Role of Prion Protein Aggregation in Neurotoxicity



Abstract
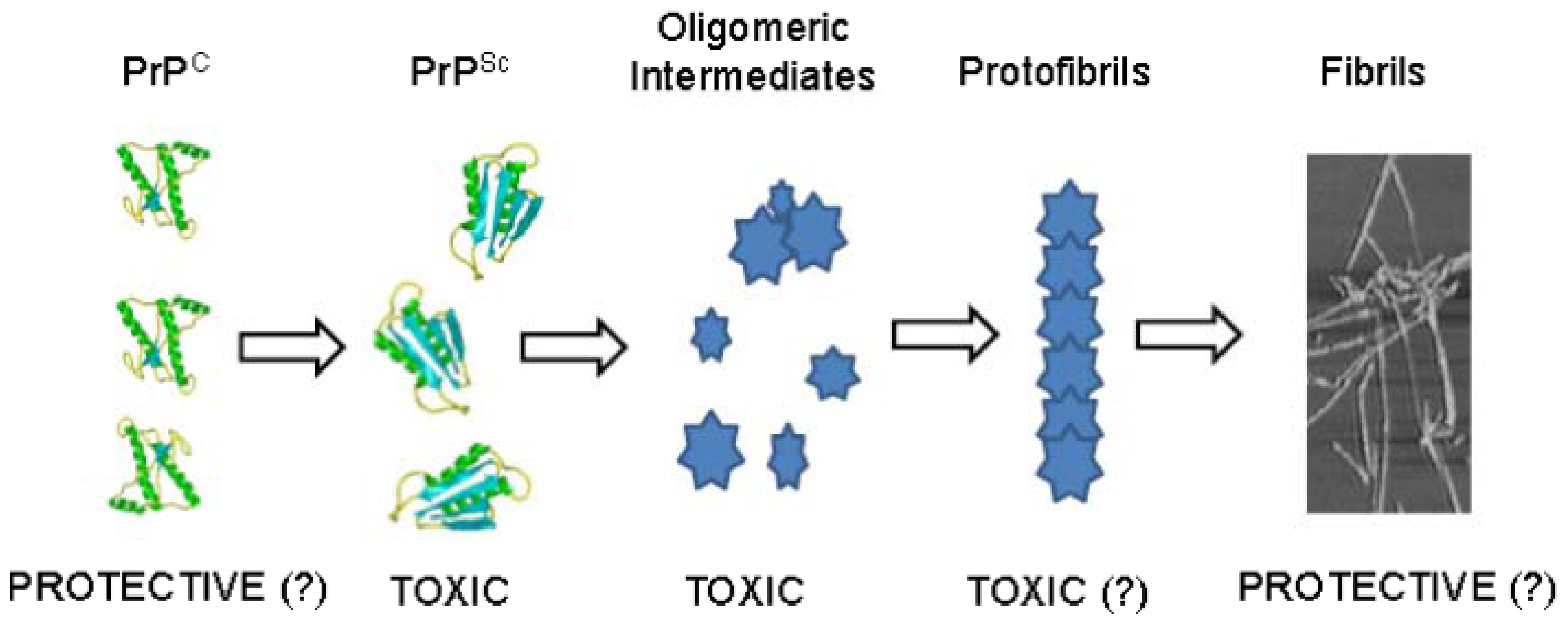
:1. Introduction
2. Transmissible Spongiform Encephalopathies (Prion Diseases)
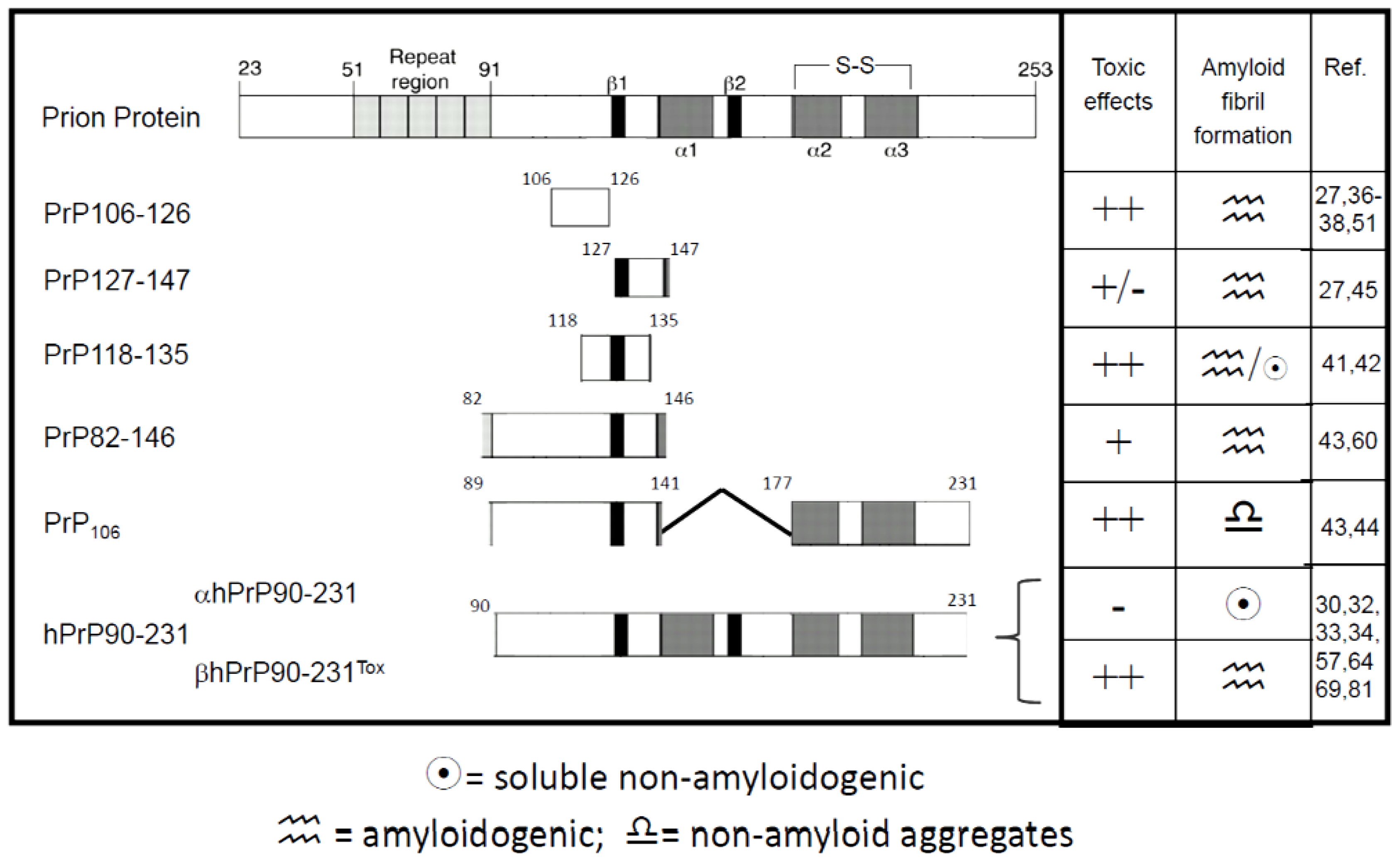
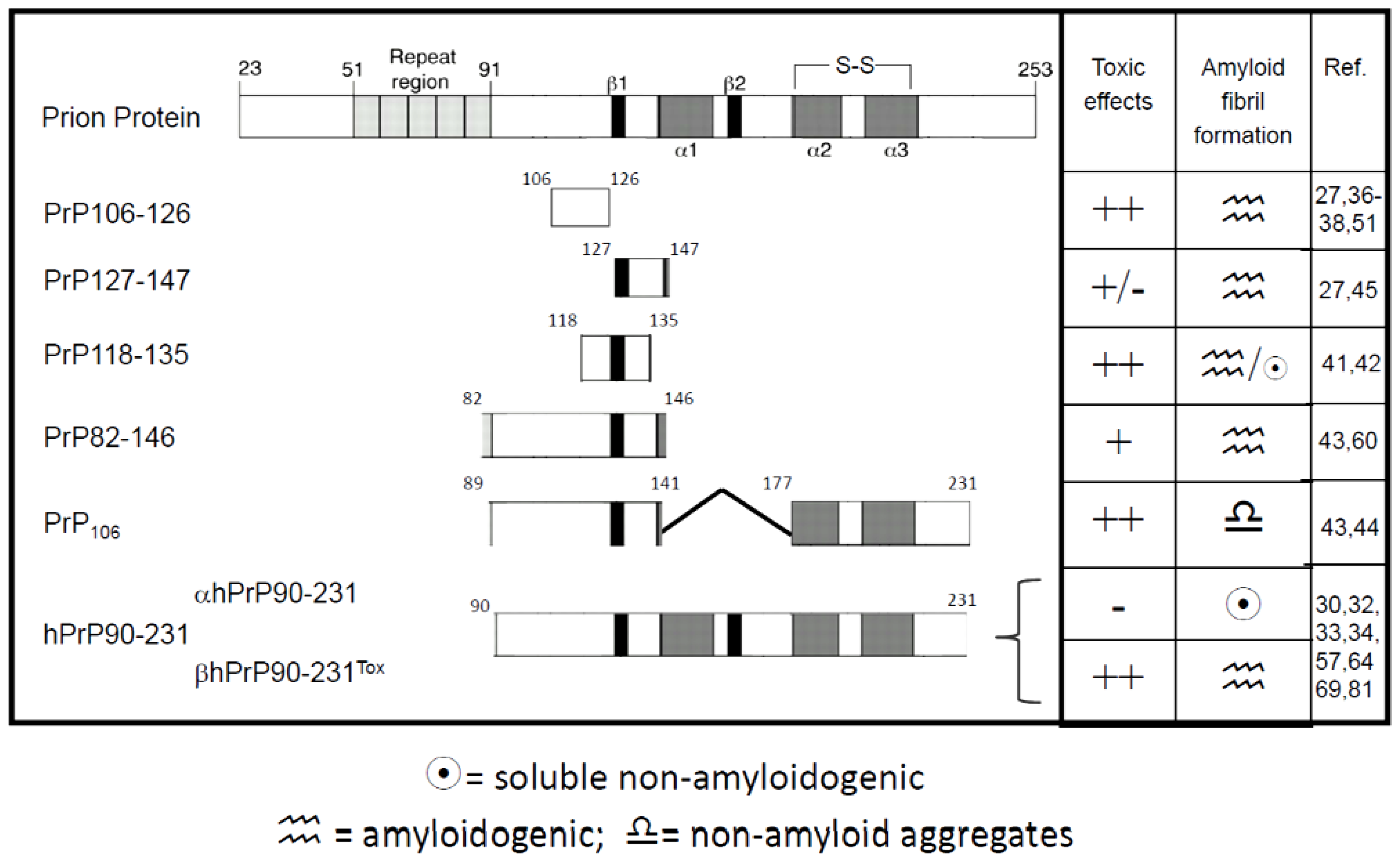
3. PrP-Derived Peptides as Models to Describe Prion Protein Aggregation Process
4. Synthetic PrP 106-126 Peptide
5. PrP106-126 Aggregation Is Not a Prerequisite for Its Toxic Activity
6. Recombinant PrP-Derived Peptides
7. Determinants of Structural Misfolding of Recombinant PrP Fragments
8. Recombinant Prion Peptides Infectivity
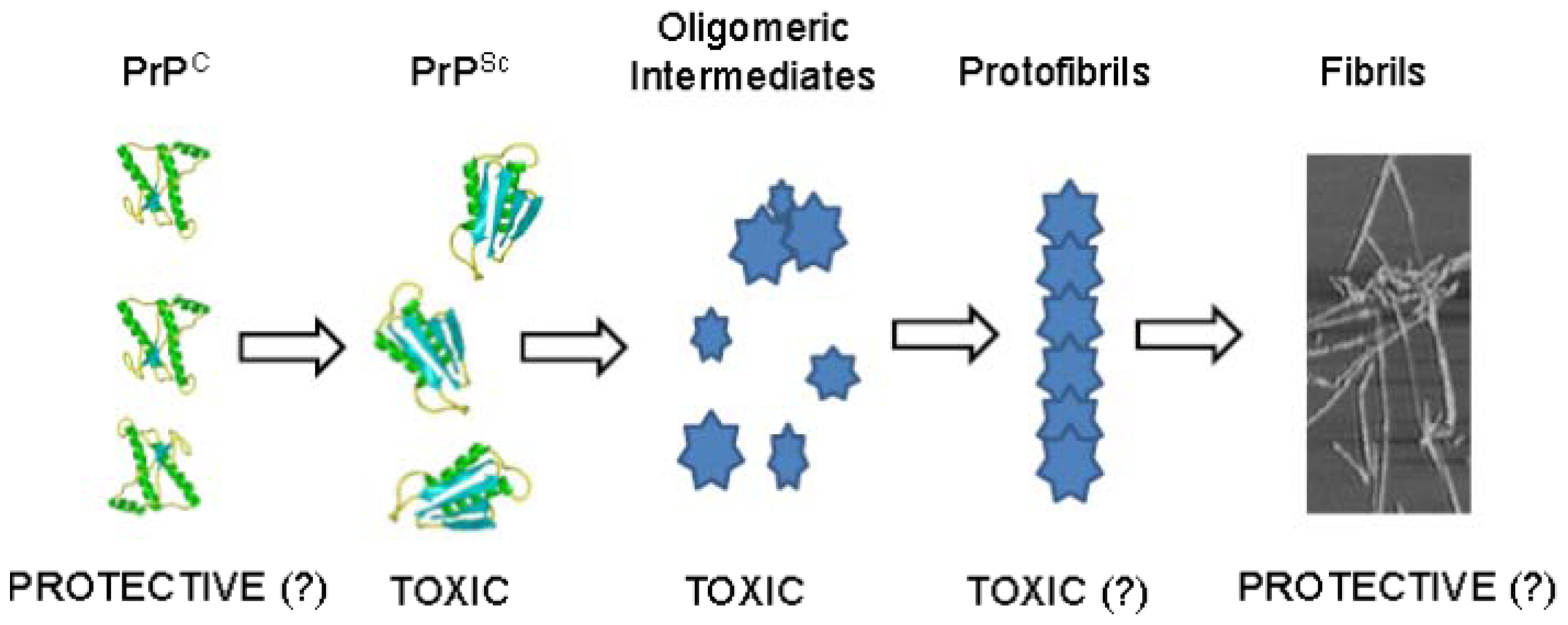
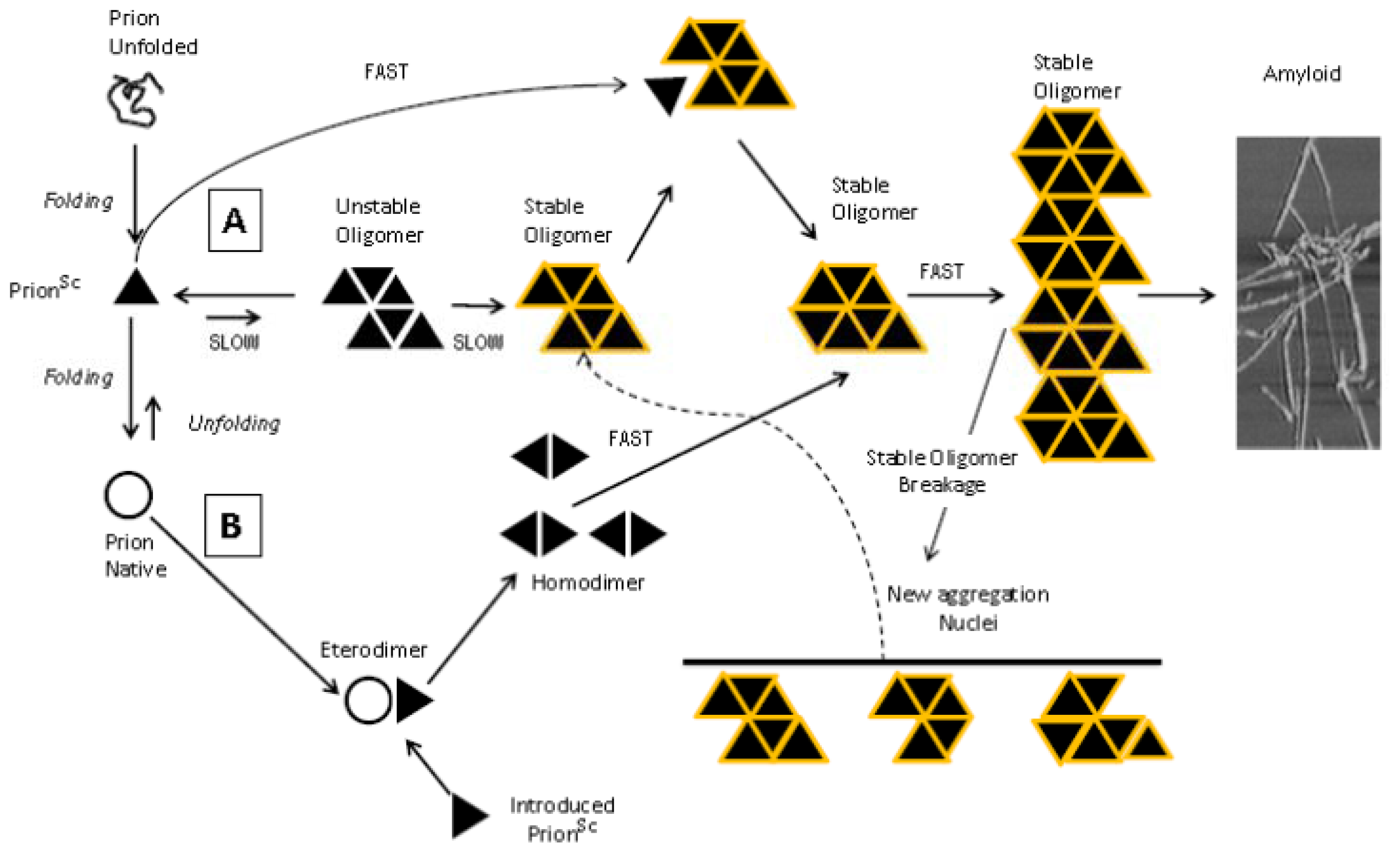
9. Mechanisms of Aggregation and Neurotoxicity of Recombinant Prion Peptides
10. Misfolding/Aggregation Pathways of E200K or D202N Disease-Related Mutations of the Human Prion Protein
11. Effect of Soil Composition on Recombinant PrP Aggregation and Neurotoxicity
12. Conclusions
Acknowledgments
References
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol 2009, 16, 574–581. [Google Scholar]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med 2004, 10, S10–S17. [Google Scholar]
- Norrby, E. Prions and protein-folding diseases. J. Int. Med 2011, 270, 1–14. [Google Scholar]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol 2010, 11, 301–307. [Google Scholar]
- Cohen, A.S.; Calkins, E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 1959, 183, 1202–1203. [Google Scholar]
- Eanes, E.D.; Glenner, G.G. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem 1968, 16, 673–677. [Google Scholar]
- Shewmaker, F.; McGlinchey, R.P.; Wickner, R.B. Structural insights into functional and pathological amyloid. J. Biol. Chem 2011, 286, 16533–16540. [Google Scholar]
- Tzotzos, S.; Doig, A.J. Amyloidogenic sequences in native protein structures. Protein Sci 2010, 19, 327–348. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
- Malchiodi-Albedi, F.; Paradisi, S.; Matteucci, A.; Frank, C.; Diociaiuti, M. Amyloid oligomer neurotoxicity, calcium dysregulation, and lipid rafts. Int. J. Alzheimer’s Dis 2011, 2011. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar]
- Kayed, R.; Pensalfini, A.; Margol, L.; Sokolov, Y.; Sarsoza, F.; Head, E.; Hall, J.; Glabe, C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem 2009, 284, 4230–4237. [Google Scholar]
- Prusiner, S.B. Shattuck lecture--neurodegenerative diseases and prions. N. Engl. J. Med 2001, 344, 1516–1526. [Google Scholar]
- Aguzzi, A. Prion diseases of humans and farm animals: Epidemiology, genetics, and pathogenesis. J. Neurochem 2006, 97, 1726–1739. [Google Scholar]
- Cohen, F.E.; Prusiner, S.B. Pathologic conformations of prion proteins. Annu. Rev. Biochem 1998, 67, 793–819. [Google Scholar]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar]
- Abid, K.; Soto, C. The intriguing prion disorders. Cell Mol. Life Sci 2006, 63, 2342–2351. [Google Scholar]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar]
- Caughey, B. Interactions between prion protein isoforms: The kiss of death? Trends Biochem. Sci 2001, 26, 235–242. [Google Scholar]
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar]
- Wroe, S.J.; Pal, S.; Siddique, D.; Hyare, H.; Macfarlane, R.; Joiner, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.; Hewitt, P.; et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: A case report. Lancet 2006, 368, 2061–2067. [Google Scholar]
