Role of the Blood-Brain Barrier in the Formation of Brain Metastases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
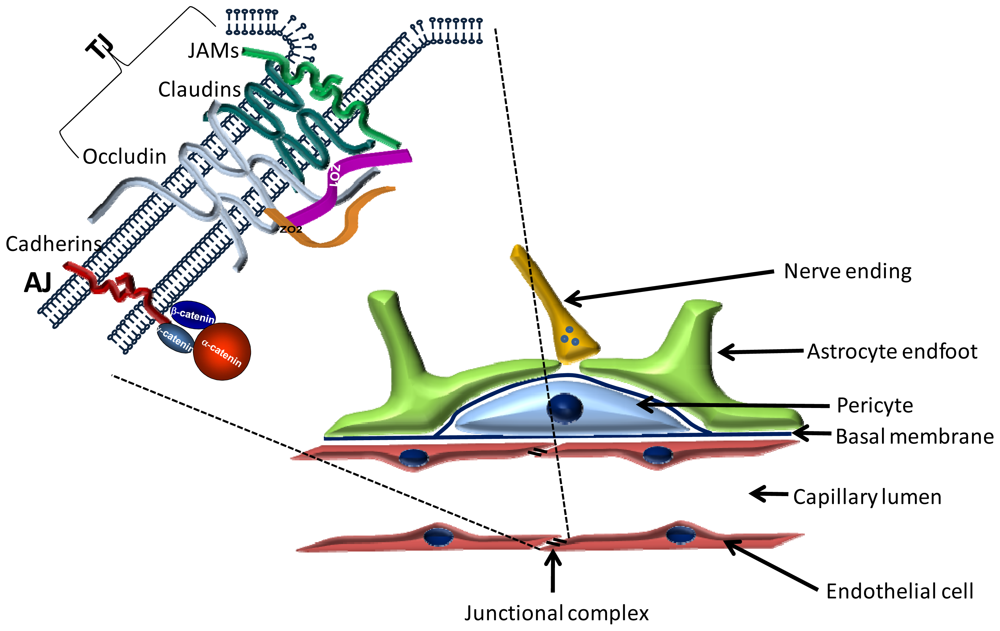
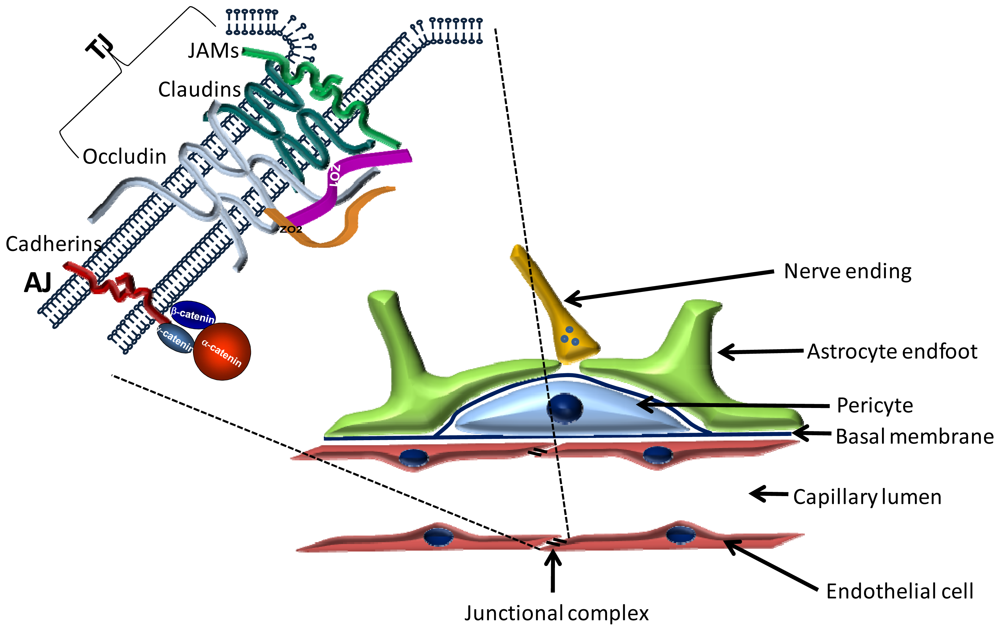
2. The Blood-Brain Barrier
2.1. Cellular Structure of the BBB
2.1.1. Endothelial Cells
2.1.2. Pericytes
2.1.3. Astrocytes
2.1.4. Other Cells of the Neurovascular Unit
2.1.5. The Basement Membrane
2.2. Molecular Structure of the BBB
2.2.1. Transmembrane TJ Proteins
2.2.1.1. Occludin
2.2.1.2. Claudins
2.2.1.3. Immunglobulin-like Molecules
2.2.1.4. Other Transmembrane Proteins of the TJs
2.2.2. Peripheral Proteins of Tight Junctions
2.2.2.1. PDZ Domain Containing Proteins
2.2.2.1.1. Zonula Occludens (ZO) Proteins
2.2.2.1.2. Other Proteins Containing PDZ Domain
2.2.2.2. Plaque Proteins without PDZ Domain
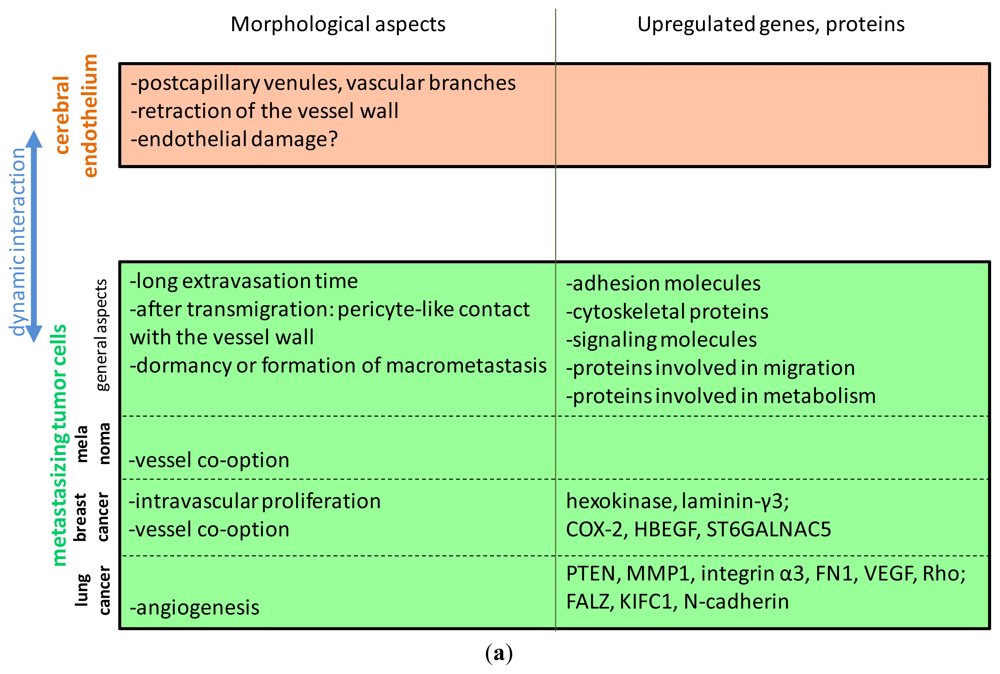
3. Mechanisms of Interaction of Tumor Cells with Brain Endothelial Cells
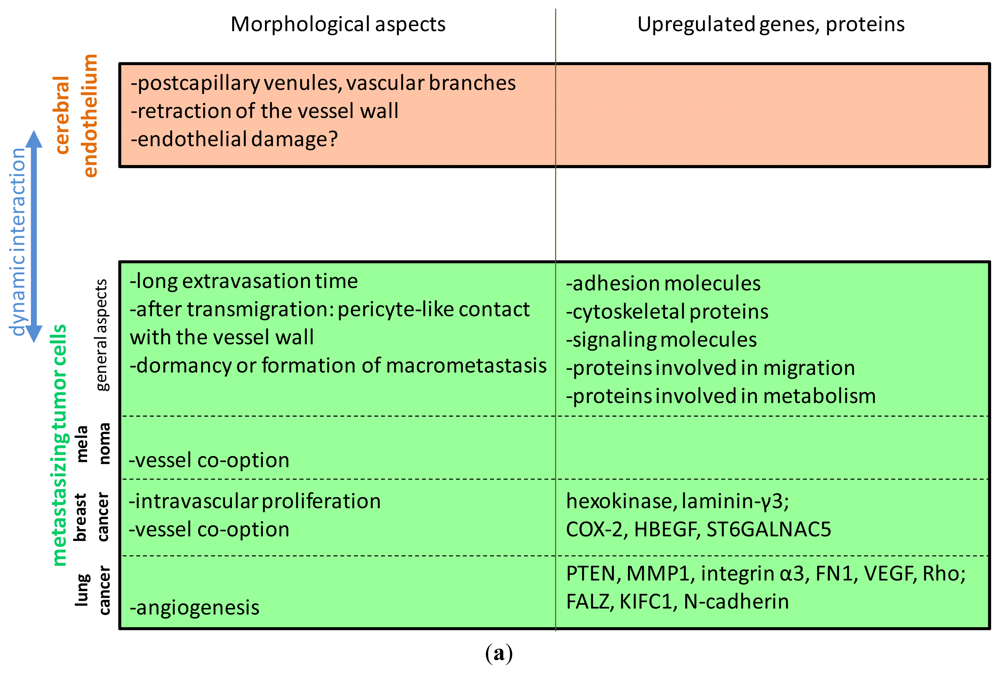
3.1. Morphological Aspects
3.2. Selectively Expressed Genes and Proteins in Brain Metastatic Cells
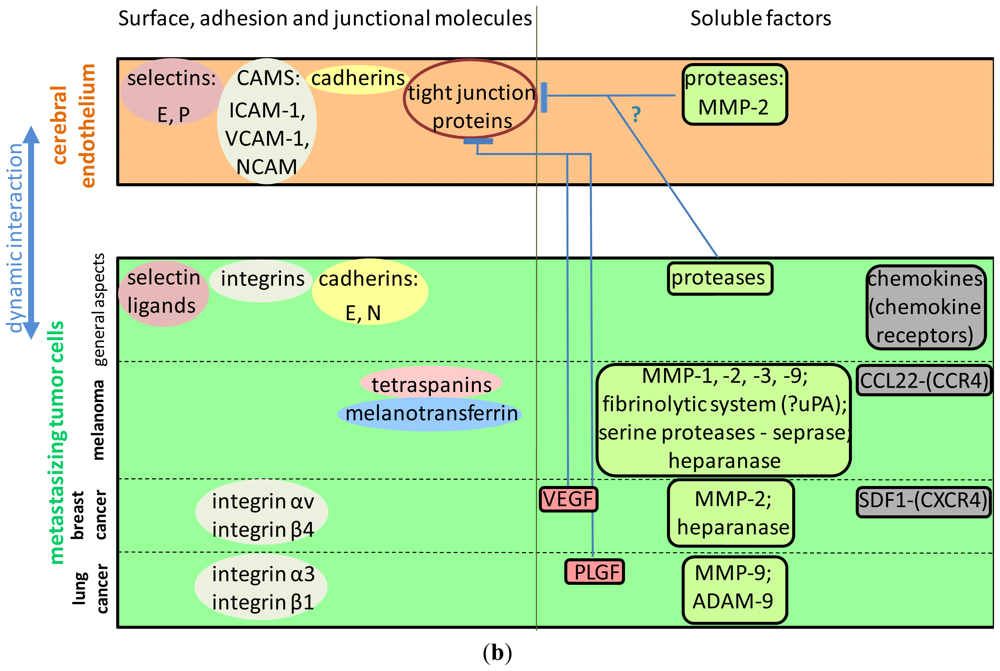
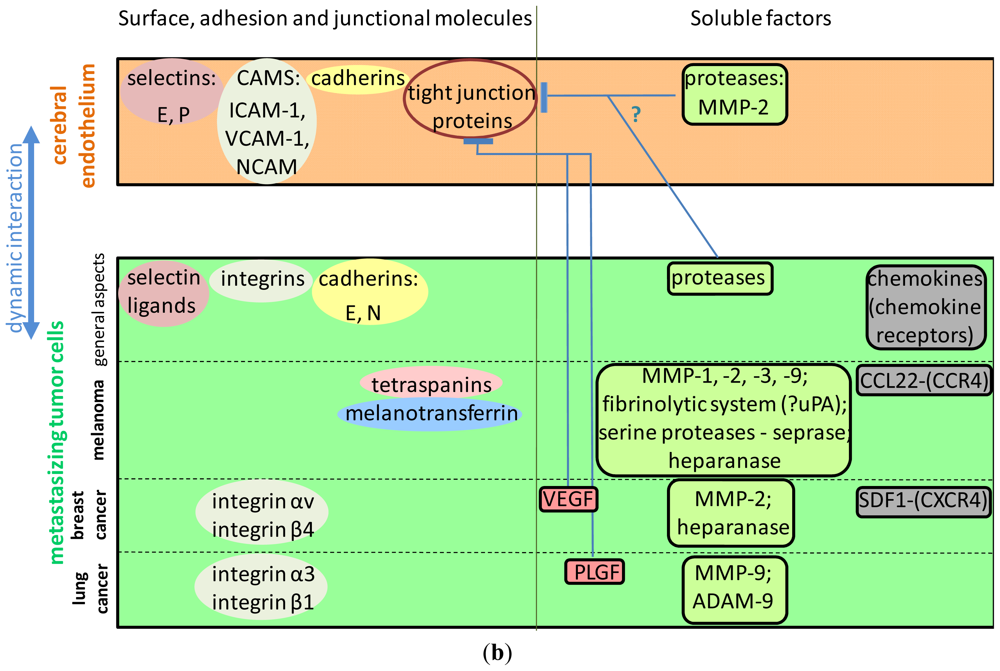
3.3. Transmigration Routes: Role of the Proteins of the Tight Junctions
3.4. Surface Molecules Mediating Different Steps of Tumor Cell Extravasation in the Brain
3.4.1. The Role of Selectins and Selectin Ligands
