The Involvement of RhoA and Wnt-5a in the Tumorigenesis and Progression of Ovarian Epithelial Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Background
Methods
Results
Conclusions
1. Introduction
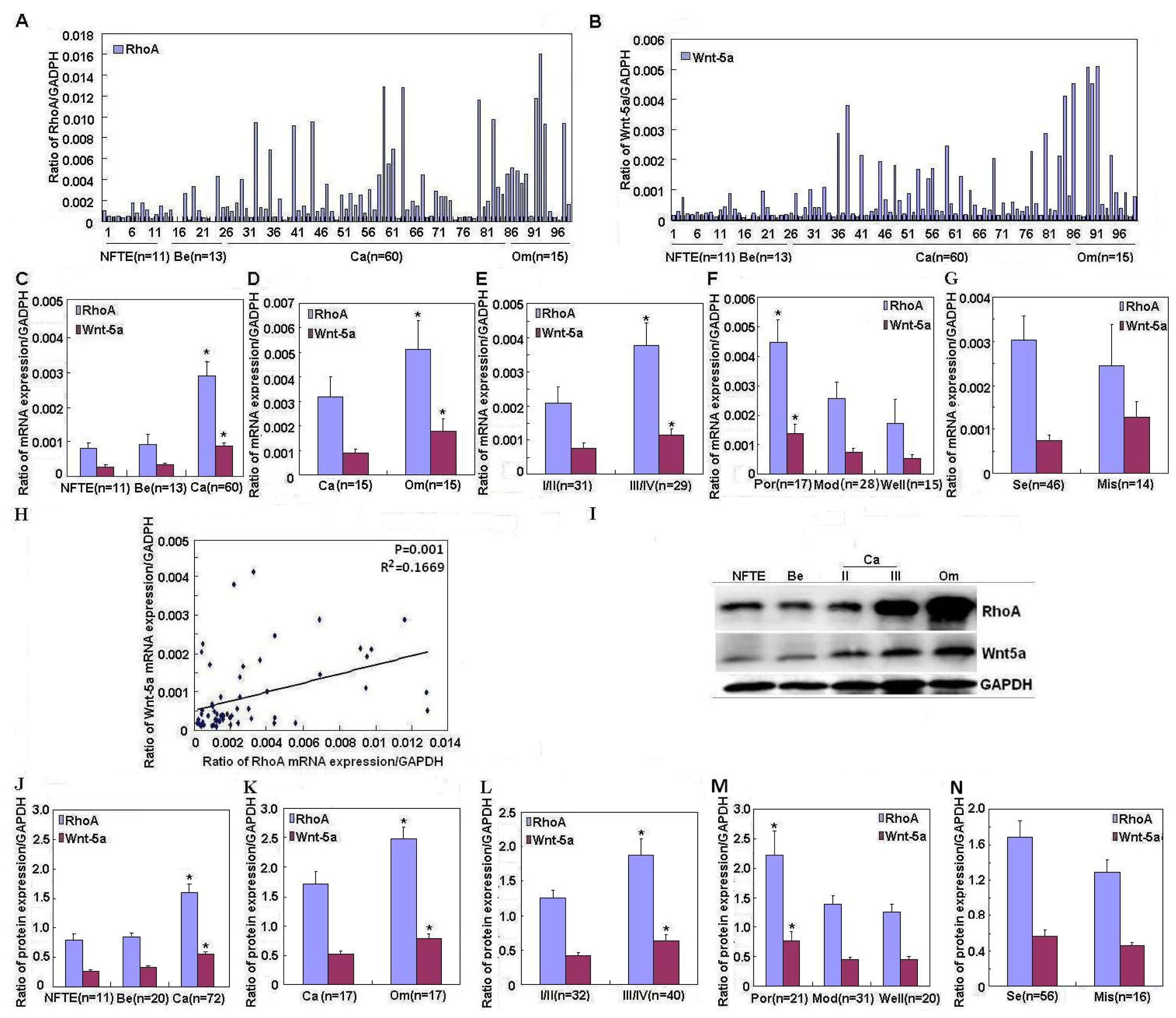
2. Results
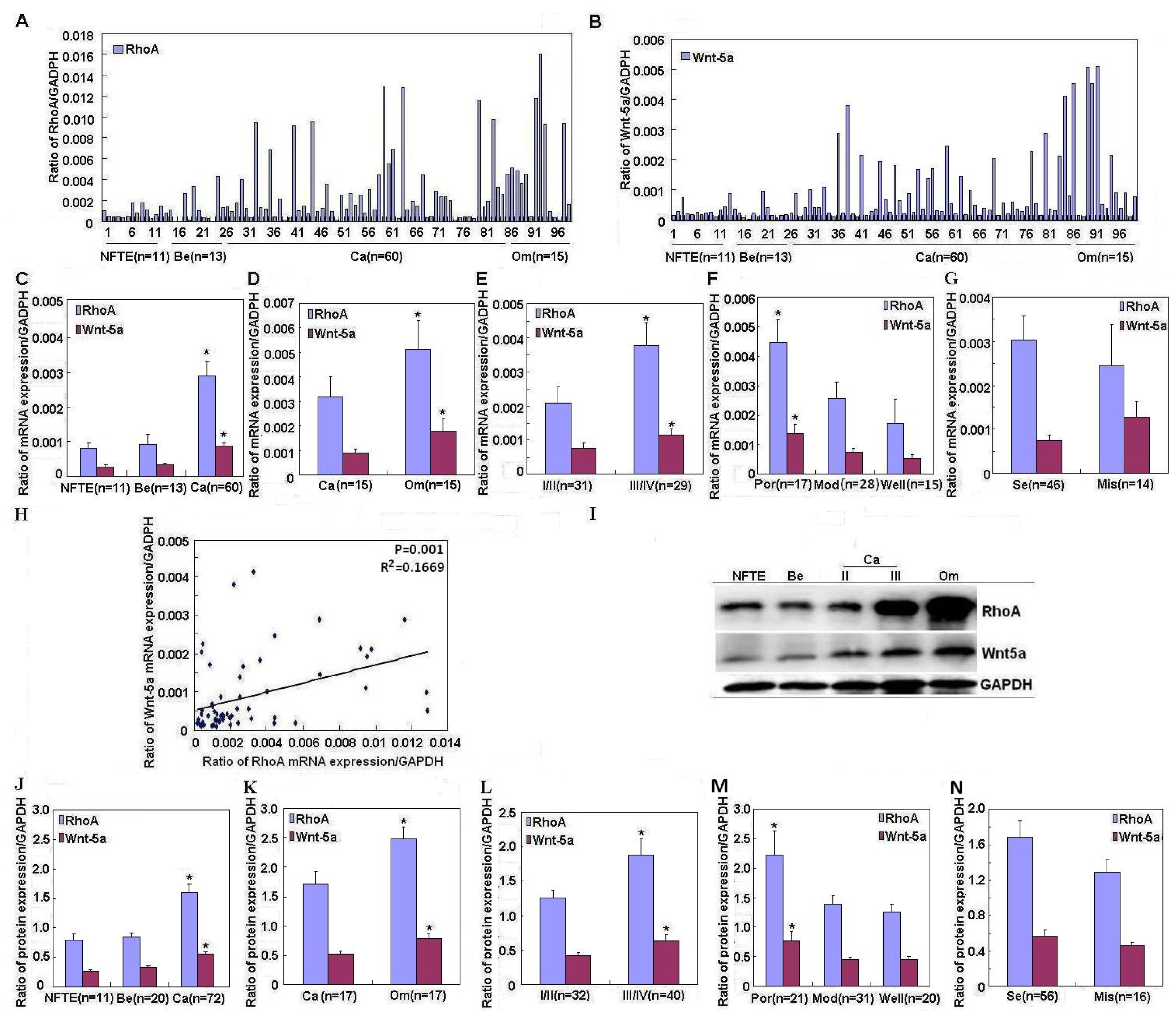
2.1. Correlation of RhoA and Wnt-5a mRNA and Protein Expression with Pathogenesis and Aggressiveness of Ovarian Carcinoma
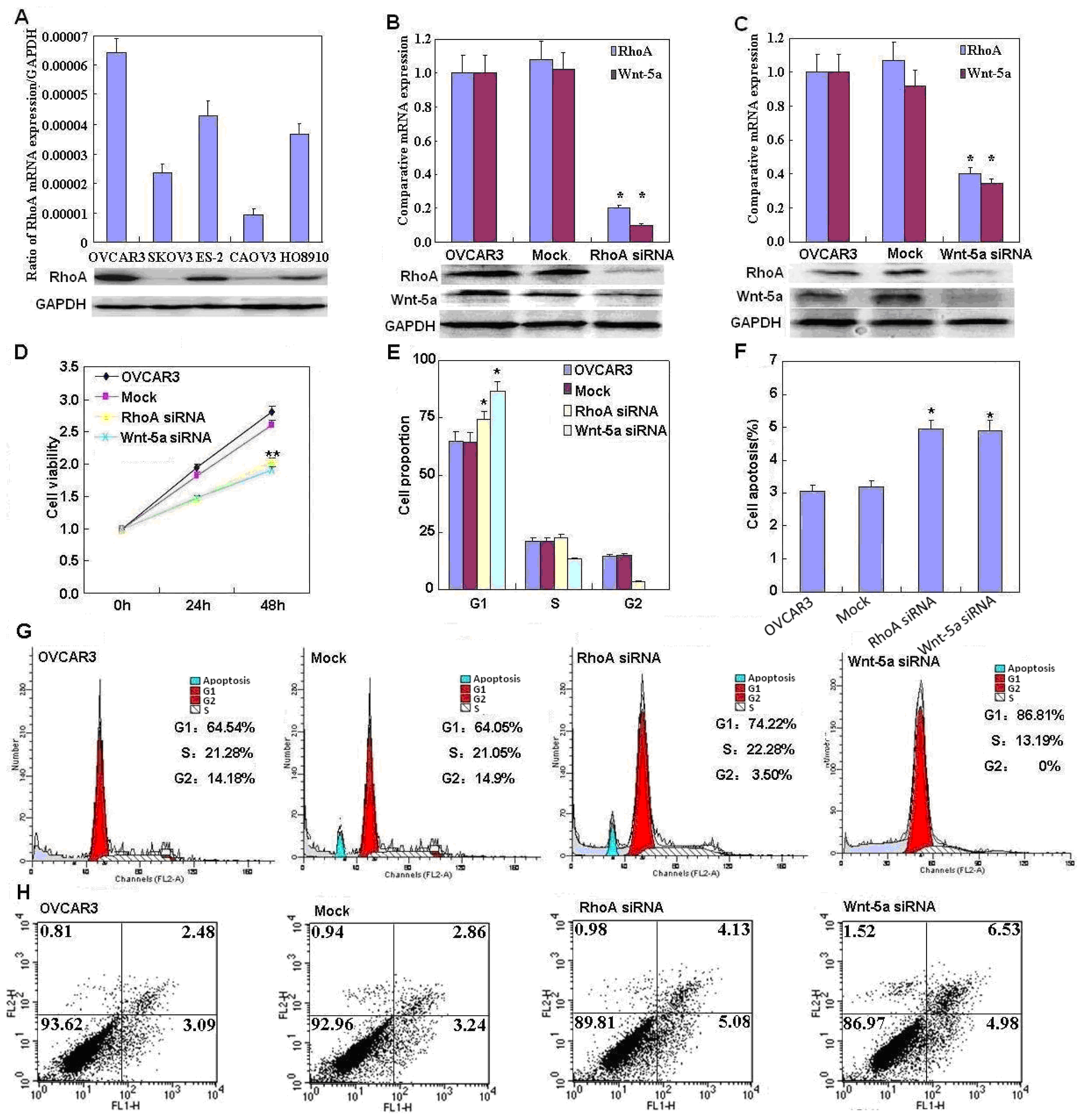
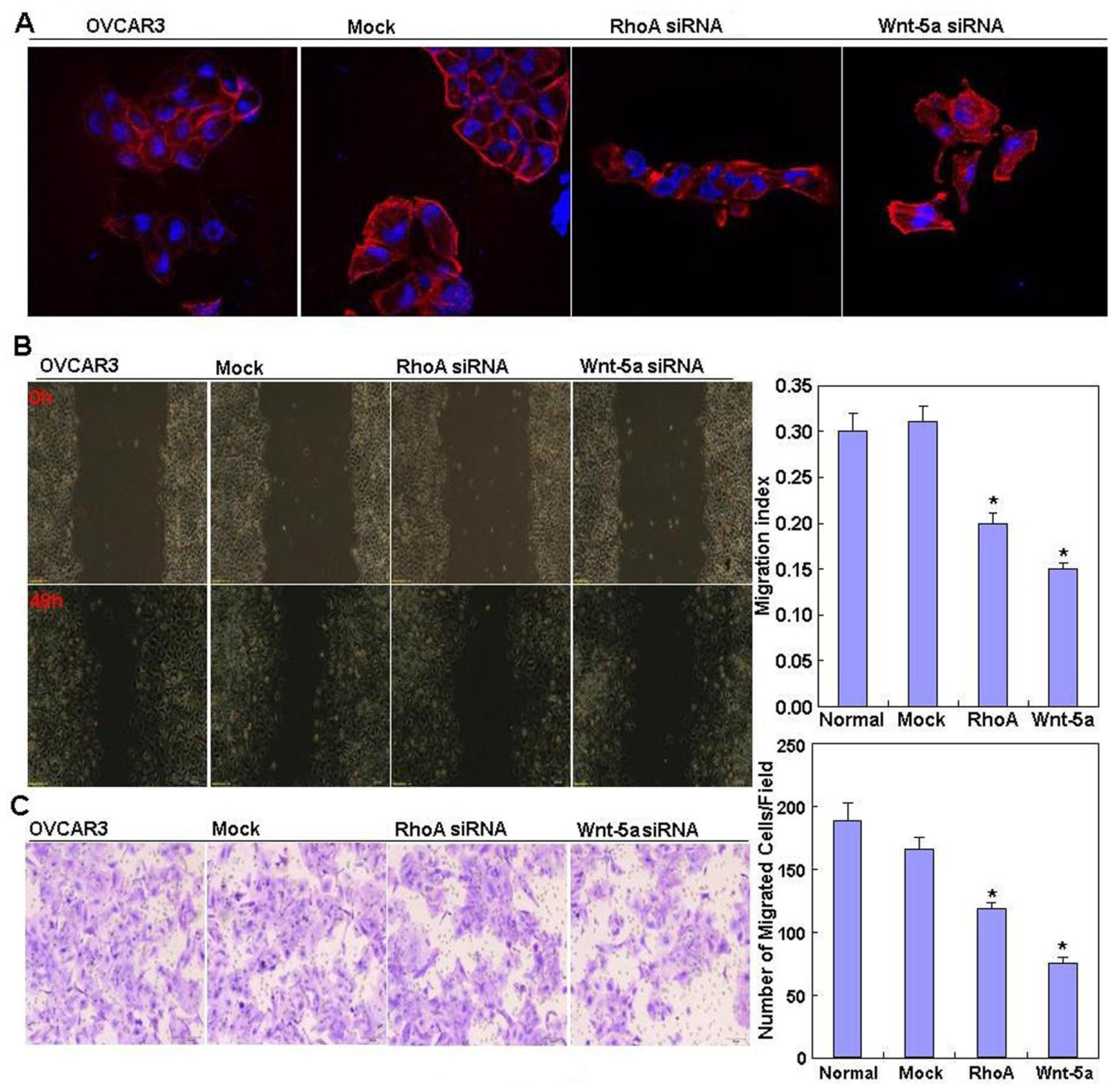
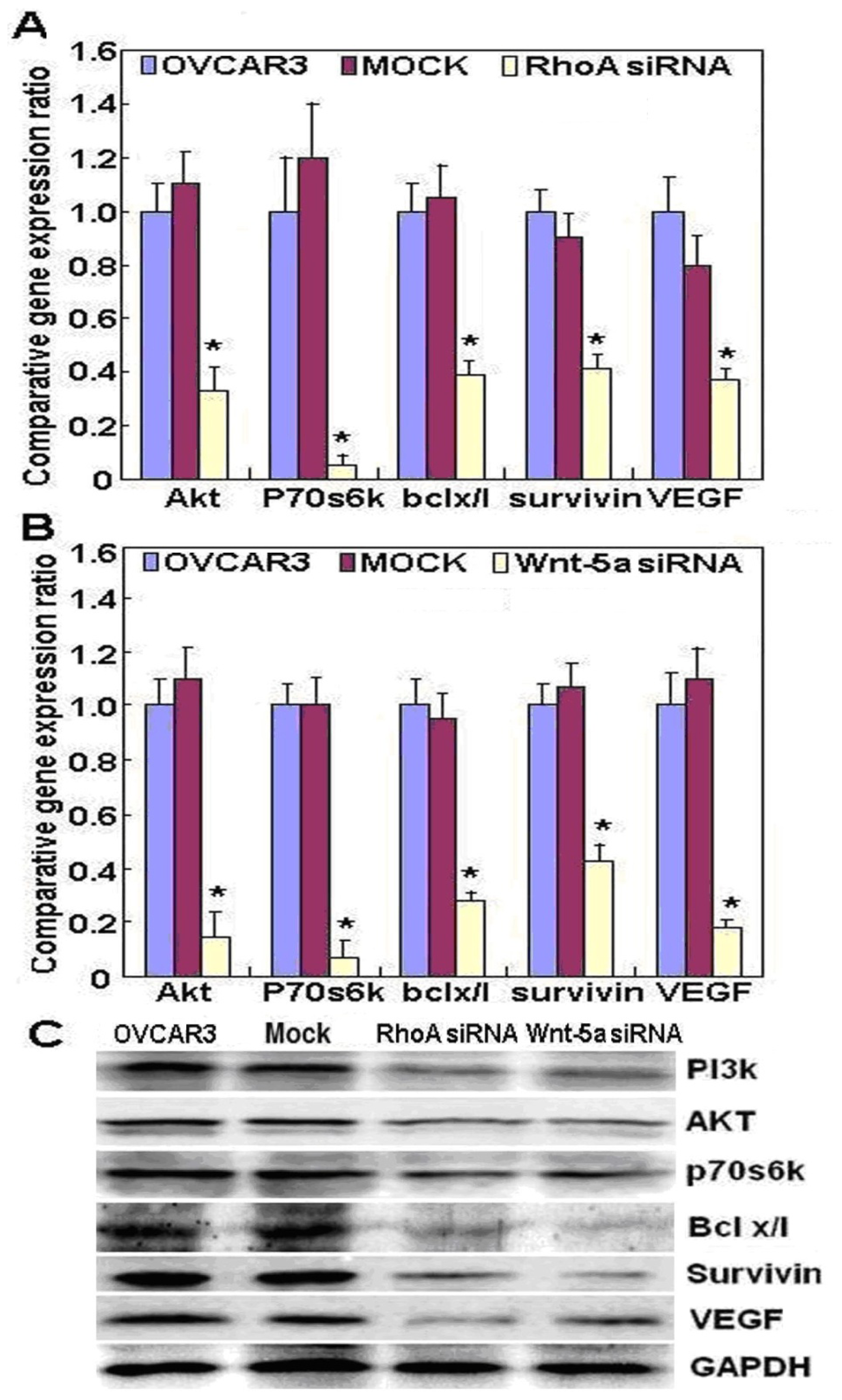
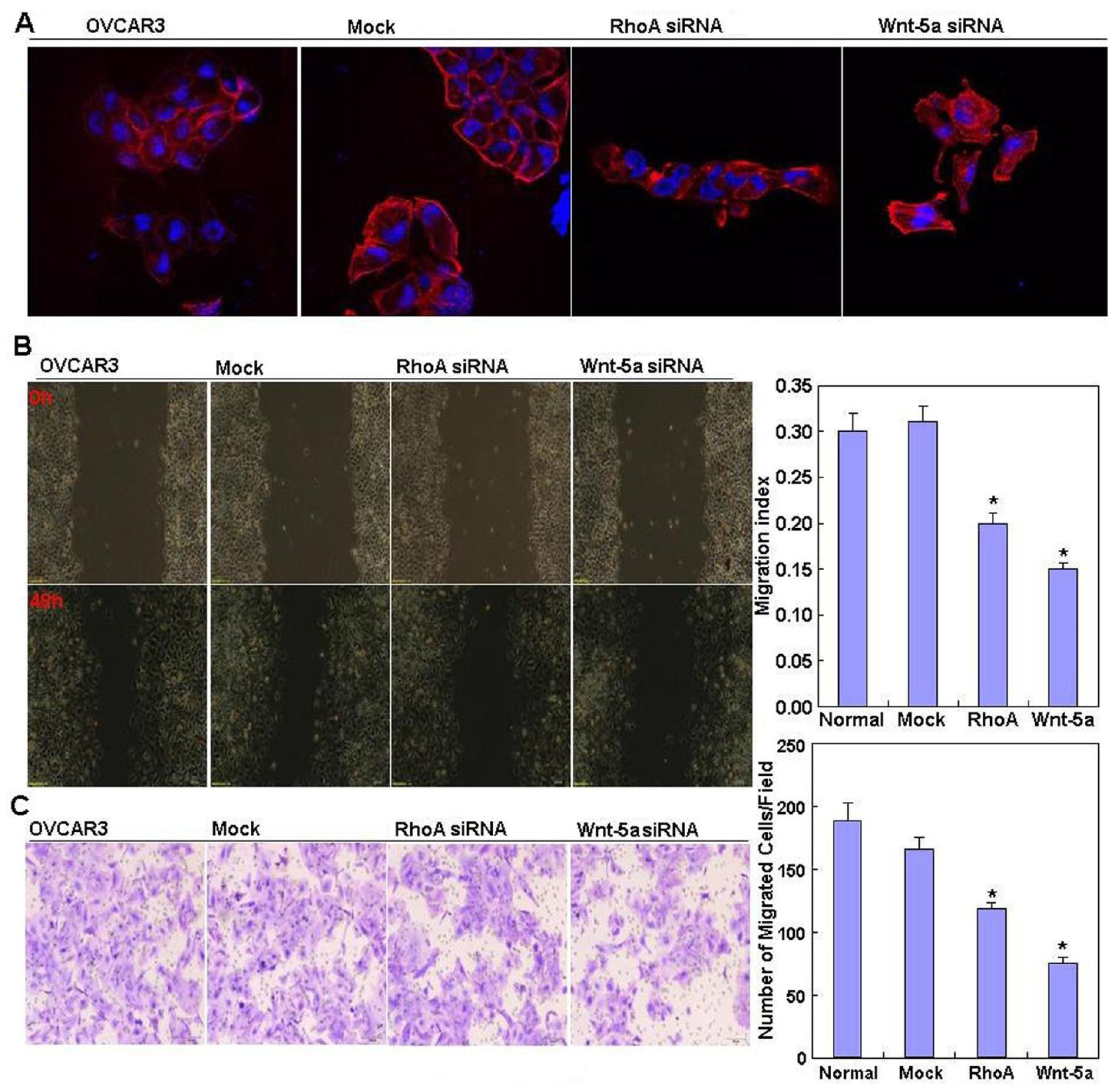
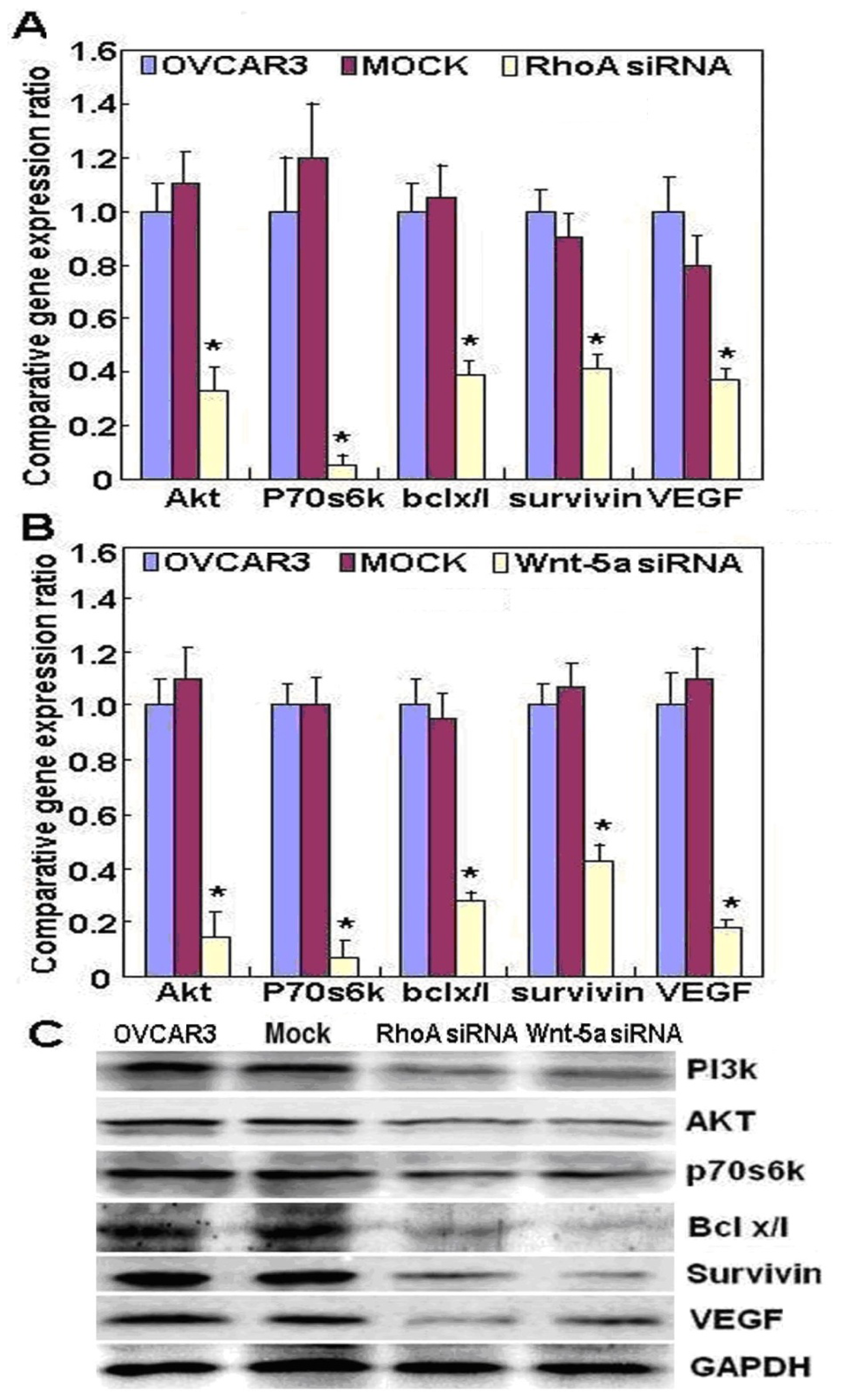
2.2. Effects of RhoA or Wnt-5a Knockdown on Phenotype and Expression of Wnt-5a Signaling-Related Molecules in Ovarian Carcinoma Cells
3. Discussion
4. Patients and Methods
4.1. Cell Culture and Transfection
4.2. Cell Viability Assay
4.3. Cell Cycle Analysis
