feuilloley, M.G.J. Gamma-aminobutyric acid acts as a specific virulence regulator in Pseudomonas aeruginosa. Microbiology 2013, 159, 339–351. [Google Scholar]
- Guthrie, G.D.; Nicholson-Guthrie, C.S. Gamma-Aminobutyric acid uptake by a bacterial system with neurotransmitter binding characteristics. Proc. Natl. Acad. Sci. USA 1989, 86, 7378–7381. [Google Scholar]
- Richard, H.; Foster, J.W. Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol 2004, 186, 6032–6041. [Google Scholar]
- Tramonti, A.; de Canio, M.; Delany, I.; Scarlato, V.; de Biase, D. Mechanisms of transcription activation exerted by GadX and GadW at the gadA and gadBC gene promoters of the glutamate-based acid resistance system in Escherichia coli. J. Bacteriol 2006, 188, 8118–8127. [Google Scholar]
- De Biase, D.; Pennacchietti, E. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: Function, distribution and biomedical implications of the gadBC operon. Mol. Microbiol 2012, 86, 770–786. [Google Scholar]
- Chevrot, R.; Rosen, R.; Haudecoeur, E.; Cirou, A.; Shelp, B.J.; Ron, E.; Faure, D. GABA controls the level of quorum-sensing signal in Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. USA 2006, 103, 7460–7464. [Google Scholar]
- Park, D.H.; Mirabella, R.; Bronstein, P.A.; Preston, G.M.; Haring, M.A.; Lim, C.K.; Collmer, A.; Schuurink, R.C. Mutations in gamma-aminobutyric acid (GABA) transaminase genes in plants or Pseudomonas syringae reduce bacterial virulence. Plant J 2010, 64, 318–330. [Google Scholar]
- Van de Mortel, J.E.; de Vos, R.C.; Dekkers, E.; Pineda, A.; Guillod, L.; Bouwmeester, K.; van Loon, J.J.; Dicke, M.; Raaijmakers, J.M. Metabolic and transcriptomic changes induced in Arabidopsis by the rhizobacterium Pseudomonas fluorescens SS101. Plant Physiol 2012, 160, 2173–2188. [Google Scholar]
- Chapalain, A.; Rossignol, G.; Lesouhaitier, O.; Merieau, A.; Gruffaz, C.; Guerillon, J.; Meyer, J.M.; Orange, N.; Feuilloley, M.G.J. Comparative study of 7 fluorescent pseudomonad clinical isolates. Can. J. Microbiol 2008, 54, 19–27. [Google Scholar]
- Mozrzymas, J.W.; Zarnowska, E.; Pytel, M.; Mercik, K. Modulation of GABAA receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J. Neurosci 2003, 23, 7981–7692. [Google Scholar]
- Shelp, B.J.; Bown, A.W.; Faure, D. Extracellular gamma-aminobutyrate mediates communication between plants and other organisms. Plant Physiol 2006, 142, 1350–1352. [Google Scholar]
- Johnson, C.R.; Muir, D.G.; Reysenbach, A.L. Characteristic bacteria associated with surfaces of coralline algae: A hypothesis for bacterial induction of marine invertebrate larvae. Mar. Ecol. Prog. Ser 1991, 74, 281–294. [Google Scholar]
- Picot, L.; Chevalier, S.; Mezghani-Abdelmoula, S.; Merieau, A.; Lesouhaitier, O.; Leroux, P.; Cazin, L.; Orange, N.; Feuilloley, M.G.J. Cytotoxic effects of the lipopolysaccharide from Pseudomonas fluorescens on neurons and glial cells. Microb. Pathog 2003, 35, 95–106. [Google Scholar]
- Angulo, M.C.; Le Meur, K.; Kozlov, A.S.; Charpak, S.; Audinat, E. GABA, a forgotten gliotransmitter. Prog. Neurobiol 2008, 86, 297–303. [Google Scholar]
- Picot, L.; Mezghani-Abdelmoula, S.; Chevalier, S.; Merieau, A.; Lesouhaitier, O.; Guerillon, J.; Cazin, L.; Orange, N.; Feuilloley, M.G.J. Regulation of the cytotoxic effects of Pseudomonas fluorescens by growth temperature. Res. Microbiol 2004, 155, 39–46. [Google Scholar]
