Revealing the Anti-Tumor Effect of Artificial miRNA p-27-5p on Human Breast Carcinoma Cell Line T-47D


Abstract
:
1. Introduction
2. Results and Discussion
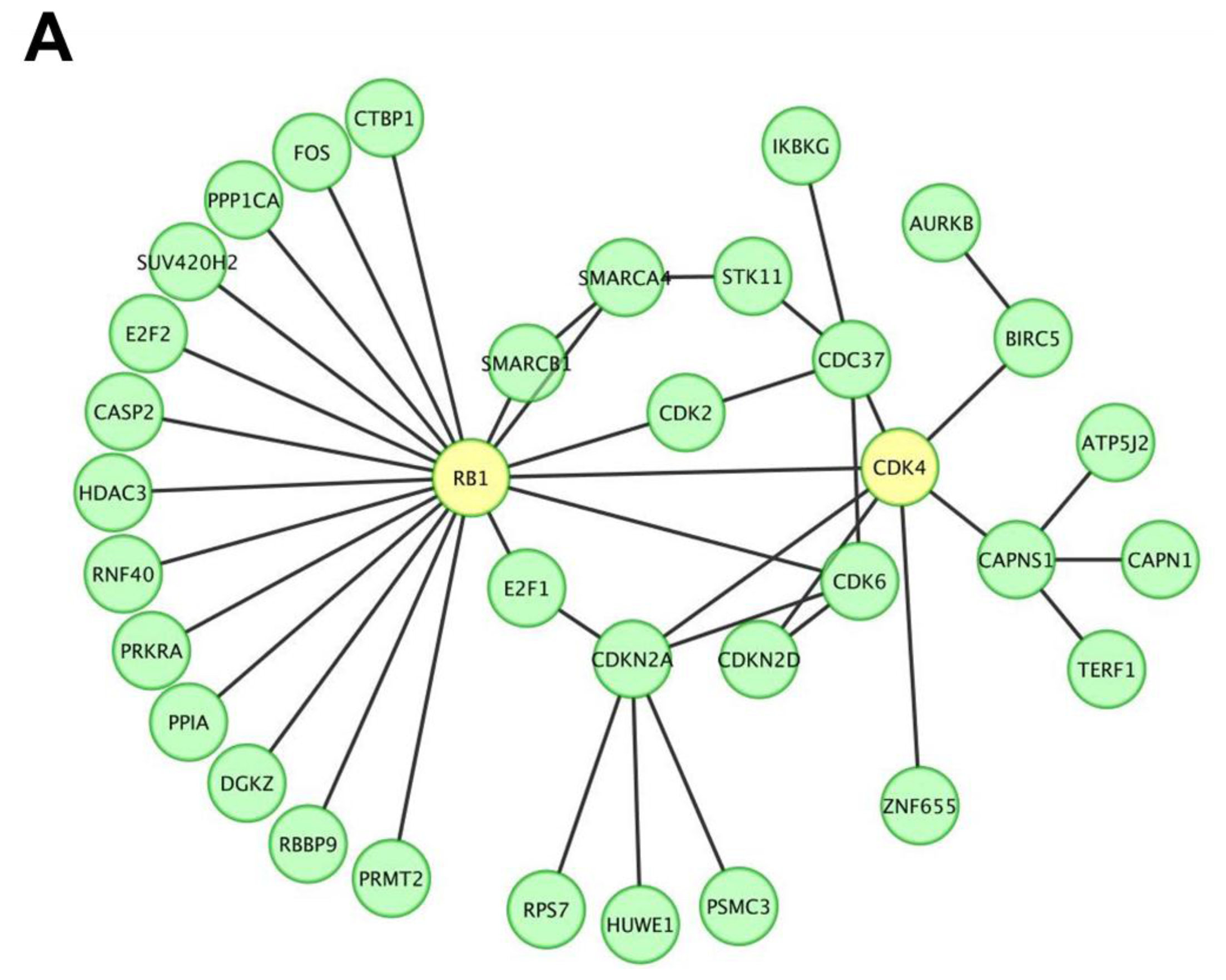
