The Effect on Proliferation and Differentiation of Cementoblast by Using Sclerostin as Inhibitor

Abstract
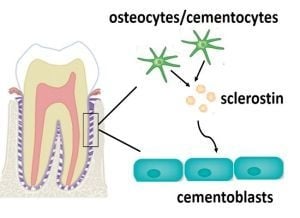
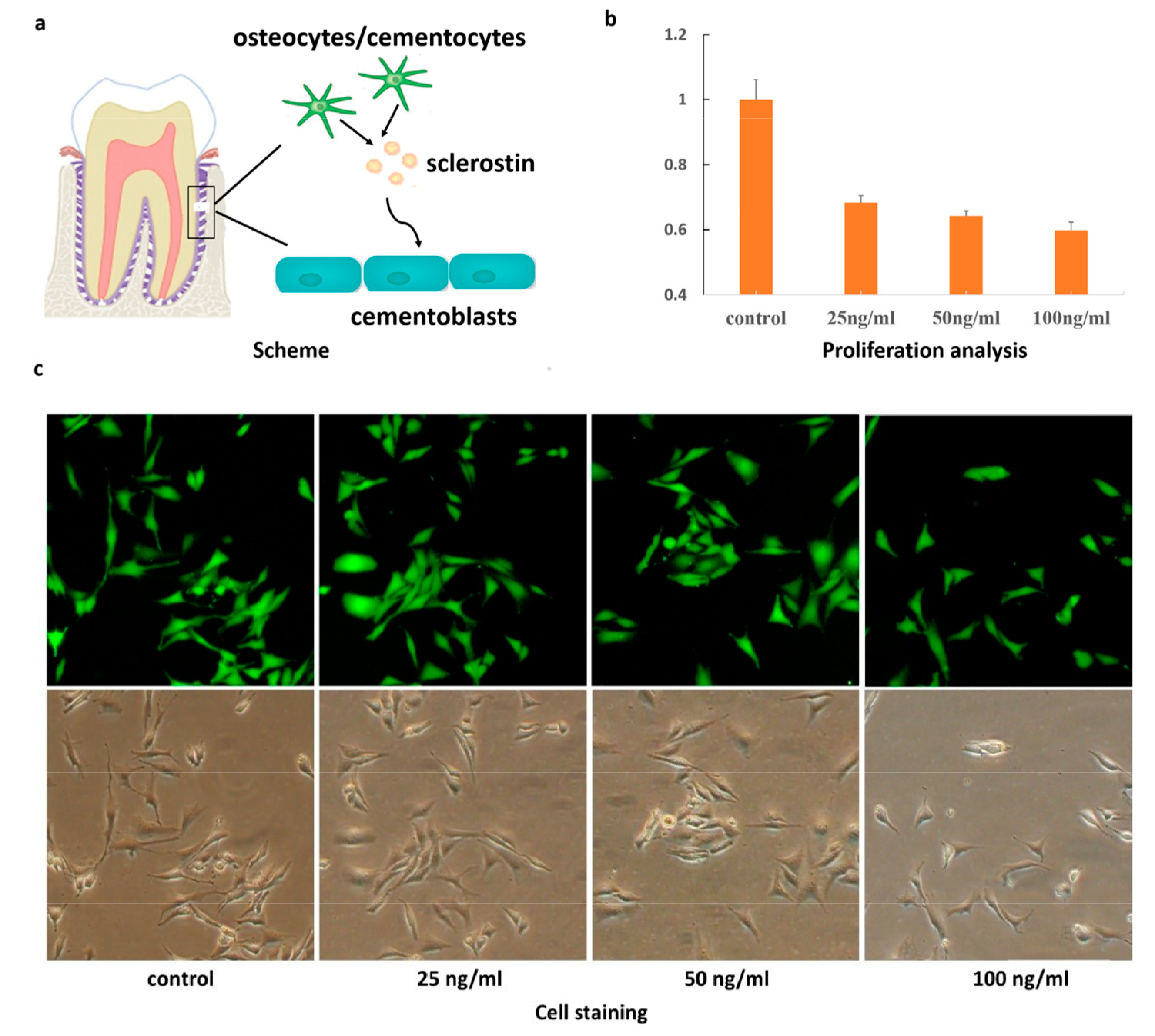
:
1. Introduction
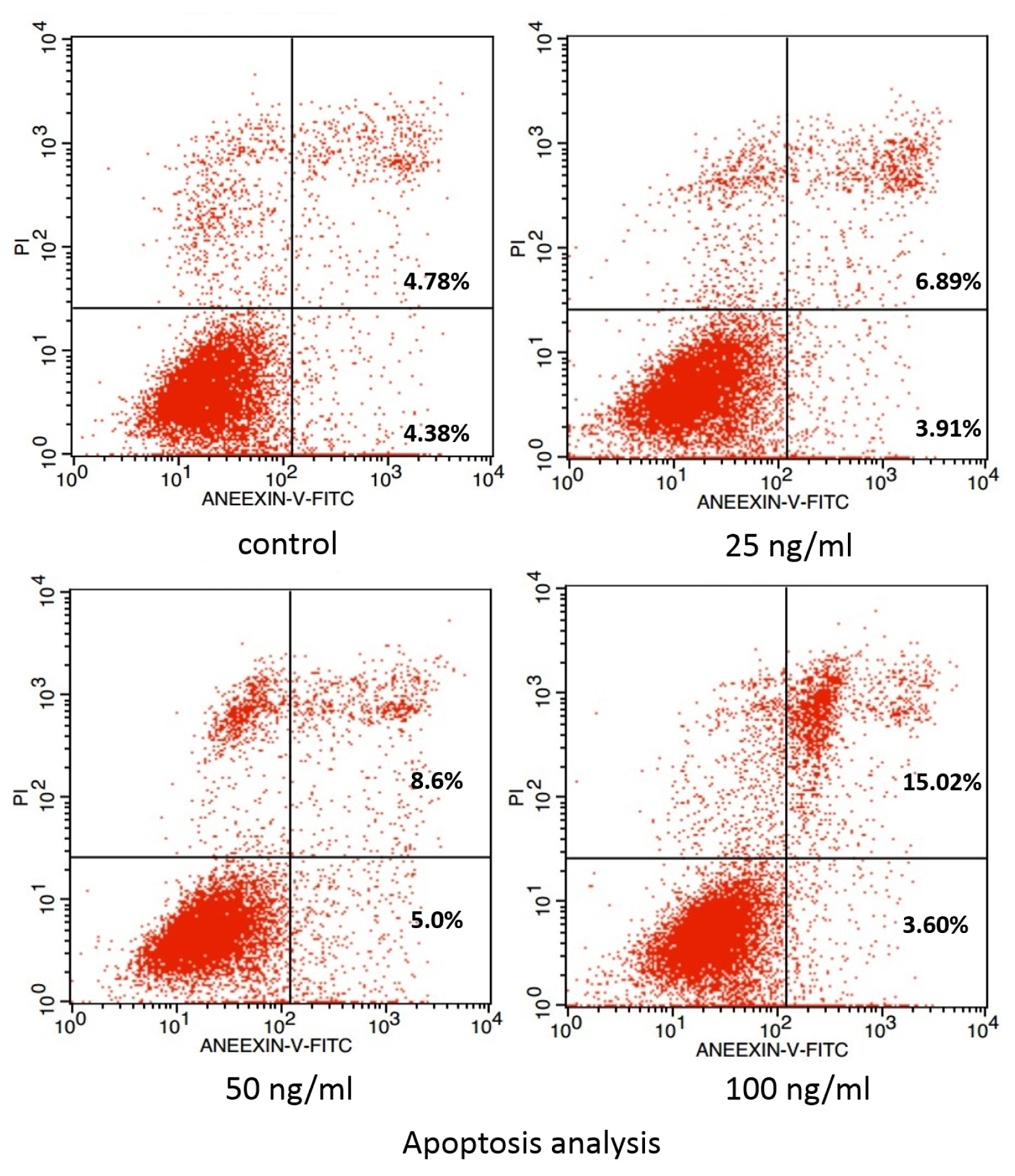
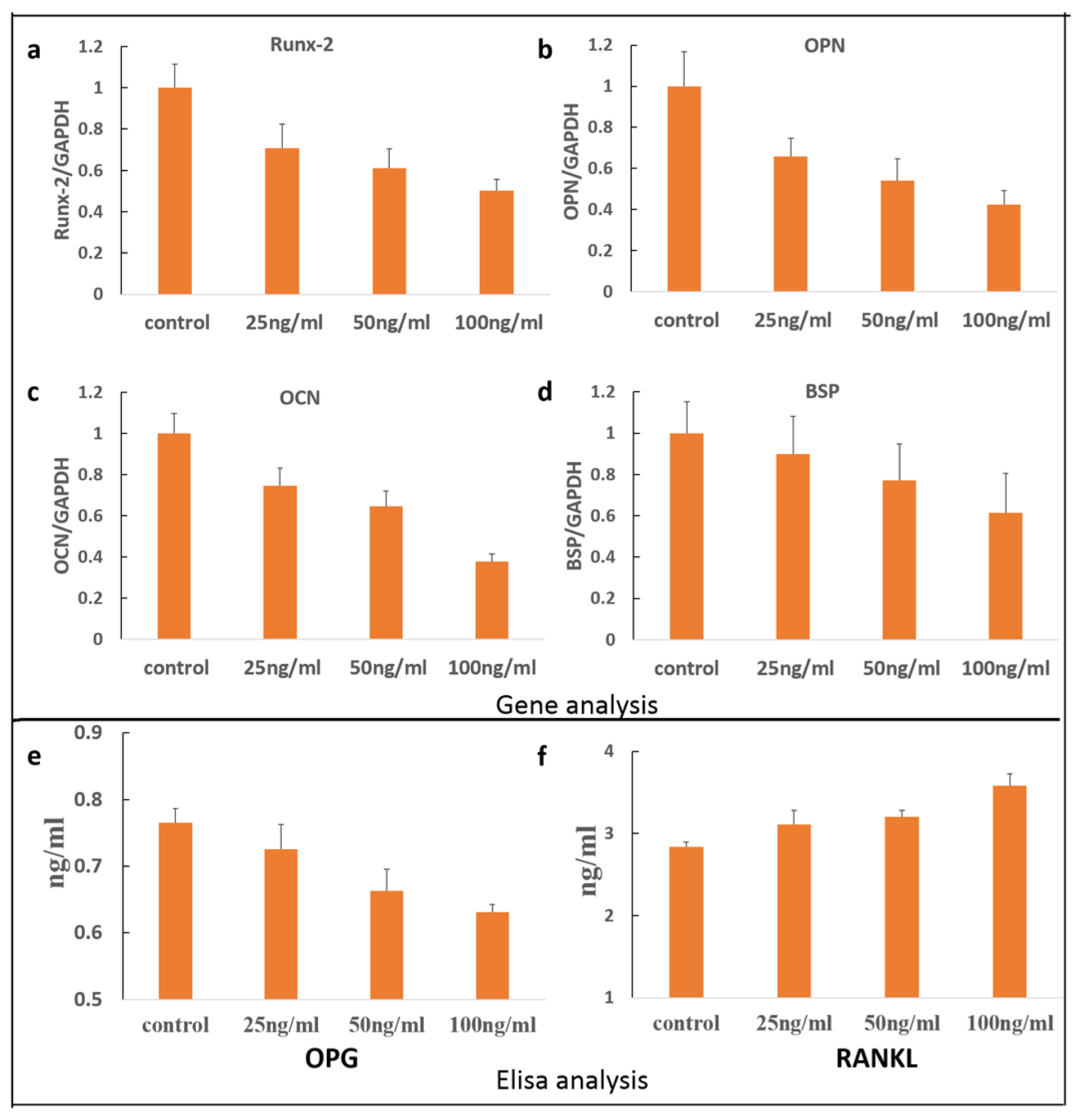
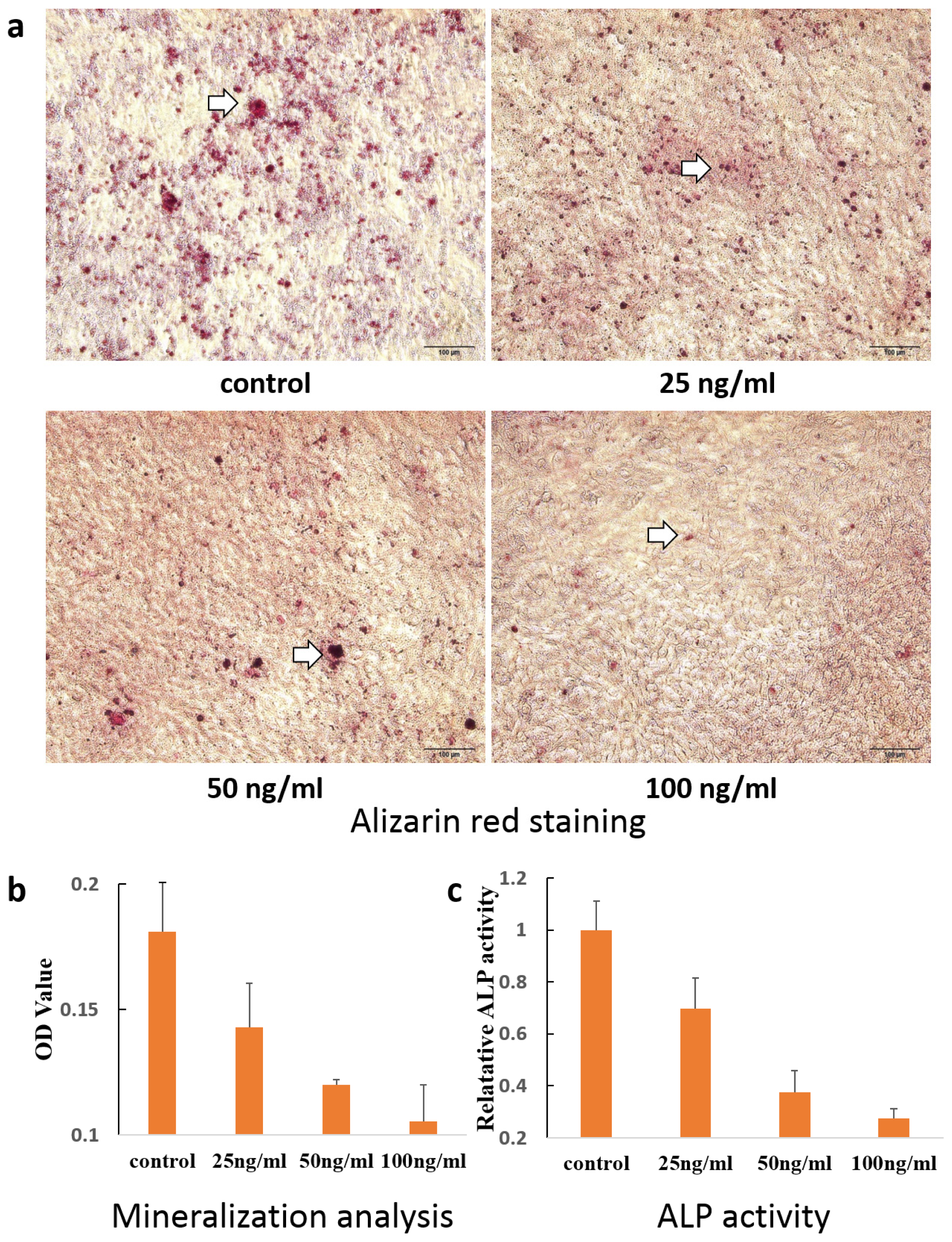
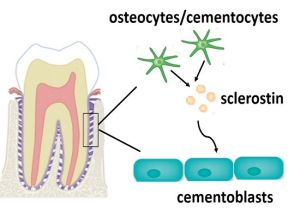
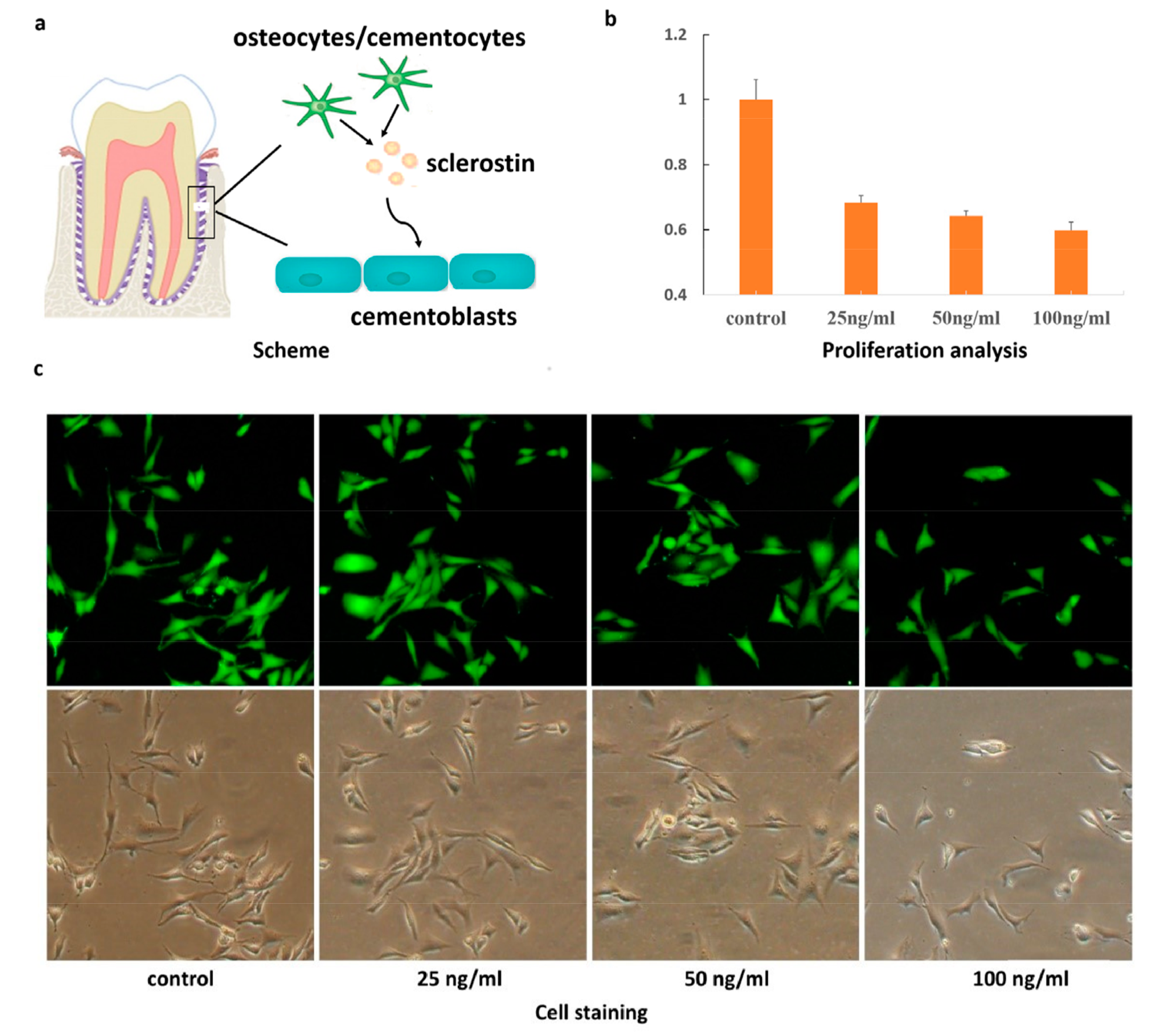
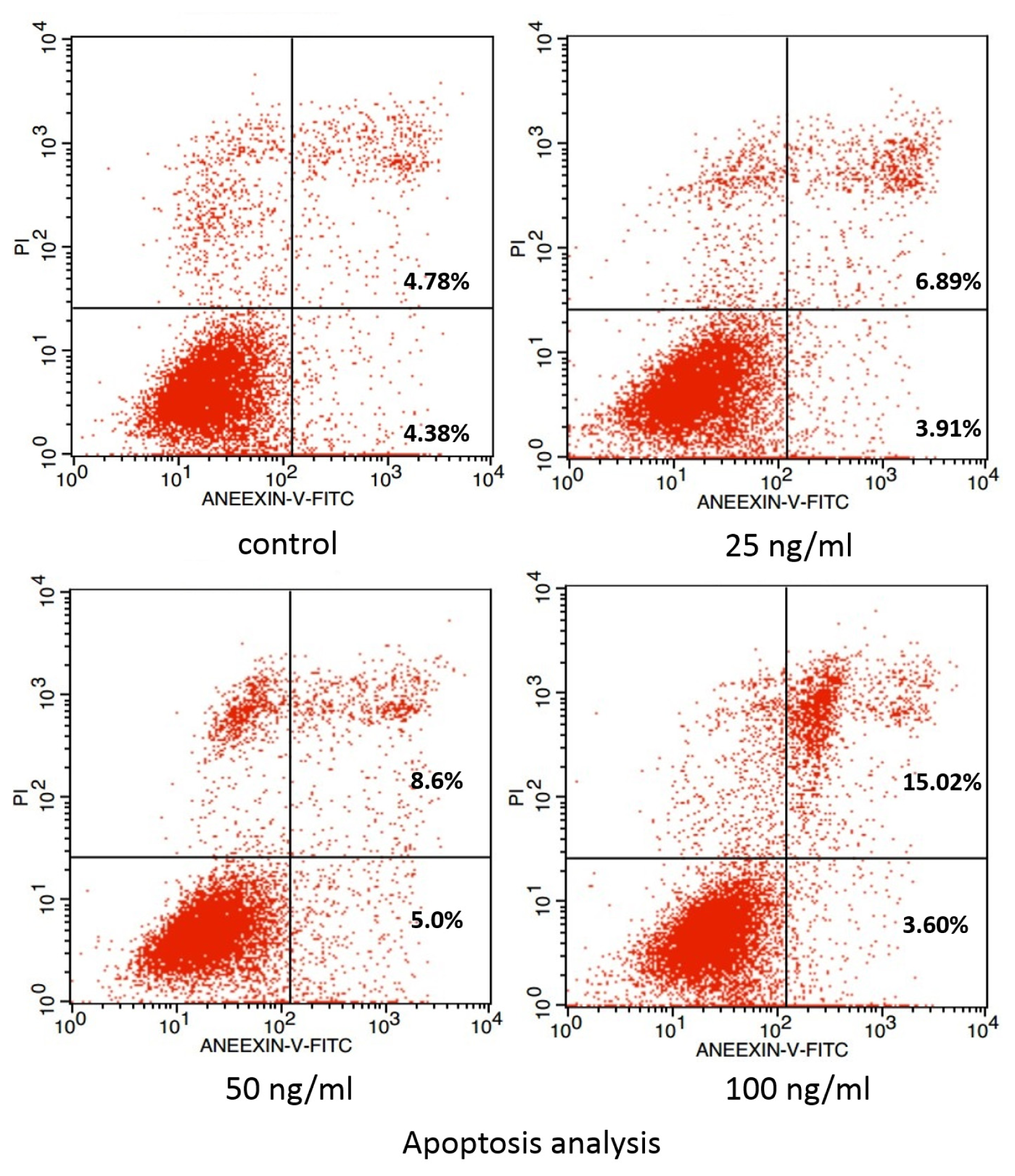
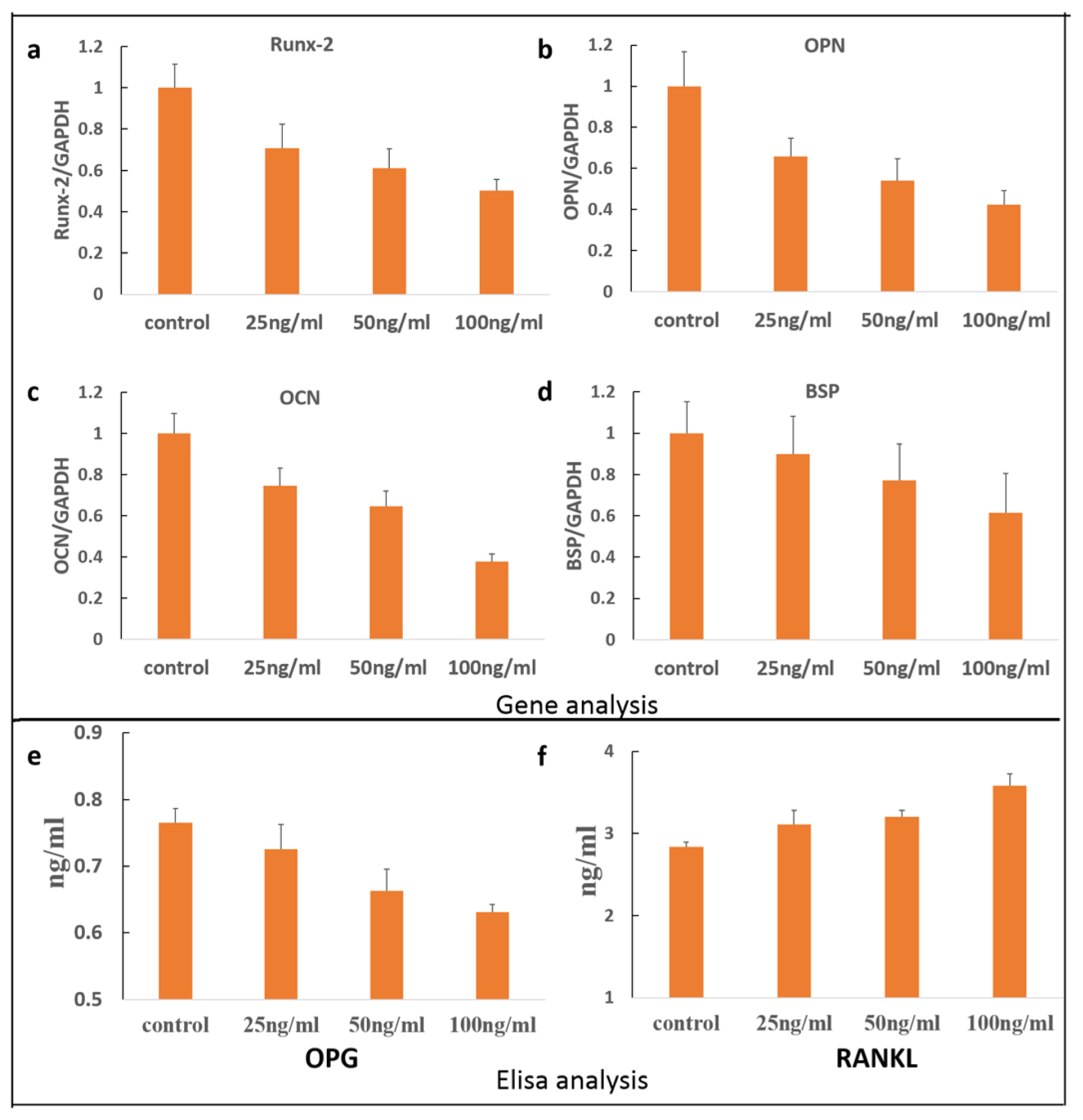
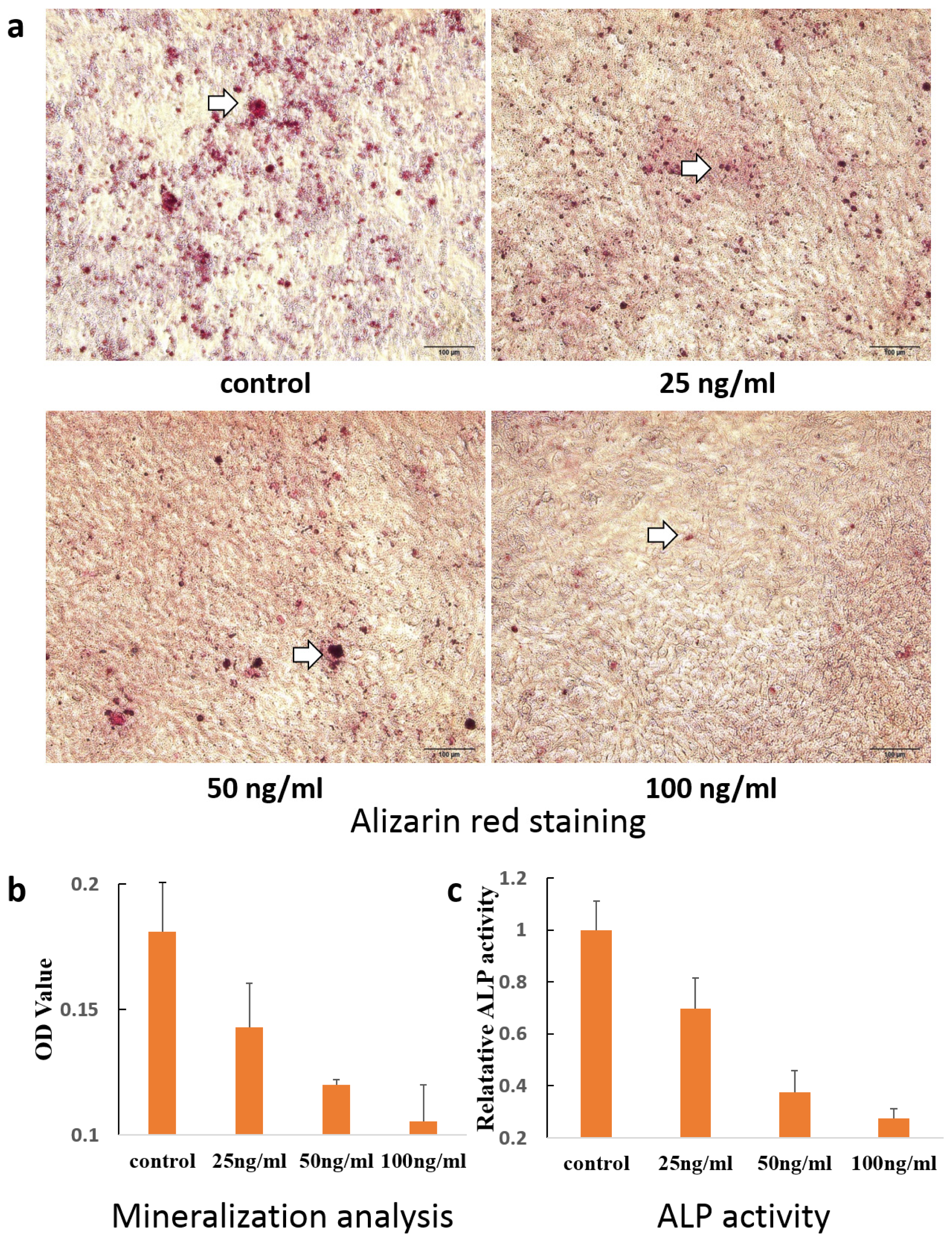
2. Results and Discussion
3. Experimental Section
3.1. Cell Culture
3.2. MTT Assay
3.3. Cell Staining
3.4. Examination of Apoptosis
3.5. Quantitative RT-PCR Analyses
3.6. OPG/RANKL Elisa Assay
3.7. Alizarin Red Staining
3.8. Alkaline Phosphatase Activity
3.9. Statistical Analysis
4. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| Runx-2 | CTTCATTCGCCTCACAAAC | CTAGCAGTGACGGTCT |
| BSP | GAGACGGCGATAGTTCC | AGTGCCGCTAACTCAA |
| OCN | TGAACAGACTCCGGCG | GATACCGTAGATGCGTTTG |
| OPN | TTTACCAGCCTGCACCC | CTAGCAGTGACGGTCT |
| GAPDH | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCTGTA |
Acknowledgments
Conflicts of Interest
References
- Bosshardt, D.D.; Selvig, K.A. Dental cementum: The dynamic tissue covering of the root. Periodontol. 2000 1997, 13, 41–75. [Google Scholar]