3.4.2. Integrins Involved in the Interaction of Tumor Cells with the Brain Endothelium
3.4.3. The Role of the Immunglobulin Superfamily of Cell Adhesion Molecules
3.4.4. Cadherins
3.4.5. Tetraspanins
3.4.6. Melanotransferrin
3.5. Soluble Factors Affecting Brain Metastasis Formation
3.5.1. Neurotrophins
3.5.2. Chemokines
3.5.3. Vascular Endothelial Growth Factor and Its Receptors
3.6. Proteases Involved in the Formation of CNS Metastases
3.6.1. The Role of Matrix Metalloproteinases in the Formation of Brain Metastases
3.6.2. Other Proteases
3.6.3. Heparanase
3.7. Signaling Pathways Involved in Tumor-endothelial Interactions in the Brain
3.7.1. Rho/Rac Signaling
3.7.2. Src Signaling
3.7.3. The PI3K-Akt-PTEN Pathway
3.7.4. Other Pathways Implicated in Brain Metastasis Formation, Transendothelial Migration or Invasion of Brain Metastatic Tumor Cells
3.8. Role of Astrocytes in Brain Metastasis Formation
4. Conclusions
Acknowledgments
References
- Platta, C.S.; Khuntia, D.; Mehta, M.P.; Suh, J.H. Current treatment strategies for brain metastasis and complications from therapeutic techniques: A review of current literature. Am. J. Clin. Oncol 2010, 33, 398–407. [Google Scholar]
- Wen, P.Y.; Black, P.M.; Loeffler, J.S. Treatment of Metastatic Cancer. In Cancer: Principles and Practice of Oncology, 6th ed; DeVita, V.T., Hellman, S., Rosenberg, S.A., Eds.; Lippincott and Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Fidler, I.J.; Schackert, G.; Zhang, R.D.; Radinsky, R.; Fujimaki, T. The biology of melanoma brain metastasis. Cancer Metastasis Rev 1999, 18, 387–400. [Google Scholar]