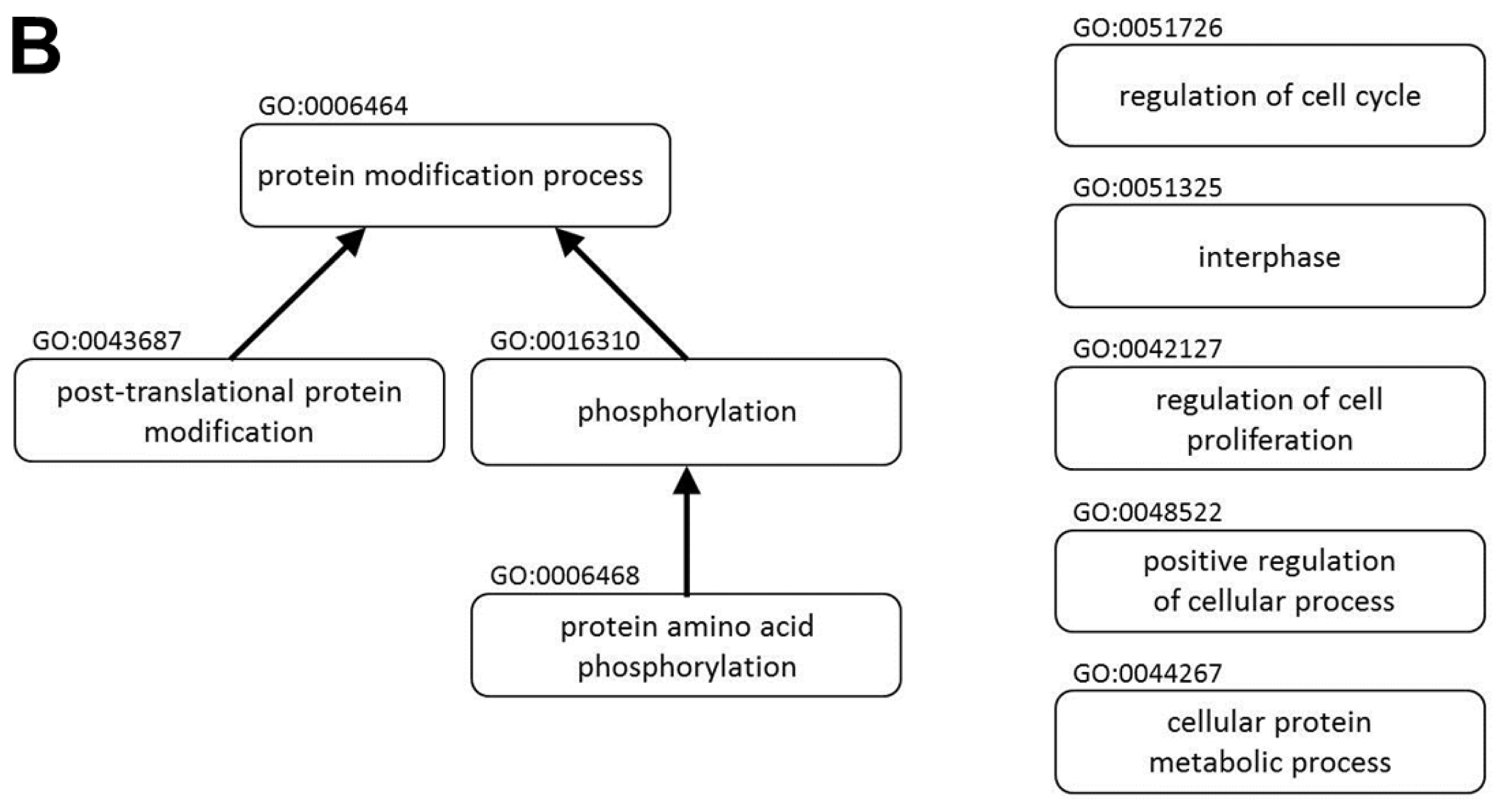
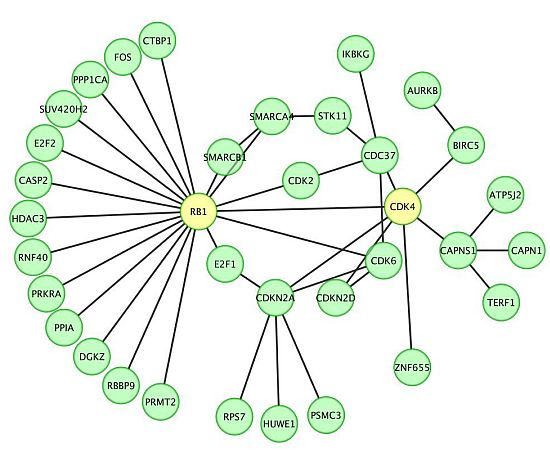