4.4. Flow Cytometric Apoptosis Assay
4.5. Wound Healing Assay
4.6. Cell Invasion Assay
4.7. Immunofluorescent Staining
4.8. Ovarian Epithelium and Carcinoma Samples
4.9. Real-Time Reverse Transcriptase PCR
4.10. Western Blotting
4.11. Statistical Analysis
5. Conclusions
Supplementary Information
ijms-14-24187-s001.pdfAcknowledgments
Conflicts of Interest
References
- Piek, J.M.; Kenemans, P.; Zweemer, R.P.; van Diest, P.J.; Verheijen, R.H. Ovarian carcinogenesis, an alternative theory. Gynecol. Oncol 2007, 107, 355. [Google Scholar]
- Piek, J.M.; van Diest, P.J.; Verheijen, R.H. Ovarian carcinogenesis: An alternative hypothesis. Adv. Exp. Med. Biol 2008, 622, 79–87. [Google Scholar]
- Yu, L.-L.; Gu, J.-Y. Advances in the role of Rho sub-family in tumor invasion. Fudan Univ. J. Med. Sci 2010, 37, 617–619. [Google Scholar]
- Hall, A.; Rho, G.T. Pases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar]
- Der Mardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol 2005, 15, 356–363. [Google Scholar]
- Olofsson, B. Rho guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell. Signal 1999, 11, 545–554. [Google Scholar]
- Liu, J.; Zhang, Y.; Xu, R.; Du, J.; Hu, Z.; Yang, L.; Chen, Y.; Zhu, Y.; Gu, L. PI3K/Akt-dependent phosphorylation of GSK3β and activation of RhoA regulate Wnt5a-induced gastric cancer cell migration. Cell. Signal 2013, 25, 447–456. [Google Scholar]
- Zhu, Y.; Tian, Y.; Du, J.; Hu, Z.; Yang, L.; Liu, J.; Gu, L. Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5a-induced breast cancer cell migration. PLoS One 2012, 7, e37823. [Google Scholar]
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. Wnt5a: Its signalling, functions and implication in diseases. Acta Physiol. (Oxf.) 2012, 204, 17–33. [Google Scholar]