- Douglas, J.G.; Margolin, K. The treatment of brain metastases from malignant melanoma. Semin. Oncol 2002, 29, 518–524. [Google Scholar]
- Pestalozzi, B.C. Brain metastases and subtypes of breast cancer. Ann. Oncol 2009, 20, 803–805. [Google Scholar]
- Meert, A.P.; Paesmans, M.; Berghmans, T.; Martin, B.; Mascaux, C.; Vallot, F.; Verdebout, J.M.; Lafitte, J.J.; Sculier, J.P. Prophylactic cranial irradiation in small cell lung cancer: A systematic review of the literature with meta-analysis. BMC Cancer 2001, 1. [Google Scholar] [CrossRef] [Green Version]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol 2008, 7, 84–96. [Google Scholar]
- Stolp, H.B.; Dziegielewska, K.M. Review: Role of Developmental inflammation and blood-brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol. Appl. Neurobiol 2009, 35, 132–146. [Google Scholar]
- Brightman, M.W.; Reese, T.S. Junctions between intimately apposed cell membranes in the vertebrate brain. J. Cell Biol 1969, 40, 648–677. [Google Scholar]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol 1977, 1, 409–417. [Google Scholar]
- The Blood-Brain Barrier, Biology and Research Protocols; Nag, S. (Ed.) Humana Press: Totowa, NJ, USA, 2003; p. 572.