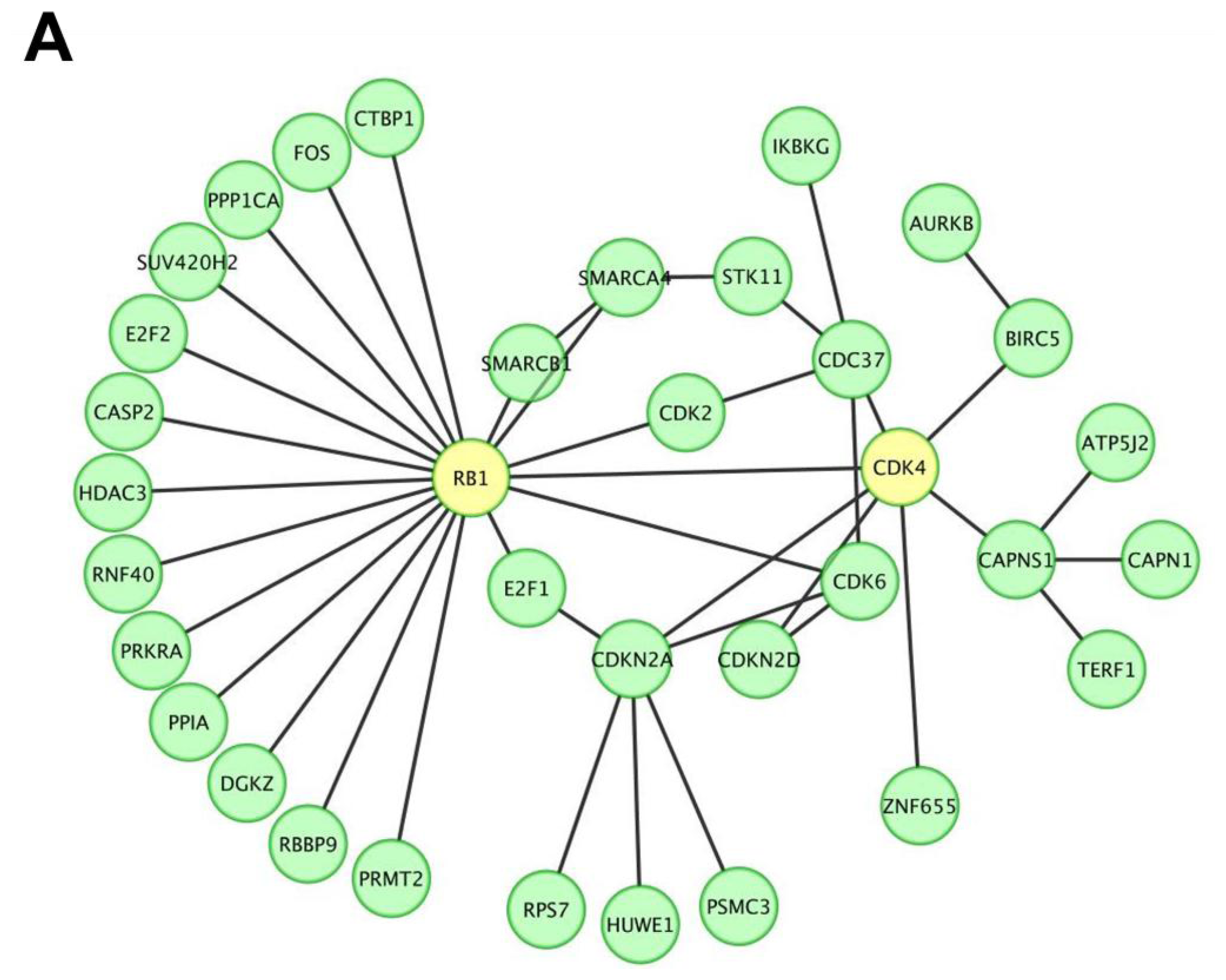
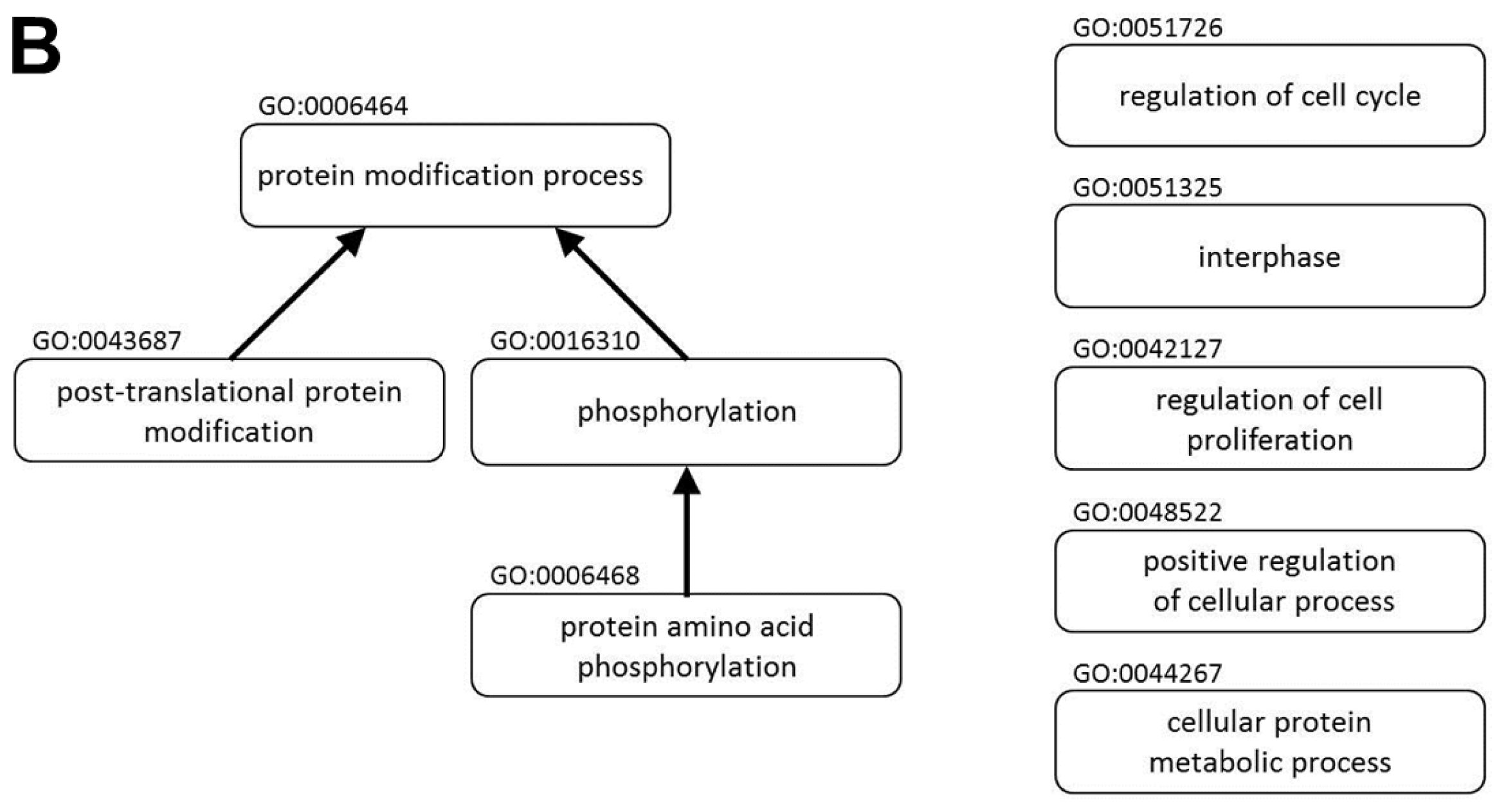