- Nanci, A.; Bosshardt, D.D. Structure of periodontal tissues in health and disease. Periodontol. 2000 2006, 40, 11–28. [Google Scholar]
- Bosshardt, D.D.; Zalzal, S.; McKee, M.D.; Nanci, A. Developmental appearance and distribution of bone sialoprotein and osteopontin in human and rat cementum. Anat. Rec 1998, 250, 13–33. [Google Scholar]
- Bosshardt, D.D. Are cementoblasts a subpopulation of osteoblasts or a unique phenotype? J. Dent. Res 2005, 84, 390–406. [Google Scholar]
- Ramanathan, C.; Hofman, Z. Root resorption in relation to orthodontic tooth movement. Acta Medica 2006, 49, 91–95. [Google Scholar]
- Weltman, B.; Vig, K.W.; Fields, H.W.; Shanker, S.; Kaizar, E.E. Root resorption associated with orthodontic tooth movement: A systematic review. Am. J. Orthod. Dentofac. Orthop 2010, 137, 462–476. [Google Scholar]
- Brezniak, N.; Wasserstein, A. Orthodontically induced inflammatory root resorption. Part ii: The clinical aspects. Angle Orthod 2002, 72, 180–184. [Google Scholar]
- Brezniak, N.; Wasserstein, A. Orthodontically induced inflammatory root resorption. Part i: The basic science aspects. Angle Orthod 2002, 72, 175–179. [Google Scholar]
- Tyrovola, J.B.; Perrea, D.; Halazonetis, D.J.; Dontas, I.; Vlachos, I.S.; Makou, M. Relation of soluble rankl and osteoprotegerin levels in blood and gingival crevicular fluid to the degree of root resorption after orthodontic tooth movement. J. Oral Sci 2010, 52, 299–311. [Google Scholar]