- Cuevas, P.; Gutierrez-Diaz, J.A.; Reimers, D.; Dujovny, M.; Diaz, F.G.; Ausman, J.I. Pericyte endothelial gap junctions in human cerebral capillaries. Anat. Embryol 1984, 170, 155–159. [Google Scholar]
- Sims, D.E. Recent Advances in pericyte biology—Implications for health and disease. Can. J. Cardiol 1991, 7, 431–443. [Google Scholar]
- Pardridge, W.M. Blood-brain barrier biology and methodology. J. Neurovirol 1999, 5, 556–569. [Google Scholar]
- Hellstrom, M.; Gerhardt, H.; Kalen, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol 2001, 153, 543–553. [Google Scholar]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar]
- Krizbai, I.; Wilhelm, I.; Bauer, H.C.; Bauer, H. The Role of Glia in the Formation and Function of the Blood-Brain Barrier. In Neuroglia, 3rd ed; Kettenmann, H., Ransom, B.R., Eds.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Kacem, K.; Lacombe, P.; Seylaz, J.; Bonvento, G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: A confocal microscopy study. Glia 1998, 23, 1–10. [Google Scholar]
- Cohen, Z.; Molinatti, G.; Hamel, E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J. Cereb. Blood Flow Metab 1997, 17, 894–904. [Google Scholar]
- Paspalas, C.D.; Papadopoulos, G.C. Ultrastructural relationships between noradrenergic nerve fibers and non-neuronal elements in the rat cerebral cortex. Glia 1996, 17, 133–146. [Google Scholar]
- Krizbai, I.A.; Deli, M.A.; Pestenacz, A.; Siklos, L.; Szabo, C.A.; Andras, I.; Joo, F. Expression of glutamate receptors on cultured cerebral endothelial cells. J. Neurosci. Res 1998, 54, 814–819. [Google Scholar]
- Nishioku, T.; Dohgu, S.; Takata, F.; Eto, T.; Ishikawa, N.; Kodama, K.B.; Nakagawa, S.; Yamauchi, A.; Kataoka, Y. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell. Mol. Neurobiol 2009, 29, 309–316. [Google Scholar]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating resident microglia after focal cerebral ischaemia in mice. J. Cereb. Blood Flow Metab 2007, 27, 1941–1953. [Google Scholar]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar]
- Tilling, T.; Engelbertz, C.; Decker, S.; Korte, D.; Huwel, S.; Galla, H.J. Expression and adhesive properties of basement membrane proteins in cerebral capillary endothelial cell cultures. Cell Tissue Res 2002, 310, 19–29. [Google Scholar]
- Savettieri, G.; di Liegro, I.; Catania, C.; Licata, L.; Pitarresi, G.L.; D’Agostino, S.; Schiera, G.; de Caro, V.; Giandalia, G.; Giannola, L.I.; et al. Neurons and ECM regulate occludin localization in brain endothelial cells. Neuroreport 2000, 11, 1081–1084. [Google Scholar]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS One 2009, 4, e5857. [Google Scholar] [CrossRef]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In vitro models of the blood-brain barrier. Acta Neurobiol. Exp 2011, 71, 113–128. [Google Scholar]
- Bauer, H.C.; Traweger, A.; Zweimueller-Mayer, J.; Lehner, C.; Tempfer, H.; Krizbai, I.; Wilhelm, I.; Bauer, H. New aspects of the molecular constituents of tissue barriers. J. Neural Transm 2011, 118, 7–21. [Google Scholar]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol 1993, 123, 1777–1788. [Google Scholar]
- Wong, V.; Gumbiner, B.M. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J. Cell Biol 1997, 136, 399–409. [Google Scholar]
- Lacaz-Vieira, F.; Jaeger, M.M.; Farshori, P.; Kachar, B. Small synthetic peptides homologous to segments of the first external loop of occludin impair tight junction resealing. J. Membr. Biol 1999, 168, 289–297. [Google Scholar]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol 1998, 141, 1539–1550. [Google Scholar]
- Piontek, J.; Winkler, L.; Wolburg, H.; Muller, S.L.; Zuleger, N.; Piehl, C.; Wiesner, B.; Krause, G.; Blasig, I.E. Formation of tight junction: Determinants of homophilic interaction between classic claudins. FASEB J 2008, 22, 146–158. [Google Scholar]
- Ohtsuki, S.; Yamaguchi, H.; Katsukura, Y.; Asashima, T.; Terasaki, T. MRNA expression Levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J. Neurochem 2008, 104, 147–154. [Google Scholar]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol 2003, 161, 653–660. [Google Scholar]
- Ouban, A.; Ahmed, A.A. Claudins in human cancer: A review. Histol. Histopathol 2010, 25, 83–90. [Google Scholar]
- Martin-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol 1998, 142, 117–127. [Google Scholar]
- Aurrand-Lions, M.; Johnson-Leger, C.; Wong, C.; Du Pasquier, L.; Imhof, B.A. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood 2001, 98, 3699–3707. [Google Scholar]
- Langer, H.F.; Orlova, V.V.; Xie, C.; Kaul, S.; Schneider, D.; Lonsdorf, A.S.; Fahrleitner, M.; Choi, E.Y.; Dutoit, V.; Pellegrini, M.; et al. A novel function of junctional adhesion molecule-c in mediating melanoma cell metastasis. Cancer Res 2011, 71, 4096–4105. [Google Scholar]
- Cangara, H.M.; Ishida, T.; Hara, T.; Sun, L.; Toh, R.; Rikitake, Y.; Kundu, R.K.; Quertermous, T.; Hirata, K.; Hayashi, Y. Role of Endothelial cell-selective adhesion molecule in hematogeneous metastasis. Microvasc. Res 2010, 80, 133–141. [Google Scholar]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol 1986, 103, 755–766. [Google Scholar]
- Gumbiner, B.; Lowenkopf, T.; Apatira, D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3460–3464. [Google Scholar]
- Haskins, J.; Gu, L.; Wittchen, E.S.; Hibbard, J.; Stevenson, B.R. ZO-3, a novel member of the maguk protein family found at the tight junction, interacts with ZO-1 and occludin. J. Cell Biol 1998, 141, 199–208. [Google Scholar]