2.1. miR P-27-5p Downregulates Gene Expressions Associated with Cell Growth, Cell Cycle, and Phosphorylation in T-47D Cells
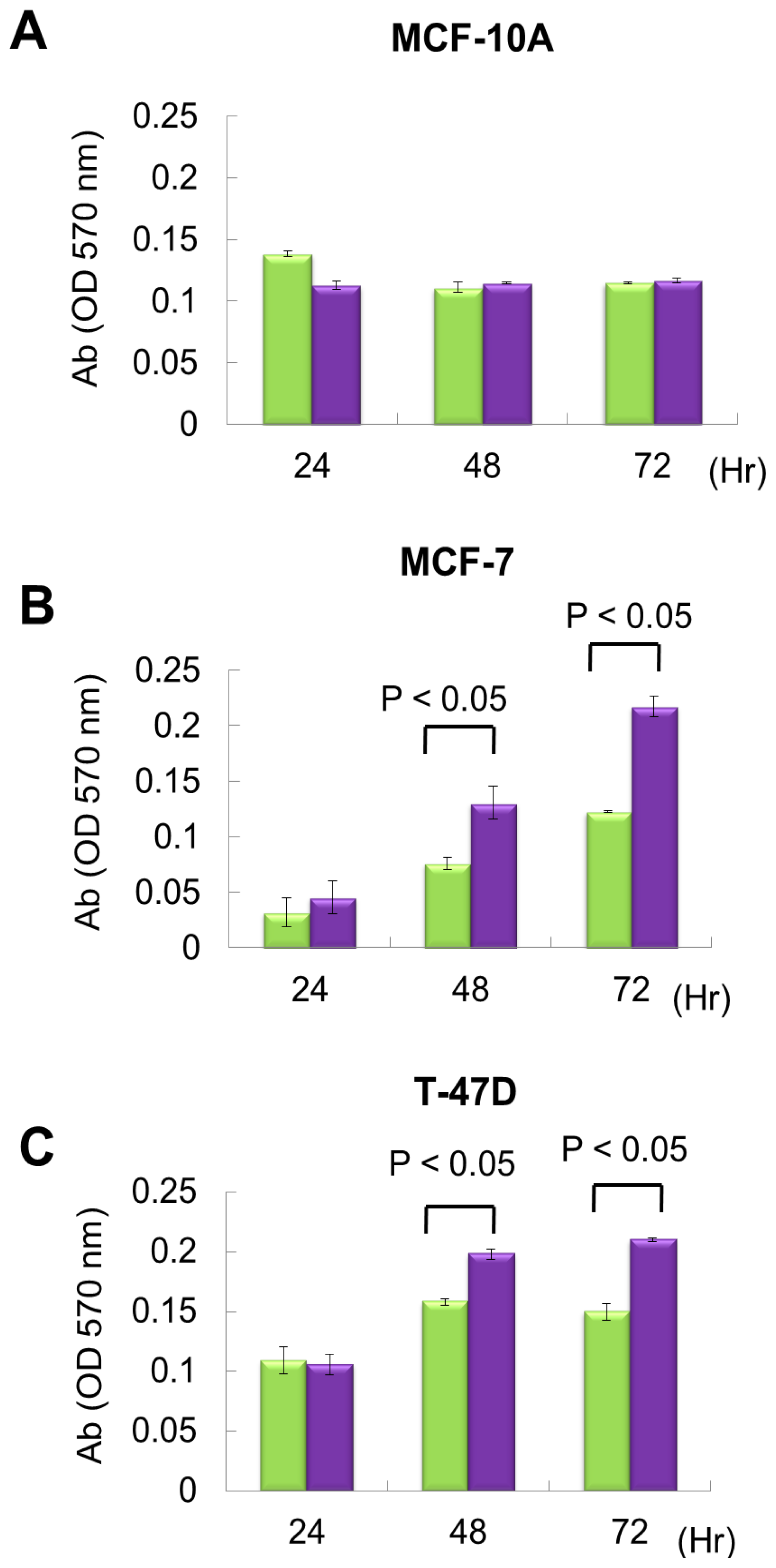