- Hsiao, K.; Prusiner, S.B. Inherited human prion diseases. Neurology 1990, 40, 1820–1827. [Google Scholar]
- Aguzzi, A.; Sigurdson, C.; Heikenwaelder, M. Molecular mechanisms of prion pathogenesis. Annu. Rev. Pathol 2008, 3, 11–40. [Google Scholar]
- Brown, P.; Gibbs, C.J., Jr; Rodgers-Johnson, P.; Asher, D.M.; Sulima, M.P.; Bacote, A.; Goldfarb, L.G.; Gajdusek, D.C. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol 1994, 35, 513–529. [Google Scholar]
- Collinge, J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum. Mol. Genet 1997, 6, 1699–1705. [Google Scholar]
- Margalith, I.; Suter, C.; Ballmer, B.; Schwarz, P.; Tiberi, C.; Sonati, T.; Falsig, J.; Nystrom, S.; Hammarstrom, P.; Aslund, A.; et al. Polythiophenes inhibit prion propagation by stabilizing PrP aggregates. J. Biol. Chem 2012, 287, 18872–18887. [Google Scholar]
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546. [Google Scholar]
- Florio, T.; Grimaldi, M.; Scorziello, A.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Forloni, G.; Schettini, G. Intracellular calcium rise through L-type calcium channels, as molecular mechanism for prion protein fragment 106–126-induced astroglial proliferation. Biochem. Biophys. Res. Commun 1996, 228, 397–405. [Google Scholar]
- Baskakov, I.V.; Legname, G.; Baldwin, M.A.; Prusiner, S.B.; Cohen, F.E. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem 2002, 277, 21140–21148. [Google Scholar]
- Baskakov, I.V.; Legname, G.; Gryczynski, Z.; Prusiner, S.B. The peculiar nature of unfolding of the human prion protein. Protein Sci 2004, 13, 586–595. [Google Scholar]
- Morillas, M.; Vanik, D.L.; Surewicz, W.K. On the mechanism of alpha-helix to beta-sheet transition in the recombinant prion protein. Biochemistry 2001, 40, 6982–6987. [Google Scholar]
- Swietnicki, W.; Morillas, M.; Chen, S.G.; Gambetti, P.; Surewicz, W.K. Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry 2000, 39, 424–431. [Google Scholar]
- Swietnicki, W.; Petersen, R.; Gambetti, P.; Surewicz, W.K. pH-dependent stability and conformation of the recombinant human prion protein PrP(90-231). J. Biol. Chem 1997, 272, 27517–27520. [Google Scholar]
- Corsaro, A.; Paludi, D.; Villa, V.; D’Arrigo, C.; Chiovitti, K.; Thellung, S.; Russo, C.; di Cola, D.; Ballerini, P.; Patrone, E.; et al. Conformation dependent pro-apoptotic activity of the recombinant human prion protein fragment 90–231. Int. J. Immunopathol. Pharmacol 2006, 19, 339–356. [Google Scholar]
- Thellung, S.; Villa, V.; Corsaro, A.; Arena, S.; Millo, E.; Damonte, G.; Benatti, U.; Tagliavini, F.; Florio, T.; Schettini, G. p38 MAP kinase mediates the cell death induced by PrP106-126 in the SH-SY5Y neuroblastoma cells. Neurobiol. Dis 2002, 9, 69–81. [Google Scholar]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347. [Google Scholar]
- Jobling, M.F.; Stewart, L.R.; White, A.R.; McLean, C.; Friedhuber, A.; Maher, F.; Beyreuther, K.; Masters, C.L.; Barrow, C.J.; Collins, S.J.; et al. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106–126. J. Neurochem 1999, 73, 1557–1565. [Google Scholar]
- O’Donovan, C.N.; Tobin, D.; Cotter, T.G. Prion protein fragment PrP-(106–126) induces apoptosis via mitochondrial disruption in human neuronal SH-SY5Y cells. J. Biol. Chem 2001, 276, 43516–43523. [Google Scholar]