- Jager, A.; Kunert, D.; Friesen, T.; Zhang, D.; Lossdorfer, S.; Gotz, W. Cellular and extracellular factors in early root resorption repair in the rat. Eur. J. Orthod 2008, 30, 336–345. [Google Scholar]
- Owman-Moll, P.; Kurol, J.; Lundgren, D. Repair of orthodontically induced root resorption in adolescents. Angle Orthod 1995, 65, 403–408, discussion 409–410. [Google Scholar]
- Tian, Y.; Huang, L.; Guo, Y.W.; Cao, L.; Wang, Y.T.; Xu, H.; Bai, D. Expression of alkaline phosphatase in immortalized murine cementoblasts in response to compression-force. Saudi Med. J 2011, 32, 1235–1240. [Google Scholar]
- Kim, H.S.; Lee, D.S.; Lee, J.H.; Kang, M.S.; Lee, N.R.; Kim, H.J.; Ko, J.S.; Cho, M.I.; Park, J.C. The effect of odontoblast conditioned media and dentin non-collagenous proteins on the differentiation and mineralization of cementoblasts in vitro. Arch. Oral Biol. 2009, 54, 71–79. [Google Scholar]
- Rego, E.B.; Inubushi, T.; Kawazoe, A.; Miyauchi, M.; Tanaka, E.; Takata, T.; Tanne, K. Effect of pge(2) induced by compressive and tensile stresses on cementoblast differentiation in vitro. Arch. Oral Biol. 2011, 56, 1238–1246. [Google Scholar]