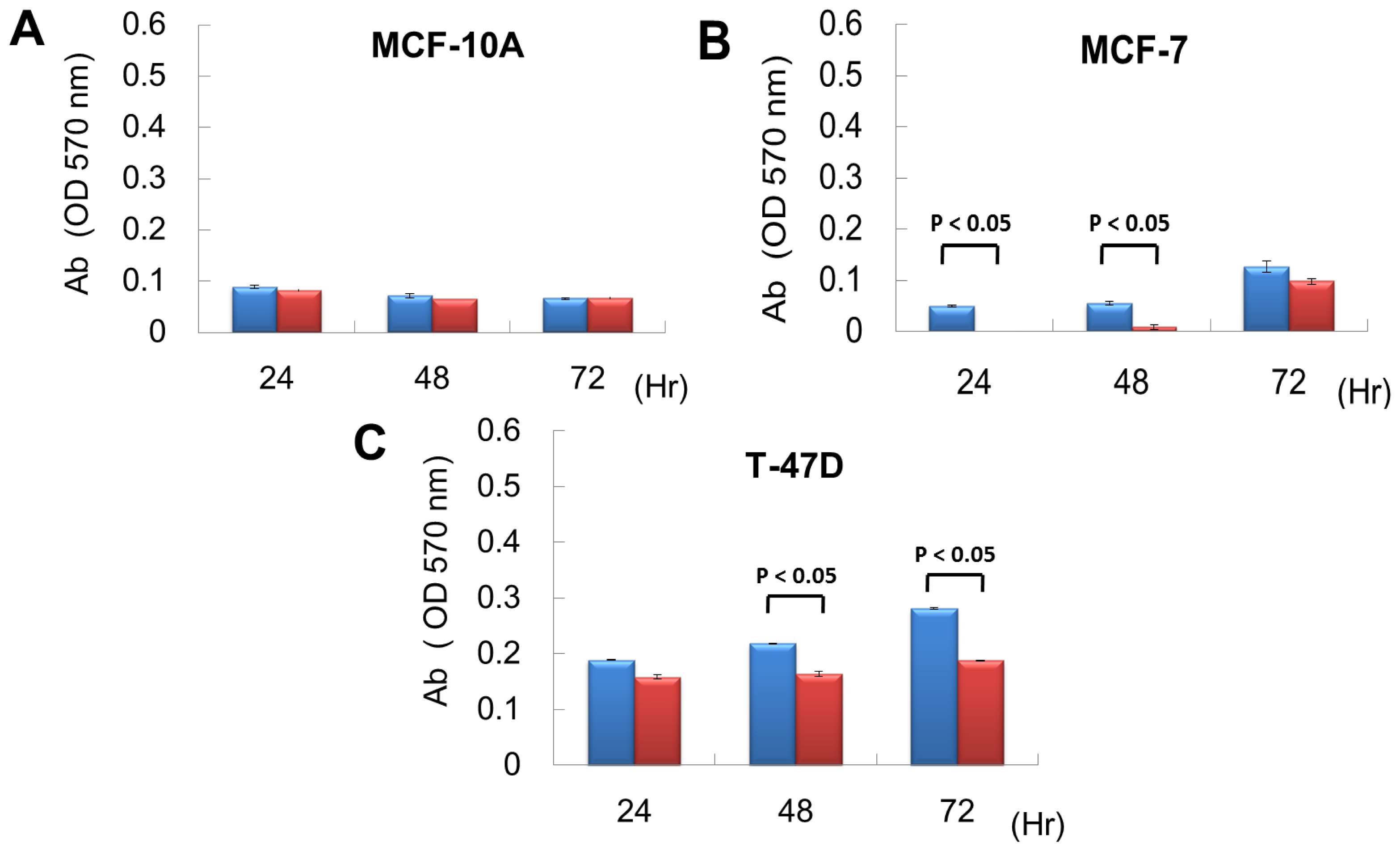
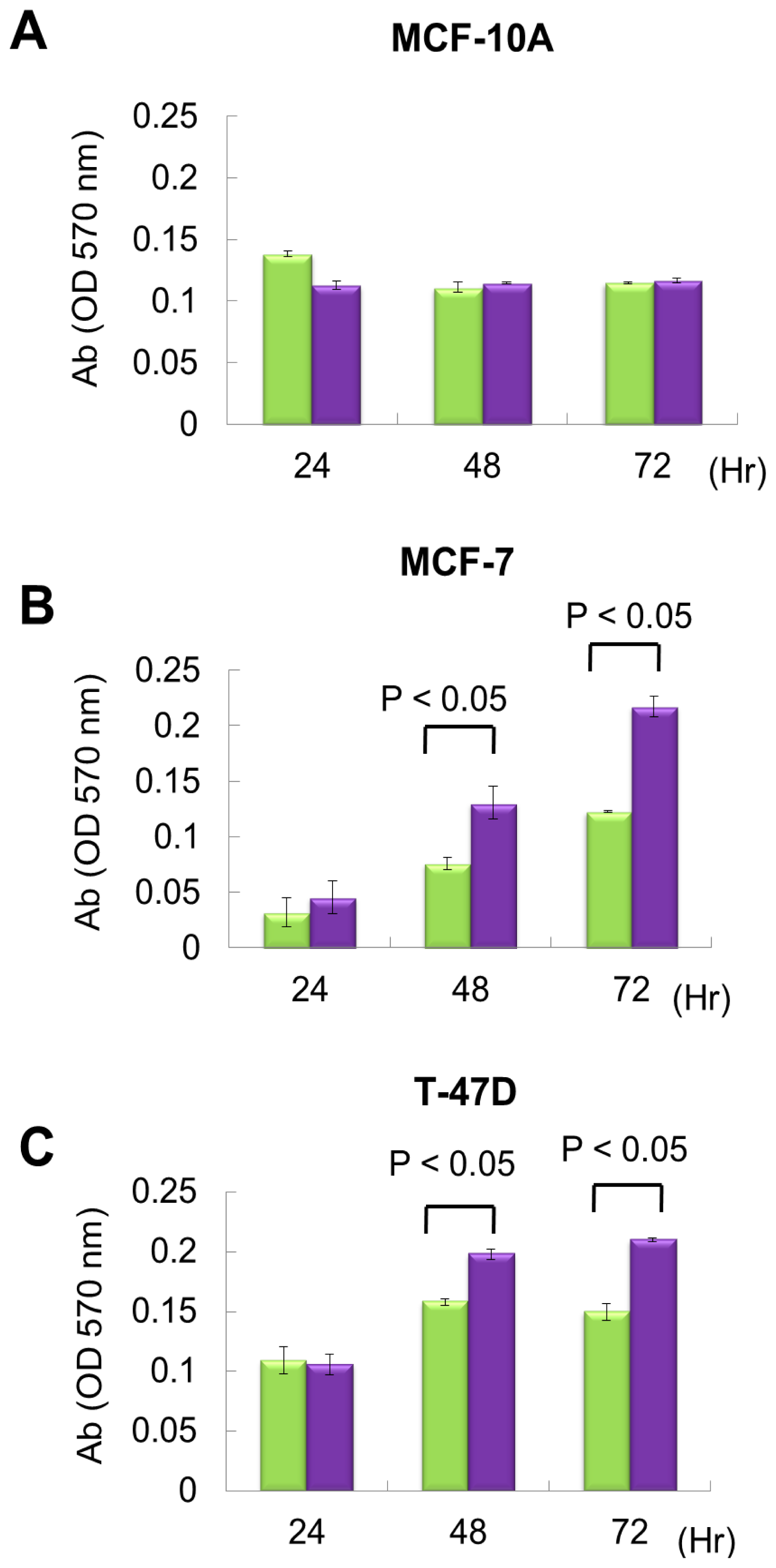
2.2. miR P-27-5p Overexpression