- Bellon-Fontaine, M.N.; Rault, J.; van Oss, C.J. Microbial adhesion to solvents: A novel method to determine the electron-donor/electron-acceptor or Lewis acid-base properties of microbial cells. Colloids Surf. B Biointerfaces 1996, 7, 47–53. [Google Scholar]
- Cornelis, P. Iron uptake and metabolism in pseudomonads. Appl. Microbiol. Biotechnol 2010, 86, 1637–1645. [Google Scholar]
- Veron, W.; Lesouhaiter, O.; Pennanec, X.; Rehel, K.; Leroux, P.; Orange, N.; Feuilloley, M.G.J. Natriuretic peptides affect Pseudomonas aeruginosa and specifically modify LPS biosynthesis. FEBS J 2007, 274, 5852–5864. [Google Scholar]
- Owens, D.F.; Kriegstein, A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci 2002, 3, 715–727. [Google Scholar]
- Bouché, N.; Lacombe, B.; Fromm, H. GABA signaling: A conserved and ubiquitous mechanism. Trends Cell. Biol 2003, 13, 607–610. [Google Scholar]
- Mezghani-Abdelmoula, S.; Khemiri, A.; Lesouhaitier, O.; Chevalier, S.; Orange, N.; Cazin, L.; Feuilloley, M.G.J. Sequential activation of constitutive and inducible nitric oxide synthase (NOS) in rat cerebellar granule neurons by Pseudomonas fluorescens and invasive behaviour of the bacteria. Microbiol. Res 2004, 159, 355–363. [Google Scholar]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun 2005, 73, 1907–1916. [Google Scholar]
- Veron, W.; Orange, N.; Feuilloley, M.G.J.; Lesouhaitier, O. Natriuretic peptide modify Pseudomonas fluorescens cytotoxicity by regulating cyclic nucleotides and modifying LPS structure. BMC Microbiol 2008, 8, 114. [Google Scholar]
- Guthrie, G.D.; Nicholson-Guthrie, C.S.; Leary, H.L. A bacterial high-affinity GABA binding protein: Isolation and characterization. Biochem. Biophys. Res. Commun 2000, 268, 65–68. [Google Scholar]
- Haas, D.; Defago, G. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol 2005, 3, 307–319. [Google Scholar]
- Bown, A.W.; MacGregor, K.B.; Shelp, B.J. Gamma-aminobutyrate: Defense against invertebrate pests? Trends Plant Sci 2006, 11, 424–427. [Google Scholar]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible GABAergic mechanism in the neuroprotective effect of gabapentin and lamotrigine against 3-nitropropionic acid induced neurotoxicity. Eur. J. Pharmacol 2012, 674, 265–274. [Google Scholar]
- Davey, M.E.; O’Toole, G.A. Microbial biofilms: From ecology to molecular genetics. Microbiol. Mol. Biol. Rev 2000, 64, 847–867. [Google Scholar]
- Ito, K.; Tanaka, K.; Nishibe, Y.; Hasegawa, J.; Ueno, H. GABA-synthesizing enzyme, GAD67, from dermal fibroblasts: Evidence for a new skin function. Biochim. Biophys. Acta 2007, 1770, 291–296. [Google Scholar]
- Grice, E.A.; Kong, H.H.; Renaud, G.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Wolfsberg, T.G.; Turner, M.L.; Segre, J.A. A diversity profile of the human skin microbiota. Genome Res 2008, 18, 1043–1050. [Google Scholar]
- Gao, Z.; Tseng, C.; Pei, Z.; Blaser, M.J. Molecular analysis of human forearm superfical skin bacterial biota. Proc. Natl. Acad. Sci. USA 2007, 104, 2927–2932. [Google Scholar]
- Raaijmakers, J.M.; De Bruijn, I.; Nybroe, O.; Ongena, M. Natural functions of lipopeptides from Bacillus and Pseudomonas: More than surfactants and antibiotics. FEMS Microbiol. Rev 2010, 34, 1037–1062. [Google Scholar]
- Becerra, C.; Albesa, I.; Eraso, A.J. Leukotoxicity of pyoverdin, production of reactive oxygen species, and effect of UV radiation. Biochem. Biophys. Res. Commun 2001, 285, 414–418. [Google Scholar]