- Rymer, D.L.; Good, T.A. The role of prion peptide structure and aggregation in toxicity and membrane binding. J. Neurochem 2000, 75, 2536–2545. [Google Scholar]
- Corsaro, A.; Thellung, S.; Villa, V.; Principe, D.R.; Paludi, D.; Arena, S.; Millo, E.; Schettini, D.; Damonte, G.; Aceto, A.; et al. Prion protein fragment 106–126 induces a p38 MAP kinase-dependent apoptosis in SH-SY5Y neuroblastoma cells independently from the amyloid fibril formation. Ann. N. Y. Acad. Sci 2003, 1010, 610–622. [Google Scholar]
- Chabry, J.; Ratsimanohatra, C.; Sponne, I.; Elena, P.P.; Vincent, J.P.; Pillot, T.; Drouet, B.; Pincon-Raymond, M.; Vandekerckhove, J.; Rosseneu, M.; et al. In vivo and in vitro neurotoxicity of the human prion protein (PrP) fragment P118-135 independently of PrP expression. J. Neurosci 2003, 23, 462–469. [Google Scholar]
- Pillot, T.; Drouet, B.; Pincon-Raymond, M.; Vandekerckhove, J.; Rosseneu, M.; Chambaz, J. A nonfibrillar form of the fusogenic prion protein fragment [118–135] induces apoptotic cell death in rat cortical neurons. J. Neurochem 2000, 75, 2298–2308. [Google Scholar]
- Bate, C.; Salmona, M.; Diomede, L.; Williams, A. Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J. Biol. Chem 2004, 279, 14983–14990. [Google Scholar]
- Bonetto, V.; Massignan, T.; Chiesa, R.; Morbin, M.; Mazzoleni, G.; Diomede, L.; Angeretti, N.; Colombo, L.; Forloni, G.; Tagliavini, F.; et al. Synthetic miniprion PrP106. J. Biol. Chem 2002, 277, 31327–31334. [Google Scholar]
- Tagliavini, F.; Prelli, F.; Verga, L.; Giaccone, G.; Sarma, R.; Gorevic, P.; Ghetti, B.; Passerini, F.; Ghibaudi, E.; Forloni, G.; et al. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 9678–9682. [Google Scholar]
- De Gioia, L.; Selvaggini, C.; Ghibaudi, E.; Diomede, L.; Bugiani, O.; Forloni, G.; Tagliavini, F.; Salmona, M. Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. J. Biol. Chem 1994, 269, 7859–7862. [Google Scholar]
- Selvaggini, C.; de Gioia, L.; Cantu, L.; Ghibaudi, E.; Diomede, L.; Passerini, F.; Forloni, G.; Bugiani, O.; Tagliavini, F.; Salmona, M. Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. Biochem. Biophys. Res. Commun 1993, 194, 1380–1386. [Google Scholar]
- Florio, T.; Paludi, D.; Villa, V.; Principe, D.R.; Corsaro, A.; Millo, E.; Damonte, G.; D’Arrigo, C.; Russo, C.; Schettini, G.; et al. Contribution of two conserved glycine residues to fibrillogenesis of the 106–126 prion protein fragment. Evidence that a soluble variant of the 106–126 peptide is neurotoxic. J. Neurochem 2003, 85, 62–72. [Google Scholar]
- Tagliavini, F.; Prelli, F.; Porro, M.; Rossi, G.; Giaccone, G.; Farlow, M.R.; Dlouhy, S.R.; Ghetti, B.; Bugiani, O.; Frangione, B. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell 1994, 79, 695–703. [Google Scholar]
- Thellung, S.; Florio, T.; Corsaro, A.; Arena, S.; Merlino, M.; Salmona, M.; Tagliavini, F.; Bugiani, O.; Forloni, G.; Schettini, G. Intracellular mechanisms mediating the neuronal death and astrogliosis induced by the prion protein fragment 106–126. Int. J. Dev. Neurosci 2000, 18, 481–492. [Google Scholar]
- Thellung, S.; Florio, T.; Villa, V.; Corsaro, A.; Arena, S.; Amico, C.; Robello, M.; Salmona, M.; Forloni, G.; Bugiani, O.; et al. Apoptotic cell death and impairment of L-type voltage-sensitive calcium channel activity in rat cerebellar granule cells treated with the prion protein fragment 106–126. Neurobiol. Dis 2000, 7, 299–309. [Google Scholar]