- Inoko, A.; Itoh, M.; Tamura, A.; Matsuda, M.; Furuse, M.; Tsukita, S. Expression and distribution of ZO-3, a tight junction MAGUK protein, in mouse tissues. Genes Cells 2003, 8, 837–845. [Google Scholar]
- Balda, M.S.; Matter, K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J 2000, 19, 2024–2033. [Google Scholar]
- Traweger, A.; Fuchs, R.; Krizbai, I.A.; Weiger, T.M.; Bauer, H.C.; Bauer, H. The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J. Biol. Chem 2003, 278, 2692–2700. [Google Scholar]
- Strell, C.; Entschladen, F. Extravasation of leukocytes in comparison to tumor cells. Cell. Commun. Signal. 2008, 6. [Google Scholar] [CrossRef]
- Lorger, M.; Felding-Habermann, B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol 2010, 176, 2958–2971. [Google Scholar]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med 2010, 16, 116–122. [Google Scholar]
- Paku, S.; Dome, B.; Toth, R.; Timar, J. Organ-specificity of the extravasation process: An ultrastructural study. Clin. Exp. Metastasis 2000, 18, 481–492. [Google Scholar]
- Fazakas, C.; Wilhelm, I.; Nagyoszi, P.; Farkas, A.E.; Hasko, J.; Molnar, J.; Bauer, H.; Bauer, H.C.; Ayaydin, F.; Dung, N.T.; et al. Transmigration of melanoma cells through the blood-brain barrier: Role of endothelial tight junctions and melanoma-released serine proteases. PLoS One 2011, 6, e20758. [Google Scholar] [CrossRef]
- Fidler, I.J. The role of the organ microenvironment in brain metastasis. Semin. Cancer Biol 2011, 21, 107–112. [Google Scholar]
- Lu, W.; Bucana, C.D.; Schroit, A.J. Pathogenesis and vascular integrity of breast cancer brain metastasis. Int. J. Cancer 2007, 120, 1023–1026. [Google Scholar]
- Barnhill, R.L.; Benson, P.J.; Lugassy, C. Conspicuous angiotropism of malignant melanoma involving the brain: Implications for extravascular migratory metastasis. Am. J. Dermatopathol 2009, 31, 205–208. [Google Scholar]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res 2010, 16, 5664–5678. [Google Scholar]
- Kikuchi, T.; Daigo, Y.; Ishikawa, N.; Katagiri, T.; Tsunoda, T.; Yoshida, S.; Nakamura, Y. Expression profiles of metastatic brain tumor from lung adenocarcinomas on cDNA microarray. Int. J. Oncol 2006, 28, 799–805. [Google Scholar]
- Zohrabian, V.M.; Nandu, H.; Gulati, N.; Khitrov, G.; Zhao, C.; Mohan, A.; Demattia, J.; Braun, A.; Das, K.; Murali, R.; et al. Gene expression profiling of metastatic brain cancer. Oncol. Rep 2007, 18, 321–328. [Google Scholar]
- Grinberg-Rashi, H.; Ofek, E.; Perelman, M.; Skarda, J.; Yaron, P.; Hajduch, M.; Jacob-Hirsch, J.; Amariglio, N.; Krupsky, M.; Simansky, D.A.; et al. The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain. Clin. Cancer Res 2009, 15, 1755–1761. [Google Scholar]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar]
- Klein, A.; Olendrowitz, C.; Schmutzler, R.; Hampl, J.; Schlag, P.M.; Maass, N.; Arnold, N.; Wessel, R.; Ramser, J.; Meindl, A.; et al. Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett 2009, 276, 212–220. [Google Scholar]
- Palmieri, D.; Fitzgerald, D.; Shreeve, S.M.; Hua, E.; Bronder, J.L.; Weil, R.J.; Davis, S.; Stark, A.M.; Merino, M.J.; Kurek, R.; et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer. Res 2009, 7, 1438–1445. [Google Scholar]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar]
- Wolburg, H.; Wolburg-Buchholz, K.; Engelhardt, B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 2005, 109, 181–190. [Google Scholar]
- Reijerkerk, A.; Kooij, G.; van der Pol, S.M.; Khazen, S.; Dijkstra, C.D.; de Vries, H.E. Diapedesis of monocytes is associated with mmp-mediated occludin disappearance in brain endothelial cells. FASEB J 2006, 20, 2550–2552. [Google Scholar]
- Dejana, E. The transcellular railway: Insights into leukocyte diapedesis. Nat. Cell Biol 2006, 8, 105–107. [Google Scholar]
- Carman, C.V. Mechanisms for transcellular diapedesis: Probing and pathfinding by ‘invadosomelike protrusions’. J. Cell. Sci 2009, 122, 3025–3035. [Google Scholar]
- Khuon, S.; Liang, L.; Dettman, R.W.; Sporn, P.H.; Wysolmerski, R.B.; Chew, T.L. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: A three-dimensional FRET study. J. Cell. Sci 2010, 123, 431–440. [Google Scholar]
- Konstantopoulos, K.; Thomas, S.N. Cancer cells in transit: The vascular interactions of tumor cells. Annu. Rev. Biomed. Eng 2009, 11, 177–202. [Google Scholar]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar]
- Brayton, J.; Qing, Z.; Hart, M.N.; VanGilder, J.C.; Fabry, Z. Influence of adhesion molecule expression by human brain microvessel endothelium on cancer cell adhesion. J. Neuroimmunol 1998, 89, 104–112. [Google Scholar]
- Maraveyas, A.; Johnson, M.J.; Xiao, Y.P.; Noble, S. Malignant melanoma as a target malignancy for the study of the anti-metastatic properties of the heparins. Cancer Metastasis Rev 2010, 29, 777–784. [Google Scholar]
- Lorger, M.; Krueger, J.S.; O’Neal, M.; Staflin, K.; Felding-Habermann, B. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proc. Natl. Acad. Sci. USA 2009, 106, 10666–10671. [Google Scholar]
- Wu, Y.J.; Muldoon, L.L.; Gahramanov, S.; Kraemer, D.F.; Marshall, D.J.; Neuwelt, E.A. Targeting alpha(V)-integrins decreased metastasis and increased survival in a nude rat breast cancer brain metastasis model. J. Neurooncol 2012, 110, 27–36. [Google Scholar]
- Fan, J.; Cai, B.; Zeng, M.; Hao, Y.; Giancotti, F.G.; Fu, B.M. Integrin beta4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann. Biomed. Eng 2011, 39, 2223–2241. [Google Scholar]