- Hakki, S.S.; Foster, B.L.; Nagatomo, K.J.; Bozkurt, S.B.; Hakki, E.E.; Somerman, M.J.; Nohutcu, R.M. Bone morphogenetic protein-7 enhances cementoblast function in vitro. J. Periodontol. 2010, 81, 1663–1674. [Google Scholar]
- Bozic, D.; Grgurevic, L.; Erjavec, I.; Brkljacic, J.; Orlic, I.; Razdorov, G.; Grgurevic, I.; Vukicevic, S.; Plancak, D. The proteome and gene expression profile of cementoblastic cells treated by bone morphogenetic protein-7 in vitro. J. Clin. Periodontol. 2012, 39, 80–90. [Google Scholar]
- Oka, H.; Miyauchi, M.; Sakamoto, K.; Kitagawa, M.; Noguchi, K.; Somerman, M.J.; Takata, T. Prostaglandin e2 inhibits mineralization and enhances matrix metalloproteinase-13 in mature cementoblasts mainly via the ep4 pathway. Arch. Oral Biol 2008, 53, 243–249. [Google Scholar]
- Zhang, H.; Tompkins, K.; Garrigues, J.; Snead, M.L.; Gibson, C.W.; Somerman, M.J. Full length amelogenin binds to cell surface lamp-1 on tooth root/periodontium associated cells. Arch. Oral Biol 2010, 55, 417–425. [Google Scholar]
- Moester, M.J.; Papapoulos, S.E.; Lowik, C.W.; van Bezooijen, R.L. Sclerostin: Current knowledge and future perspectives. Calcif. Tissue Int 2010, 87, 99–107. [Google Scholar]
- Atkins, G.J.; Findlay, D.M. Osteocyte regulation of bone mineral: A little give and take. Osteoporos. Int 2012, 23, 2067–2079. [Google Scholar]
- Van Bezooijen, R.L.; ten Dijke, P.; Papapoulos, S.E.; Lowik, C.W. Sost/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev 2005, 16, 319–327. [Google Scholar]