- Ellenbroek, S.I.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar]
- Horiuchi, A.; Imai, T.; Wang, C.; Ohira, S.; Feng, Y.; Nikaido, T.; Konishi, I. Up-regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab. Investig 2003, 83, 861–870. [Google Scholar]
- Peacock, J.G.; Miller, A.L.; Bradley, W.D.; Rodriguez, O.C.; Webb, D.J.; Koleske, A.J. The Abl-related gene tyrosine kinase acts through p190RhoGAP to inhibit actomyosin contractility and regulate focal adhesion dynamics upon adhesion to fibronectin. Mol. Biol. Cell 2007, 18, 3860–3872. [Google Scholar]
- Merajver, S.D.; Usmani, S.Z. Multifaceted role of Rho proteins in angiogenesis. J. Mammary Gland Biol. Neoplasia 2005, 10, 291–298. [Google Scholar]
- Ip, C.K.M.; Cheung, A.N.Y.; Ngan, H.Y.S.; Wong, A.S.T. p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene 2011, 30, 2420–2432. [Google Scholar] [Green Version]
- Denoyelle, C.; Albanese, P.; Uzan, G.; Hong, L.; Vannier, J.P.; Soria, J.; Soria, C. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell Signal 2003, 15, 327–338. [Google Scholar]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res 2006, 66, 10439–10448. [Google Scholar]
- Li, X.; Jia, Y.; Zhang, W.; Zhang, Y.; Li, B.; Huang, M.; Bao, F.; Wu, J.; Lou, Y. The Research progress about Wnt pathway on lung cancer stem cells. Chin. J. Lung Cancer 2011, 14, 695–698. [Google Scholar]
- Dejmek, J.; Dejmek, A.; Safholm, A.; Sjolander, A.; Andresson, T. Wnt-5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer Res 2005, 65, 9142–9146. [Google Scholar]
- Kremenevskaja, N.; von Wasielewski, R.; Rao, A.S.; Schöfl, C.; Andersson, T.; Brabant, G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005, 24, 2144–2154. [Google