- Yoshimasu, T.; Sakurai, T.; Oura, S.; Hirai, I.; Tanino, H.; Kokawa, Y.; Naito, Y.; Okamura, Y.; Ota, I.; Tani, N.; et al. Increased expression of integrin alpha3beta1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer. Sci 2004, 95, 142–148. [Google Scholar]
- Langley, R.R.; Carlisle, R.; Ma, L.; Specian, R.D.; Gerritsen, M.E.; Granger, D.N. Endothelial expression of vascular cell adhesion molecule-1 correlates with metastatic pattern in spontaneous melanoma. Microcirculation 2001, 8, 335–345. [Google Scholar]
- Sipos, E.; Chen, L.; Andras, I.E.; Wrobel, J.; Zhang, B.; Pu, H.; Park, M.; Eum, S.Y.; Toborek, M. Proinflammatory adhesion molecules facilitate polychlorinated biphenyl-mediated enhancement of brain metastasis formation. Toxicol. Sci 2012, 126, 362–371. [Google Scholar]
- Onodera, H.; Nagayama, S.; Tachibana, T.; Fujimoto, A.; Imamura, M. Brain metastasis from colorectal cancer. Int. J. Colorectal Dis 2005, 20, 57–61. [Google Scholar]
- Duenisch, P.; Reichart, R.; Mueller, U.; Brodhun, M.; Bjerkvig, R.; Romeike, B.; Walter, J.; Herbold, C.; Regenbrecht, C.R.; Kalff, R.; et al. Neural cell adhesion molecule isoform 140 declines with rise of who grade in human gliomas and serves as indicator for the invasion zone of multiform glioblastomas and brain metastases. J. Cancer Res. Clin. Oncol 2011, 137, 399–414. [Google Scholar]
- Shabani, H.K.; Kitange, G.; Tsunoda, K.; Anda, T.; Tokunaga, Y.; Shibata, S.; Kaminogo, M.; Hayashi, T.; Ayabe, H.; Iseki, M. Immunohistochemical expression of e-cadherin in metastatic brain tumors. Brain Tumor Pathol 2003, 20, 7–12. [Google Scholar]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer. 2010, 9. [Google Scholar] [CrossRef]
- Zeljko, M.; Pecina-Slaus, N.; Martic, T.N.; Kusec, V.; Beros, V.; Tomas, D. Molecular alterations of E-cadherin and beta-catenin in brain metastases. Front. Biosci 2011, 3, 616–624. [Google Scholar]
- Yoo, J.Y.; Yang, S.H.; Lee, J.E.; Cho, D.G.; Kim, H.K.; Kim, S.H.; Kim, I.S.; Hong, J.T.; Sung, J.H.; Son, B.C.; et al. E-cadherin as a predictive marker of brain metastasis in non-small-cell lung cancer, and its regulation by pioglitazone in a preclinical model. J. Neurooncol 2012, 109, 219–227. [Google Scholar]
- Qi, J.; Chen, N.; Wang, J.; Siu, C.H. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol. Biol. Cell 2005, 16, 4386–4397. [Google Scholar]
- Qi, J.; Wang, J.; Romanyuk, O.; Siu, C.H. Involvement of Src family kinases in N-cadherin phosphorylation and beta-catenin dissociation during transendothelial migration of melanoma cells. Mol. Biol. Cell 2006, 17, 1261–1272. [Google Scholar]
- Longo, N.; Yanez-Mo, M.; Mittelbrunn, M.; de la Rosa, G.; Munoz, M.L.; Sanchez-Madrid, F.; Sanchez-Mateos, P. Regulatory role of tetraspanin CD9 in tumor-endothelial cell interaction during transendothelial invasion of melanoma cells. Blood 2001, 98, 3717–3726. [Google Scholar]
- Takeda, Y.; Li, Q.; Kazarov, A.R.; Epardaud, M.; Elpek, K.; Turley, S.J.; Hemler, M.E. Diminished metastasis in tetraspanin CD151-knockout mice. Blood 2011, 118, 464–472. [Google Scholar]
- Rolland, Y.; Demeule, M.; Fenart, L.; Beliveau, R. Inhibition of melanoma brain metastasis by targeting melanotransferrin at the cell surface. Pigment Cell. Melanoma Res 2009, 22, 86–98. [Google Scholar]
- Aucoin, R.; Marchetti, D. Brain metastases in melanoma: Roles of neurotrophins. Neuro Oncol 2004, 6, 154–165. [Google Scholar]
- Nicolson, G.L.; Menter, D.G. Trophic factors and central nervous system metastasis. Cancer Metastasis Rev 1995, 14, 303–321. [Google Scholar]
- Kim, H.; Li, Q.; Hempstead, B.L.; Madri, J.A. Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J. Biol. Chem 2004, 279, 33538–33546. [Google Scholar]
- Lecht, S.; Arien-Zakay, H.; Kohan, M.; Lelkes, P.I.; Lazarovici, P. Angiostatic effects of K252a, a Trk inhibitor, in murine brain capillary endothelial cells. Mol. Cell. Biochem 2010, 339, 201–213. [Google Scholar]
- Shimizu, F.; Sano, Y.; Abe, M.A.; Maeda, T.; Ohtsuki, S.; Terasaki, T.; Kanda, T. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J. Cell. Physiol 2011, 226, 255–266. [Google Scholar]
- Izraely, S.; Klein, A.; Sagi-Assif, O.; Meshel, T.; Tsarfaty, G.; Hoon, D.S.; Witz, I.P. Chemokine-chemokine receptor axes in melanoma brain metastasis. Immunol. Lett 2010, 130, 107–114. [Google Scholar]
- Klein, A.; Sagi-Assif, O.; Izraely, S.; Meshel, T.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.O.; Erez, N.; Hoon, D.S.; Witz, I.P. The metastatic microenvironment: Brain-derived soluble factors alter the malignant phenotype of cutaneous and brain-metastasizing melanoma cells. Int. J. Cancer 2012, 131, 2509–2518. [Google Scholar]
- Lee, B.C.; Lee, T.H.; Avraham, S.; Avraham, H.K. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res 2004, 2, 327–338. [Google Scholar]
- Lee, T.H.; Avraham, H.K.; Jiang, S.; Avraham, S. Vascular endothelial growth factor modulates the transendothelial migration of MDA-MB-231 breast cancer cells through regulation of brain microvascular endothelial cell permeability. J. Biol. Chem 2003, 278, 5277–5284. [Google Scholar]
- Li, B.; Wang, C.; Zhang, Y.; Zhao, X.Y.; Huang, B.; Wu, P.F.; Li, Q.; Li, H.; Liu, Y.S.; Cao, L.Y.; et al. Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene 2012. [Google Scholar] [CrossRef]
- Tester, A.M.; Waltham, M.; Oh, S.J.; Bae, S.N.; Bills, M.M.; Walker, E.C.; Kern, F.G.; Stetler-Stevenson, W.G.; Lippman, M.E.; Thompson, E.W. Pro-matrix metalloproteinase-2 transfection increases orthotopic primary growth and experimental metastasis of MDA-MB-231 human breast cancer cells in nude mice. Cancer Res 2004, 64, 652–658. [Google Scholar]