- Morse, L.R.; Sudhakar, S.; Lazzari, A.A.; Tun, C.; Garshick, E.; Zafonte, R.; Battaglino, R.A. Sclerostin: A candidate biomarker of sci-induced osteoporosis. Osteoporos. Int 2013, 24, 961–968. [Google Scholar]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res 2008, 23, 860–869. [Google Scholar]
- Canalis, E. Wnt signalling in osteoporosis: Mechanisms and novel therapeutic approaches. Nat. Rev. Endocrinol 2013, 9, 575–583. [Google Scholar]
- O’Brien, C.A.; Nakashima, T.; Takayanagi, H. Osteocyte control of osteoclastogenesis. Bone 2013, 54, 258–263. [Google Scholar]
- Galli, C.; Passeri, G.; Macaluso, G.M. Osteocytes and wnt: The mechanical control of bone formation. J. Dent. Res 2010, 89, 331–343. [Google Scholar]
- Burgers, T.A.; Williams, B.O. Regulation of wnt/β-catenin signaling within and from osteocytes. Bone 2013, 54, 244–249. [Google Scholar]
- Zhao, L.; Shim, J.W.; Dodge, T.R.; Robling, A.G.; Yokota, H. Inactivation of lrp5 in osteocytes reduces young’s modulus and responsiveness to the mechanical loading. Bone 2013, 54, 35–43. [Google Scholar]
- Taut, A.D.; Jin, Q.; Chung, J.H.; Galindo-Moreno, P.; Yi, E.S.; Sugai, J.V.; Ke, H.Z.; Liu, M.; Giannobile, W.V. Sclerostin antibody stimulates bone regeneration following experimental periodontitis. J. Bone Miner. Res 2013. [Google Scholar] [CrossRef]