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol 1998, 8, 449–457. [Google Scholar]
- Hafiz, F.B.; Brown, D.R. A model for the mechanism of astrogliosis in prion disease. Mol. Cell Neurosci 2000, 16, 221–232. [Google Scholar]
- McHattie, S.J.; Brown, D.R.; Bird, M.M. Cellular uptake of the prion protein fragment PrP106-126 in vitro. J. Neurocytol 1999, 28, 149–159. [Google Scholar]
- Salmona, M.; Malesani, P.; de Gioia, L.; Gorla, S.; Bruschi, M.; Molinari, A.; Della Vedova, F.; Pedrotti, B.; Marrari, M.A.; Awan, T.; et al. Molecular determinants of the physicochemical properties of a critical prion protein region comprising residues 106–126. Biochem. J 1999, 342, 207–214. [Google Scholar]
- Walsh, P.; Neudecker, P.; Sharpe, S. Structural properties and dynamic behavior of nonfibrillar oligomers formed by PrP(106–126). J. Am. Chem. Soc 2010, 132, 7684–7695. [Google Scholar]
- Jackson, G.S.; Hosszu, L.; Power, A.; Hill, A.F.; Kenney, J.; Saibil, H.; Craven, C.; Waltho, J.P.; Clarke, A.R; Collinge, J. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 1999, 283, 1935–1937. [Google Scholar]
- Hornemann, S.; Schorn, C.; Wuthrich, K. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 2004, 5, 1159–1164. [Google Scholar]
- Corsaro, A.; Thellung, S.; Chiovitti, K.; Villa, V.; Simi, A.; Raggi, F.; Paludi, D.; Russo, C.; Aceto, A.; Florio, T. Dual modulation of ERK1/2 and p38 MAP kinase activities induced by minocycline reverses the neurotoxic effects of the prion protein fragment 90–231. Neurotox. Res 2009, 15, 138–154. [Google Scholar]
- Fioriti, L.; Angeretti, N.; Colombo, L.; De Luigi, A.; Colombo, A; Manzoni, C.; Morbin, M.; Tagliavini, F.; Salmona, M.; Chiesa, R.; et al. Neurotoxic and gliotrophic activity of a synthetic peptide homologous to GSS disease amyloid protein. J. Neurosci 2007, 27, 1576–1583. [Google Scholar]
- Thellung, S.; Corsaro, A.; Villa, V.; Simi, A.; Vella, S.; Pagano, A.; Florio, T. Human PrP90-231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death Dis 2011, 2, e138. [Google Scholar]
- Thellung, S.; Corsaro, A.; Villa, V.; Venezia, V.; Nizzari, M.; Bisaglia, M.; Russo, C.; Schettini, G.; Aceto, A.; Florio, T. Amino-terminally truncated prion protein PrP90-231 induces microglial activation in vitro. Ann. N. Y. Acad. Sci 2007, 1096, 258–270. [Google Scholar]
- Thellung, S.; Villa, V.; Corsaro, A.; Pellistri, F.; Venezia, V.; Russo, C.; Aceto, A.; Robello, M.; Florio, T. ERK1/2 and p38 MAP kinases control prion protein fragment 90-231-induced astrocyte proliferation and microglia activation. Glia 2007, 55, 1469–1485. [Google Scholar]
- Villa, V.; Tonelli, M.; Thellung, S.; Corsaro, A.; Tasso, B.; Novelli, F.; Canu, C.; Pino, A.; Chiovitti, K.; Paludi, D.; et al. Efficacy of novel acridine derivatives in the inhibition of hPrP90-231 prion protein fragment toxicity. Neurotox. Res 2011, 19, 556–574. [Google Scholar]
- Zou, W.Q.; Capellari, S.; Parchi, P.; Sy, M.S.; Gambetti, P.; Chen, S.G. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J. Biol. Chem 2003, 278, 40429–40436. [Google Scholar]
- Mange, A.; Beranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell Auspices Eur. Cell Biol. Org 2004, 96, 125–132. [Google Scholar]
- Lewis, V.; Hill, A.F.; Haigh, C.L.; Klug, G.M.; Masters, C.L.; Lawson, V.A.; Collins, S.J. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J. Neuropathol. Exp. Neurol 2009, 68, 1125–1135. [Google Scholar]