- Mendes, O.; Kim, H.T.; Stoica, G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin. Exp. Metastasis 2005, 22, 237–246. [Google Scholar]
- Mendes, O.; Kim, H.T.; Lungu, G.; Stoica, G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin. Exp. Metastasis 2007, 24, 341–351. [Google Scholar]
- Xie, T.X.; Huang, F.J.; Aldape, K.D.; Kang, S.H.; Liu, M.; Gershenwald, J.E.; Xie, K.; Sawaya, R.; Huang, S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res 2006, 66, 3188–3196. [Google Scholar]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS One 2011, 6, e20599. [Google Scholar] [CrossRef]
- Stark, A.M.; Anuszkiewicz, B.; Mentlein, R.; Yoneda, T.; Mehdorn, H.M.; Held-Feindt, J. Differential expression of matrix metalloproteinases in brain- and bone-seeking clones of metastatic MDA-MB-231 breast cancer cells. J. Neurooncol 2007, 81, 39–48. [Google Scholar]
- Rodrigues-Ferreira, S.; Abdelkarim, M.; Dillenburg-Pilla, P.; Luissint, A.C.; di-Tommaso, A.; Deshayes, F.; Pontes, C.L.; Molina, A.; Cagnard, N.; Letourneur, F.; et al. Angiotensin II facilitates breast cancer cell migration and metastasis. PLoS One 2012, 7, e35667. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Zhang, J.; Zhu, H.; Min, J.; Feng, Y.; Zhang, H. Biological characteristics of a specific brain metastatic cell line derived from human lung adenocarcinoma. Med. Oncol 2010, 27, 708–714. [Google Scholar]
- Lee, K.Y.; Kim, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, H.J. Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier. Anticancer Res 2011, 31, 4307–4313. [Google Scholar]
- Shintani, Y.; Higashiyama, S.; Ohta, M.; Hirabayashi, H.; Yamamoto, S.; Yoshimasu, T.; Matsuda, H.; Matsuura, N. Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res 2004, 64, 4190–4196. [Google Scholar]
- Perides, G.; Zhuge, Y.; Lin, T.; Stins, M.F.; Bronson, R.T.; Wu, J.K. The fibrinolytic system facilitates tumor cell migration across the blood-brain barrier in experimental melanoma brain metastasis. BMC Cancer 2006, 6. [Google Scholar] [CrossRef]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol 2006, 38, 2018–2039. [Google Scholar]
- Marchetti, D.; Nicolson, G.L. Human heparanase: A molecular determinant of brain metastasis. Adv. Enzyme Regul 2001, 41, 343–359. [Google Scholar]
- Roy, M.; Marchetti, D. Cell surface heparan sulfate released by heparanase promotes melanoma cell migration and angiogenesis. J. Cell. Biochem 2009, 106, 200–209. [Google Scholar]
- Zhang, L.; Sullivan, P.; Suyama, J.; Marchetti, D. Epidermal growth factor-induced heparanase nucleolar localization augments DNA topoisomerase I activity in brain metastatic breast cancer. Mol. Cancer. Res 2010, 8, 278–290. [Google Scholar]
- Ridgway, L.D.; Wetzel, M.D.; Marchetti, D. Modulation of GEF-H1 induced signaling by heparanase in brain metastatic melanoma cells. J. Cell. Biochem 2010, 111, 1299–1309. [Google Scholar]
- Marchetti, D.; Li, J.; Shen, R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res 2000, 60, 4767–4770. [Google Scholar]
- Deli, M.A.; Joo, F.; Krizbai, I.; Lengyel, I.; Nunzi, M.G.; Wolff, J.R. Calcium/calmodulin-stimulated protein kinase II is present in primary cultures of cerebral endothelial cells. J. Neurochem 1993, 60, 1960–1963. [Google Scholar]
- Krizbai, I.; Szabo, G.; Deli, M.; Maderspach, K.; Lehel, C.; Olah, Z.; Wolff, J.R.; Joo, F. Expression of protein kinase C family members in the cerebral endothelial cells. J. Neurochem 1995, 65, 459–462. [Google Scholar]
- Fabian, G.; Szabo, C.A.; Bozo, B.; Greenwood, J.; Adamson, P.; Deli, M.A.; Joo, F.; Krizbai, I.A.; Szucs, M. Expression of G-protein subtypes in cultured cerebral endothelial cells. Neurochem. Int 1998, 33, 179–185. [Google Scholar]
- Krizbai, I.A.; Deli, M.A. Signalling pathways regulating the tight junction permeability in the blood-brain barrier. Cell. Mol. Biol 2003, 49, 23–31. [Google Scholar]
- Krizbai, I.A.; Bauer, H.; Amberger, A.; Hennig, B.; Szabo, H.; Fuchs, R.; Bauer, H.C. Growth factor-induced morphological, physiological and molecular characteristics in cerebral endothelial cells. Eur. J. Cell Biol 2000, 79, 594–600. [Google Scholar]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Brocker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol 2003, 160, 267–277. [Google Scholar]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for rho/rock signalling and extracellular proteolysis. Nat. Cell Biol 2003, 5, 711–719. [Google Scholar]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar]
- Li, B.; Zhao, W.D.; Tan, Z.M.; Fang, W.G.; Zhu, L.; Chen, Y.H. Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett 2006, 580, 4252–4260. [Google Scholar]
- Ahn, J.; Sanz-Moreno, V.; Marshall, C.J. The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J. Cell. Sci 2012, 125, 1814–1826. [Google Scholar]
- Farkas, A.; Szatmari, E.; Orbok, A.; Wilhelm, I.; Wejksza, K.; Nagyoszi, P.; Hutamekalin, P.; Bauer, H.; Bauer, H.C.; Traweger, A.; et al. Hyperosmotic mannitol in-duces Src kinase-dependent phosphorylation of beta-catenin in cerebral endothelial cells. J. Neurosci. Res 2005, 80, 855–861. [Google Scholar]
- Takenaga, Y.; Takagi, N.; Murotomi, K.; Tanonaka, K.; Takeo, S. Inhibition of Src activity decreases tyrosine phosphorylation of occludin in brain capillaries and attenuates increase in permeability of the blood-brain barrier after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab 2009, 29, 1099–1108. [Google Scholar]