- McColm, J.; Hu, L.; Womack, T.; Tang, C.C.; Chiang, A.Y. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J. Bone Miner. Res 2013. [Google Scholar] [CrossRef]
- Jager, A.; Gotz, W.; Lossdorfer, S.; Rath-Deschner, B. Localization of sost/sclerostin in cementocytes in vivo and in mineralizing periodontal ligament cells in vitro. J. Periodontal Res. 2010, 45, 246–254. [Google Scholar]
- Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Turcott, E.; Skonier, J.E.; Winkler, D.G.; Latham, J.A. Sclerostin promotes the apoptosis of human osteoblastic cells: A novel regulation of bone formation. Bone 2004, 35, 828–835. [Google Scholar]
- Nemoto, E.; Koshikawa, Y.; Kanaya, S.; Tsuchiya, M.; Tamura, M.; Somerman, M.J.; Shimauchi, H. Wnt signaling inhibits cementoblast differentiation and promotes proliferation. Bone 2009, 44, 805–812. [Google Scholar]
- Hakki, S.S.; Bozkurt, S.B.; Hakki, E.E.; Belli, S. Effects of mineral trioxide aggregate on cell survival, gene expression associated with mineralized tissues, and biomineralization of cementoblasts. J. Endod 2009, 35, 513–519. [Google Scholar]
- Hakki, S.S.; Nohutcu, R.M.; Hakki, E.E.; Berry, J.E.; Akkaya, M.S.; Somerman, M.J. Dexamethasone and basic-fibroblast growth factor regulate markers of mineralization in cementoblasts in vitro. J. Periodontol. 2005, 76, 1550–1558. [Google Scholar]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a rankl-dependent pathway. PLoS One 2011, 6, e25900. [Google Scholar]