- Dron, M.; Moudjou, M.; Chapuis, J.; Salamat, M.K.; Bernard, J.; Cronier, S.; Langevin, C.; Laude, H. Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J. Biol. Chem 2010, 285, 10252–10264. [Google Scholar]
- Leffers, K.W.; Schell, J.; Jansen, K.; Lucassen, R.; Kaimann, T.; Nagel-Steger, L.; Tatzelt, J.; Riesner, D. The structural transition of the prion protein into its pathogenic conformation is induced by unmasking hydrophobic sites. J. Mol. Biol 2004, 344, 839–853. [Google Scholar]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wuthrich, K. NMR structure of the mouse prion protein domain PrP(121–321). Nature 1996, 382, 180–182. [Google Scholar]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar]
- Peretz, D.; Williamson, R.A.; Matsunaga, Y.; Serban, H.; Pinilla, C.; Bastidas, R.B.; Rozenshteyn, R.; James, T.L.; Houghten, R.A.; Cohen, F.E.; et al. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J. Mol. Biol 1997, 273, 614–622. [Google Scholar]
- Salmona, M.; Morbin, M.; Massignan, T.; Colombo, L.; Mazzoleni, G.; Capobianco, R.; Diomede, L.; Thaler, F.; Mollica, L.; Musco, G.; et al. Structural properties of Gerstmann-Straussler-Scheinker disease amyloid protein. J. Biol. Chem 2003, 278, 48146–48153. [Google Scholar]
- Holscher, C.; Delius, H.; Burkle, A. Overexpression of nonconvertible PrPc delta114-121 in scrapie-infected mouse neuroblastoma cells leads to trans-dominant inhibition of wild-type PrP(Sc) accumulation. J. Virol 1998, 72, 1153–1159. [Google Scholar]
- Norstrom, E.M.; Mastrianni, J.A. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J. Biol. Chem 2005, 280, 27236–27243. [Google Scholar]
- Gallo, M.; Paludi, D.; Cicero, D.O.; Chiovitti, K.; Millo, E.; Salis, A.; Damonte, G.; Corsaro, A.; Thellung, S.; Schettini, G.; et al. Identification of a conserved N-capping box important for the structural autonomy of the prion alpha 3-helix: The disease associated D202N mutation destabilizes the helical conformation. Int. J. Immunopathol. Pharmacol 2005, 18, 95–112. [Google Scholar]
- Baskakov, I.V.; Legname, G.; Prusiner, S.B.; Cohen, F.E. Folding of prion protein to its native alpha-helical conformation is under kinetic control. J. Biol. Chem 2001, 276, 19687–19690. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol 2005, 346, 645–659. [Google Scholar]
- Torrent, J.; Alvarez-Martinez, M.T.; Harricane, M.C.; Heitz, F.; Liautard, J.P.; Balny, C.; Lange, R. High pressure induces scrapie-like prion protein misfolding and amyloid fibril formation. Biochemistry 2004, 43, 7162–7170. [Google Scholar]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar]
- Corsaro, A.; Thellung, S.; Russo, C.; Villa, V.; Arena, S.; D’Adamo, M.C.; Paludi, D.; Rossi Principe, D.; Damonte, G.; Benatti, U.; et al. Expression in E. coli and purification of recombinant fragments of wild type and mutant human prion protein. Neurochem. Int 2002, 41, 55–63. [Google Scholar]
- Villa, V.; Corsaro, A.; Thellung, S.; Paludi, D.; Chiovitti, K.; Venezia, V.; Nizzari, M.; Russo, C.; Schettini, G.; Aceto, A.; Florio, T. Characterization of the proapoptotic intracellular mechanisms induced by a toxic conformer of the recombinant human prion protein fragment 90–231. Ann. N. Y. Acad. Sci 2006, 1090, 276–291. [Google Scholar]