- Nanni, P.; Nicoletti, G.; Palladini, A.; Croci, S.; Murgo, A.; Ianzano, M.L.; Grosso, V.; Stivani, V.; Antognoli, A.; Lamolinara, A.; et al. Multiorgan metastasis of human HER-2(+) breast cancer in Rag2(−)/(−);Il2rg(−)/(−) mice and treatment with PI3K inhibitor. PLoS One 2012, 7, e39626. [Google Scholar] [CrossRef]
- Davies, M.A.; Stemke-Hale, K.; Lin, E.; Tellez, C.; Deng, W.; Gopal, Y.N.; Woodman, S.E.; Calderone, T.C.; Ju, Z.; Lazar, A.J.; et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin. Cancer Res 2009, 15, 7538–7546. [Google Scholar]
- Anfuso, C.D.; Giurdanella, G.; Motta, C.; Muriana, S.; Lupo, G.; Ragusa, N.; Alberghina, M. PKCalpha-MAPK/ERK-phospholipase A2 signaling is required for human melanoma-enhanced brain endothelial cell proliferation and motility. Microvasc. Res 2009, 78, 338–357. [Google Scholar]
- Mierke, C.T. Cancer cells regulate biomechanical properties of human microvascular endothelial cells. J. Biol. Chem 2011, 286, 40025–40037. [Google Scholar]
- Peng, H.H.; Hodgson, L.; Henderson, A.J.; Dong, C. Involvement of phospholipase C signaling in melanoma cell-induced endothelial junction disassembly. Front. Biosci 2005, 10, 1597–1606. [Google Scholar]
- Cruz-Munoz, W.; Jaramillo, M.L.; Man, S.; Xu, P.; Banville, M.; Collins, C.; Nantel, A.; Francia, G.; Morgan, S.S.; Cranmer, L.D.; et al. Roles for endothelin receptor B and BCL2A1 in spontaneous CNS metastasis of melanoma. Cancer Res 2012, 72, 4909–4919. [Google Scholar]
- Zhang, C.; Zhang, F.; Tsan, R.; Fidler, I.J. Transforming growth factor-beta2 is a molecular determinant for site-specific melanoma metastasis in the brain. Cancer Res 2009, 69, 828–835. [Google Scholar]
- Palmieri, D.; Bronder, J.L.; Herring, J.M.; Yoneda, T.; Weil, R.J.; Stark, A.M.; Kurek, R.; Vega-Valle, E.; Feigenbaum, L.; Halverson, D.; et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res 2007, 67, 4190–4198. [Google Scholar]
- Hicks, D.G.; Short, S.M.; Prescott, N.L.; Tarr, S.M.; Coleman, K.A.; Yoder, B.J.; Crowe, J.P.; Choueiri, T.K.; Dawson, A.E.; Budd, G.T.; et al. Breast cancers with brain metastases are more likely to be estrogen receptor negative, express the basal cytokeratin CK5/6, and overexpress HER2 or EGFR. Am. J. Surg. Pathol 2006, 30, 1097–1104. [Google Scholar]
- Heon, S.; Yeap, B.Y.; Lindeman, N.I.; Joshi, V.A.; Butaney, M.; Britt, G.J.; Costa, D.B.; Rabin, M.S.; Jackman, D.M.; Johnson, B.E. The impact of initial gefitinib or erlotinib versus chemotherapy on central nervous system progression in advanced non-small cell lung cancer with EGFR mutations. Clin. Cancer Res 2012, 18, 4406–4414. [Google Scholar]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massague, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009, 138, 51–62. [Google Scholar]
- Colone, M.; Calcabrini, A.; Toccacieli, L.; Bozzuto, G.; Stringaro, A.; Gentile, M.; Cianfriglia, M.; Ciervo, A.; Caraglia, M.; Budillon, A.; et al. The multidrug transporter P-glycoprotein: A mediator of melanoma invasion? J. Invest. Dermatol 2008, 128, 957–971. [Google Scholar]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res 2008, 100, 35–83. [Google Scholar]
- Wilhelm, I.; Nagyoszi, P.; Farkas, A.E.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Fazakas, C.; Dung, N.T.; Bottka, S.; Bauer, H.; et al. Hyperosmotic stress induces axl activation and cleavage in cerebral endothelial cells. J. Neurochem 2008, 107, 116–126. [Google Scholar]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 2010, 70, 1544–1554. [Google Scholar]
- Lin, Q.; Balasubramanian, K.; Fan, D.; Kim, S.J.; Guo, L.; Wang, H.; Bar-Eli, M.; Aldape, K.D.; Fidler, I.J. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 2010, 12, 748–754. [Google Scholar]
- Kim, S.J.; Kim, J.S.; Park, E.S.; Lee, J.S.; Lin, Q.; Langley, R.R.; Maya, M.; He, J.; Kim, S.W.; Weihua, Z.; et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 2011, 13, 286–298. [Google Scholar]
- Menter, D.G.; Herrmann, J.L.; Nicolson, G.L. The role of trophic factors and autocrine/paracrine growth factors in brain metastasis. Clin. Exp. Metastasis 1995, 13, 67–88. [Google Scholar]
- Seike, T.; Fujita, K.; Yamakawa, Y.; Kido, M.A.; Takiguchi, S.; Teramoto, N.; Iguchi, H.; Noda, M. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin. Exp. Metastasis 2011, 28, 13–25. [Google Scholar]
- Sierra, A.; Price, J.E.; Garcia-Ramirez, M.; Mendez, O.; Lopez, L.; Fabra, A. Astrocyte-derived cytokines contribute to the metastatic brain specificity of breast cancer cells. Lab. Invest 1997, 77, 357–368. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wilhelm, I.; Molnár, J.; Fazakas, C.; Haskó, J.; Krizbai, I.A. Role of the Blood-Brain Barrier in the Formation of Brain Metastases. Int. J. Mol. Sci. 2013, 14, 1383-1411. https://doi.org/10.3390/ijms14011383
Wilhelm I, Molnár J, Fazakas C, Haskó J, Krizbai IA. Role of the Blood-Brain Barrier in the Formation of Brain Metastases. International Journal of Molecular Sciences. 2013; 14(1):1383-1411. https://doi.org/10.3390/ijms14011383
Chicago/Turabian StyleWilhelm, Imola, Judit Molnár, Csilla Fazakas, János Haskó, and István A. Krizbai. 2013. "Role of the Blood-Brain Barrier in the Formation of Brain Metastases" International Journal of Molecular Sciences 14, no. 1: 1383-1411. https://doi.org/10.3390/ijms14011383
APA StyleWilhelm, I., Molnár, J., Fazakas, C., Haskó, J., & Krizbai, I. A. (2013). Role of the Blood-Brain Barrier in the Formation of Brain Metastases. International Journal of Molecular Sciences, 14(1), 1383-1411. https://doi.org/10.3390/ijms14011383