- Makin, S.A.; Beveridge, T.J. The influence of A-band and B-band lipopolysaccharide on the surface characteristics and adhesion of Pseudomonas aeruginosa to surfaces. Microbiology 1996, 142, 299–307. [Google Scholar]
- Yokota, S.; Fujii, N. Contributions of the lipopolysaccharide outer core oligosaccharide region on the cell surface properties of Pseudomonas aeruginosa. Comp. Immunol. Microbiol. Infect. Dis 2007, 30, 97–109. [Google Scholar]
- Mitrovic, B.; Ignarro, L.J.; Vinters, H.V.; Akers, M.-A.; Schmid, I.; Uittenbogaart, C.; Merrill, J.E. Nitric oxide induces necrotic but not apoptotic cell death in oligodendrocytes. Neuroscience 1995, 65, 531–539. [Google Scholar]
- Burini, J.F.; Gügi, B.; Merieau, A.; Guespin-Michel, J.F. Lipase and acidic phosphatase from the psychrotrophic bacterium Pseudomonas fluorescens: Two enzymes whose synthesis is regulated by the growth temperature. FEMS Microbiol. Lett 1994, 122, 13–18. [Google Scholar]
- Silby, M.W.; Cerdeño-Tárraga, A.M.; Vernikos, G.S.; Giddens, S.R.; Jackson, R.W.; Preston, G.M.; Zhang, X.X.; Moon, C.D.; Gehrig, S.M.; Godfrey, S.A.; et al. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol 2009, 10, R51. [Google Scholar]
- O’Toole, G.A.; Kolter, R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol 1998, 30, 295–304. [Google Scholar]
- Bazire, A.; Diab, F.; Jebbar, M.; Haras, D. Influence of high salinity on biofilm formation by Pseudomonas aeruginosa. J. Ind. Microbiol. Biotechnol 2007, 34, 5–8. [Google Scholar]
- Hiemenz, P.C.; Lagowski, J.J. Principles of Colloid and Surface Chemistry; Lagowski, J.J., Ed.; Marcel Dekker: New York, NY, USA, 1977; p. 209. [Google Scholar]
- Darveau, R.P.; Hancock, R.E. Procedure for isolation of bacterial lipopolysaccharides from both smooth and rough Pseudomonas aeruginosa and Salmonella typhimurium strains. J. Bacteriol 1983, 155, 831–838. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dagorn, A.; Chapalain, A.; Mijouin, L.; Hillion, M.; Duclairoir-Poc, C.; Chevalier, S.; Taupin, L.; Orange, N.; Feuilloley, M.G.J. Effect of GABA, a Bacterial Metabolite, on Pseudomonas fluorescens Surface Properties and Cytotoxicity. Int. J. Mol. Sci. 2013, 14, 12186-12204. https://doi.org/10.3390/ijms140612186
Dagorn A, Chapalain A, Mijouin L, Hillion M, Duclairoir-Poc C, Chevalier S, Taupin L, Orange N, Feuilloley MGJ. Effect of GABA, a Bacterial Metabolite, on Pseudomonas fluorescens Surface Properties and Cytotoxicity. International Journal of Molecular Sciences. 2013; 14(6):12186-12204. https://doi.org/10.3390/ijms140612186
Chicago/Turabian StyleDagorn, Audrey, Annelise Chapalain, Lily Mijouin, Mélanie Hillion, Cécile Duclairoir-Poc, Sylvie Chevalier, Laure Taupin, Nicole Orange, and Marc G. J. Feuilloley. 2013. "Effect of GABA, a Bacterial Metabolite, on Pseudomonas fluorescens Surface Properties and Cytotoxicity" International Journal of Molecular Sciences 14, no. 6: 12186-12204. https://doi.org/10.3390/ijms140612186
APA StyleDagorn, A., Chapalain, A., Mijouin, L., Hillion, M., Duclairoir-Poc, C., Chevalier, S., Taupin, L., Orange, N., & Feuilloley, M. G. J. (2013). Effect of GABA, a Bacterial Metabolite, on Pseudomonas fluorescens Surface Properties and Cytotoxicity. International Journal of Molecular Sciences, 14(6), 12186-12204. https://doi.org/10.3390/ijms140612186