Scholar]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon: Functions and mechanisms of β-catenin-independent Wnt signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar]
- Kurayoshi, M.; Yamamoto, H.; Izumi, S.; Kikuchi, A. Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signaling. Biochem. J 2007, 402, 515–523. [Google Scholar]
- Kikuchi, A.; Yamamoto, H. Tumor formation due to abnormalities in the β-catenin-independent pathway of Wnt signaling. Cancer Sci 2008, 99, 202–208. [Google Scholar]
- Peng, C.; Zhang, X.; Yu, H.; Wu, D.; Zheng, J. Wnt5a as a predictor in poor clinical outcome of patients and a mediator in chemoresistance of ovarian cancer. Int. J. Gynecol. Cancer 2011, 21, 280–288. [Google Scholar]
- Badiglian, F.L.; Oshima, C.T.; de Oliveira Lima, F.; de Oliveira Costa, H.; de Sousa Damião, R.; Gomes, T.S.; Gonçalves, W.J. Canonical and noncanonical Wnt pathway: A comparison among normal ovary, benign ovarian tumor and ovarian cancer. Oncol. Rep 2009, 21, 313–320. [Google Scholar]
- Strutt, D.; Weber, U.; Mlodzik, M. The role of RhoA in tissue polarity and Frizzled signalling. Nature 1997, 387, 292–295. [Google Scholar]
- Qian, D.; Jones, C.; Rzadzinska, A.; Mark, S.; Zhang, X.; Steel, K.P.; Dai, X.; Chen, P. Wnt5a functions in planar cell polarity regulation in mice. Dev. Biol 2007, 306, 121–133. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, S.; Wang, J.; Gou, W.-F.; Xiu, Y.-L.; Zheng, H.-C.; Zong, Z.-H.; Takano, Y.; Zhao, Y. The Involvement of RhoA and Wnt-5a in the Tumorigenesis and Progression of Ovarian Epithelial Carcinoma. Int. J. Mol. Sci. 2013, 14, 24187-24199. https://doi.org/10.3390/ijms141224187