- Paulis, D.; Maras, B.; Schinina, M.E.; di Francesco, L.; Principe, S.; Galeno, R.; Abdel-Haq, H.; Cardone, F.; Florio, T.; Pocchiari, M.; et al. The pathological prion protein forms ionic conductance in lipid bilayer. Neurochem. Int 2011, 59, 168–174. [Google Scholar]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Aceto, A.; Florio, T. Recombinant human prion protein fragment 90–231, a useful model to study prion neurotoxicity. Omics J. Integr. Biol 2012, 16, 50–59. [Google Scholar]
- May, B.C.; Govaerts, C.; Prusiner, S.B.; Cohen, F.E. Prions: So many fibers, so little infectivity. Trends Biochem. Sci 2004, 29, 162–165. [Google Scholar]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci 2003, 26, 267–298. [Google Scholar]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. (Berl) 2003, 81, 678–699. [Google Scholar]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar]
- Tzaban, S.; Friedlander, G.; Schonberger, O.; Horonchik, L.; Yedidia, Y.; Shaked, G.; Gabizon, R.; Taraboulos, A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002, 41, 12868–12875. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Salnikov, V.V.; Gill, A.C.; Baskakov, I.V. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease. Protein Sci 2005, 14, 1222–1232. [Google Scholar]
- Rezaei, H.; Eghiaian, F.; Perez, J.; Doublet, B.; Choiset, Y.; Haertle, T.; Grosclaude, J. Sequential generation of two structurally distinct ovine prion protein soluble oligomers displaying different biochemical reactivities. J. Mol. Biol 2005, 347, 665–679. [Google Scholar]
- Chiovitti, K.; Corsaro, A.; Thellung, S.; Villa, V.; Paludi, D.; D’Arrigo, C.; Russo, C.; Perico, A.; Ianieri, A.; di Cola, D.; et al. Intracellular accumulation of a mild-denatured monomer of the human PrP fragment 90–231, as possible mechanism of its neurotoxic effects. J. Neurochem 2007, 103, 2597–2609. [Google Scholar]
- Simoneau, S.; Rezaei, H.; Sales, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007, 3, e125. [Google Scholar]
- Paludi, D.; Thellung, S.; Chiovitti, K.; Corsaro, A.; Villa, V.; Russo, C.; Ianieri, A.; Bertsch, U.; Kretzschmar, H.A.; Aceto, A.; et al. Different structural stability and toxicity of PrP(ARR) and PrP(ARQ) sheep prion protein variants. J. Neurochem 2007, 103, 2291–2300. [Google Scholar]
- Baskakov, I.V.; Bocharova, O.V. In vitro conversion of mammalian prion protein into amyloid fibrils displays unusual features. Biochemistry 2005, 44, 2339–2348. [Google Scholar]
- Frankenfield, K.N.; Powers, E.T.; Kelly, J.W. Influence of the N-terminal domain on the aggregation properties of the prion protein. Protein Sci 2005, 14, 2154–2166. [Google Scholar]
- Zhou, M.; Ottenberg, G.; Sferrazza, G.F.; Lasmezas, C.I. Highly neurotoxic monomeric alpha-helical prion protein. Proc. Natl. Acad. Sci. USA 2012, 109, 3113–3118. [Google Scholar]
- Lawson, V.A.; Priola, S.A.; Wehrly, K.; Chesebro, B. N-terminal truncation of prion protein affects both formation and conformation of abnormal protease-resistant prion protein generated in vitro. J. Biol. Chem 2001, 276, 35265–35271. [Google Scholar]
- Lawson, V.A.; Priola, S.A.; Meade-White, K.; Lawson, M.; Chesebro, B. Flexible N-terminal region of prion protein influences conformation of protease-resistant prion protein isoforms associated with cross-species scrapie infection in vivo and in vitro. J. Biol. Chem 2004, 279, 13689–13695. [Google Scholar]
- Goldstein, R.F.; Stryer, L. Cooperative polymerization reactions. Analytical approximations, numerical examples, and experimental strategy. Biophys. J 1986, 50, 583–599. [Google Scholar]