- Atkins, G.J.; Rowe, P.S.; Lim, H.P.; Welldon, K.J.; Ormsby, R.; Wijenayaka, A.R.; Zelenchuk, L.; Evdokiou, A.; Findlay, D.M. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a mepe-asarm-dependent mechanism. J. Bone Miner. Res 2011, 26, 1425–1436. [Google Scholar]
- Brunkow, M.E.; Gardner, J.C.; van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the sost gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet 2001, 68, 577–589. [Google Scholar]
- Van Bezooijen, R.L.; Papapoulos, S.E.; Lowik, C.W. Bone morphogenetic proteins and their antagonists: The sclerostin paradigm. J. Endocrinol. Investig 2005, 28, 15–17. [Google Scholar]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel bmp antagonist. EMBO J 2003, 22, 6267–6276. [Google Scholar]
- Kubota, T.; Michigami, T.; Ozono, K. Wnt signaling in bone. Clin. Pediatr. Endocrinol 2010, 19, 49–56. [Google Scholar]
- Lehnen, S.D.; Gotz, W.; Baxmann, M.; Jager, A. Immunohistochemical evidence for sclerostin during cementogenesis in mice. Ann. Anat 2012, 194, 415–421. [Google Scholar]
- Van Bezooijen, R.L.; Bronckers, A.L.; Gortzak, R.A.; Hogendoorn, P.C.; van der Wee-Pals, L.; Balemans, W.; Oostenbroek, H.J.; van Hul, W.; Hamersma, H.; Dikkers, F.G.; et al. Sclerostin in mineralized matrices and van buchem disease. J. Dent. Res 2009, 88, 569–574. [Google Scholar]
- Bustin, S. Transparency of reporting in molecular diagnostics. Int. J. Mol. Sci 2013, 14, 15878–15884. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bao, X.; Liu, Y.; Han, G.; Zuo, Z.; Hu, M. The Effect on Proliferation and Differentiation of Cementoblast by Using Sclerostin as Inhibitor. Int. J. Mol. Sci. 2013, 14, 21140-21152. https://doi.org/10.3390/ijms141021140
Bao X, Liu Y, Han G, Zuo Z, Hu M. The Effect on Proliferation and Differentiation of Cementoblast by Using Sclerostin as Inhibitor. International Journal of Molecular Sciences. 2013; 14(10):21140-21152. https://doi.org/10.3390/ijms141021140
Chicago/Turabian StyleBao, Xingfu, Yuyan Liu, Guanghong Han, Zhigang Zuo, and Min Hu. 2013. "The Effect on Proliferation and Differentiation of Cementoblast by Using Sclerostin as Inhibitor" International Journal of Molecular Sciences 14, no. 10: 21140-21152. https://doi.org/10.3390/ijms141021140
APA StyleBao, X., Liu, Y., Han, G., Zuo, Z., & Hu, M. (2013). The Effect on Proliferation and Differentiation of Cementoblast by Using Sclerostin as Inhibitor. International Journal of Molecular Sciences, 14(10), 21140-21152. https://doi.org/10.3390/ijms141021140