Chen S, Wang J, Gou W-F, Xiu Y-L, Zheng H-C, Zong Z-H, Takano Y, Zhao Y. The Involvement of RhoA and Wnt-5a in the Tumorigenesis and Progression of Ovarian Epithelial Carcinoma. International Journal of Molecular Sciences. 2013; 14(12):24187-24199. https://doi.org/10.3390/ijms141224187
Chicago/Turabian StyleChen, Shuo, Jun Wang, Wen-Feng Gou, Ying-Ling Xiu, Hua-Chuan Zheng, Zhi-Hong Zong, Yasuo Takano, and Yang Zhao. 2013. "The Involvement of RhoA and Wnt-5a in the Tumorigenesis and Progression of Ovarian Epithelial Carcinoma" International Journal of Molecular Sciences 14, no. 12: 24187-24199. https://doi.org/10.3390/ijms141224187
APA StyleChen, S., Wang, J., Gou, W.-F., Xiu, Y.-L., Zheng, H.-C., Zong, Z.-H., Takano, Y., & Zhao, Y. (2013). The Involvement of RhoA and Wnt-5a in the Tumorigenesis and Progression of Ovarian Epithelial Carcinoma. International Journal of Molecular Sciences, 14(12), 24187-24199. https://doi.org/10.3390/ijms141224187