- Harper, J.D.; Lieber, C.M.; Lansbury, P.T., Jr. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-beta protein. Chem. Biol 1997, 4, 951–959. [Google Scholar]
- Ferrone, F. Analysis of protein aggregation kinetics. Methods Enzymol 1999, 309, 256–274. [Google Scholar]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar]
- Hesketh, S.; Thompsett, A.R.; Brown, D.R. Prion protein polymerisation triggered by manganese-generated prion protein seeds. J. Neurochem 2012, 120, 177–189. [Google Scholar]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci 2001, 24, 519–550. [Google Scholar]
- Rosenmann, H.; Talmor, G.; Halimi, M.; Yanai, A.; Gabizon, R.; Meiner, Z. Prion protein with an E200K mutation displays properties similar to those of the cellular isoform PrP(C). J. Neurochem 2001, 76, 1654–1662. [Google Scholar]
- Zhang, Y.; Swietnicki, W.; Zagorski, M.G.; Surewicz, W.K.; Sonnichsen, F.D. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J. Biol. Chem 2000, 275, 33650–33654. [Google Scholar]
- Hasegawa, K.; Mohri, S.; Yokoyama, T. Fragment molecular orbital calculations reveal that the E200K mutation markedly alters local structural stability in the human prion protein. Prion 2010, 4, 38–44. [Google Scholar]
- Van der Kamp, M.W.; Daggett, V. The consequences of pathogenic mutations to the human prion protein. Protein Eng. Des. Sel 2009, 22, 461–468. [Google Scholar]
- Yin, S.; Pham, N.; Yu, S.; Li, C.; Wong, P.; Chang, B.; Kang, S.C.; Biasini, E.; Tien, P.; Harris, D.A.; et al. Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc. Natl. Acad. Sci. USA 2007, 104, 7546–7551. [Google Scholar]
- Corsaro, A.; Thellung, S.; Bucciarelli, T.; Scotti, L.; Chiovitti, K.; Villa, V.; D’Arrigo, C.; Aceto, A.; Florio, T. High hydrophobic amino acid exposure is responsible of the neurotoxic effects induced by E200K or D202N disease-related mutations of the human prion protein. Int. J. Biochem. Cell Biol 2011, 43, 372–382. [Google Scholar]
- Capellari, S.; Parchi, P.; Russo, C.M.; Sanford, J.; Sy, M.S.; Gambetti, P.; Petersen, R.B. Effect of the E200K mutation on prion protein metabolism. Comparative study of a cell model and human brain. Am. J. Pathol 2000, 157, 613–622. [Google Scholar]
- Williams, E.S.; Young, S. Spongiform encephalopathies in Cervidae. Rev. Sci. Tech. (International Office of Epizootics) 1992, 11, 551–567. [Google Scholar]
- Miller, M.W.; Williams, E.S.; Hobbs, N.T.; Wolfe, L.L. Environmental sources of prion transmission in mule deer. Emerg. Infect. Dis 2004, 10, 1003–1006. [Google Scholar]
- Purdey, M. Elevated silver, barium and strontium in antlers, vegetation and soils sourced from CWD cluster areas: Do Ag/Ba/Sr piezoelectric crystals represent the transmissible pathogenic agent in TSEs? Med. Hypotheses 2004, 63, 211–225. [Google Scholar]
- Ragnarsdottir, K.V.; Hawkins, D.P. Bioavailable copper and manganese in soils from Iceland and their relationship with scrapie occurrence in sheep. J. Geochem. Explor 2006, 88, 228–234. [Google Scholar]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Prions in the environment: Occurrence, fate and mitigation. Prion 2008, 2, 162–169. [Google Scholar]
- Gough, K.C.; Maddison, B.C. Prion transmission: Prion excretion and occurrence in the environment. Prion 2010, 4, 275–282. [Google Scholar]
- Brown, P.; Gajdusek, D.C. Survival of scrapie virus after 3 years’ interment. Lancet 1991, 337, 269–270. [Google Scholar]
- Saunders, S.E.; Yuan, Q.; Bartz, J.C.; Bartelt-Hunt, S. Effects of solution chemistry and aging time on prion protein adsorption and replication of soil-bound prions. PLoS One 2011, 6, e18752. [Google Scholar]
- Saunders, S.E.; Shikiya, R.A.; Langenfeld, K.; Bartelt-Hunt, S.L.; Bartz, J.C. Replication efficiency of soil-bound prions varies with soil type. J. Virol 2011, 85, 5476–5482. [Google Scholar]
- Seidel, B.; Thomzig, A.; Buschmann, A.; Groschup, M.H.; Peters, R.; Beekes, M.; Terytze, K. Scrapie Agent (Strain 263K) can transmit disease via the oral route after persistence in soil over years. PLoS One 2007, 2, e435. [Google Scholar]
- Johnson, C.J.; Pedersen, J.A.; Chappell, R.J.; McKenzie, D.; Aiken, J.M. Oral transmissibility of prion disease is enhanced by binding to soil particles. PLoS Pathog 2007, 3, e93. [Google Scholar]
- Gonzalez-Romero, D.; Barria, M.A.; Leon, P.; Morales, R.; Soto, C. Detection of infectious prions in urine. FEBS Lett 2008, 582, 3161–3166. [Google Scholar]
- Corsaro, A.; Anselmi, C.; Polano, M.; Aceto, A.; Florio, T.; de Nobili, M. The interaction of humic substances with the human prion protein fragment 90–231 affects its protease K resistance and cell internalization. J. Biol. Regul. Homeost. Agents 2010, 24, 27–39. [Google Scholar]
- Polano, M.; Anselmi, C.; Leita, L.; Negro, A.; de Nobili, M. Organic polyanions act as complexants of prion protein in soil. Biochem. Biophys. Res. Commun 2008, 367, 323–329. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Disease | Affected Species |
|---|---|
| Kuru | Human |
| Creutzfeldt-Jakob disease (CJD) | |
| sporadic Creutzfeldt-Jakob disease (sCJD) | |
| iatrogenic Creutzfeldt-Jakob disease (iCJD) | |
| variant Creutzfeldt-Jakob disease (vCJD) | |
| familial Creutzfeldt-Jakob disease (fCJD) | |
| Gerstmann-Sträussler-Scheinker syndrome (GSS) | |
| Fatal familial insomnia (FFI) | |
| Scrapie | sheep, goat cattle elk, mule deer, moose cat |
| Bovine spongiform encephalopathy (BSE) | |
| Chronic wasting disease (CWD) | |
| Feline spongiform encephalopathy (FSE) | |
| Prion Protein Mutation | Prion Disease |
|---|---|
| P102L, P105L, A117V, G131V, F198S, D202N, Q217R, M232T | Gerstmann-Straussler-Scheinker disease (GSS) |
| D178N + (variant Val 129) V180N, M232R, T183A, E200K, R208H, V210I, M232R | Creutzfeldt-Jakob disease (CJD) |
| D178N + (variant Met 129) | Fatal familial insomnia (FFI) |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of Prion Protein Aggregation in Neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648-8669. https://doi.org/10.3390/ijms13078648
Corsaro A, Thellung S, Villa V, Nizzari M, Florio T. Role of Prion Protein Aggregation in Neurotoxicity. International Journal of Molecular Sciences. 2012; 13(7):8648-8669. https://doi.org/10.3390/ijms13078648
Chicago/Turabian StyleCorsaro, Alessandro, Stefano Thellung, Valentina Villa, Mario Nizzari, and Tullio Florio. 2012. "Role of Prion Protein Aggregation in Neurotoxicity" International Journal of Molecular Sciences 13, no. 7: 8648-8669. https://doi.org/10.3390/ijms13078648
APA StyleCorsaro, A., Thellung, S., Villa, V., Nizzari, M., & Florio, T. (2012). Role of Prion Protein Aggregation in Neurotoxicity. International Journal of Molecular Sciences, 13(7), 8648-8669. https://doi.org/10.3390/ijms13078648