Inhibits the Growth of Breast Cancer Cells
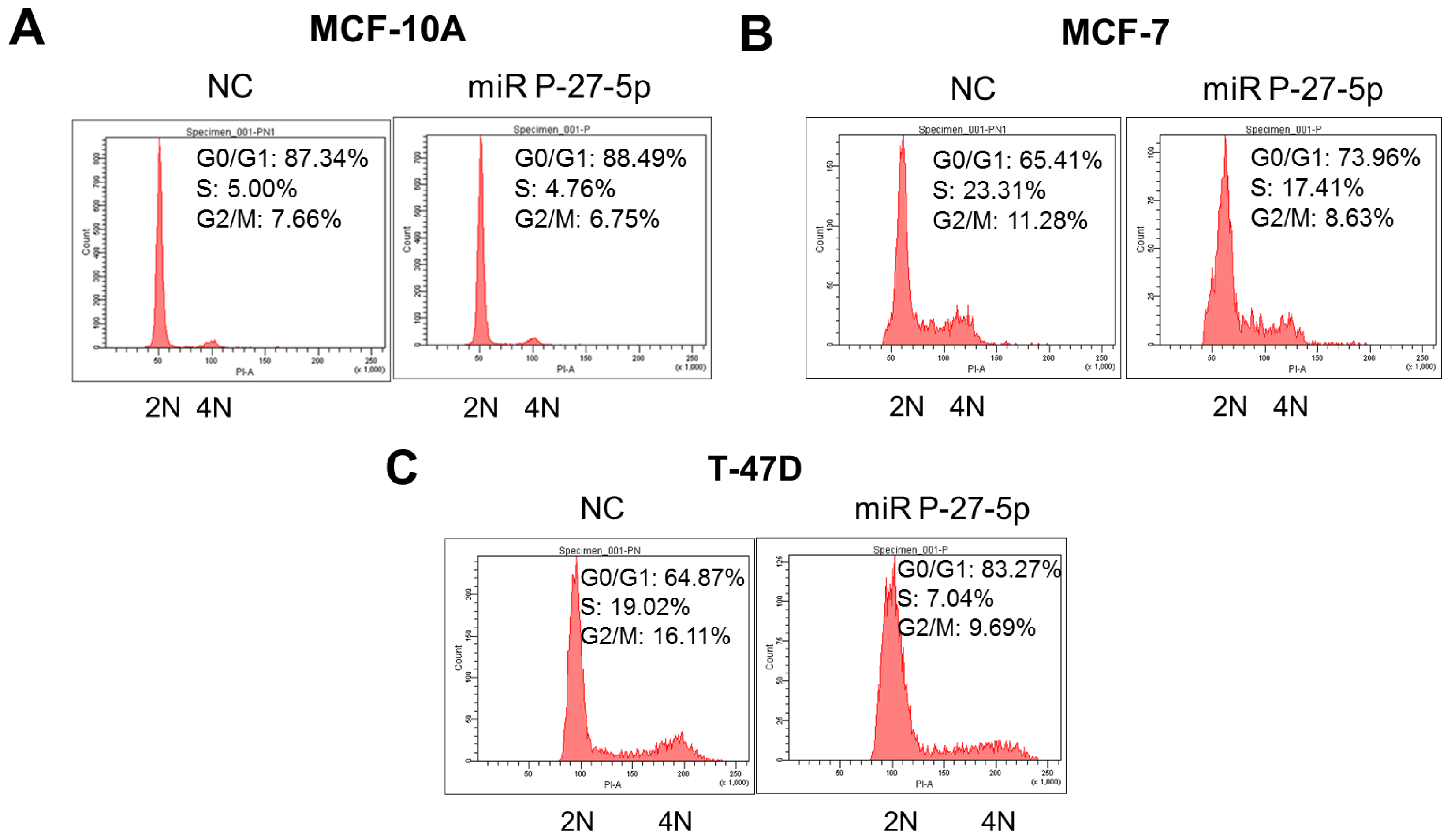
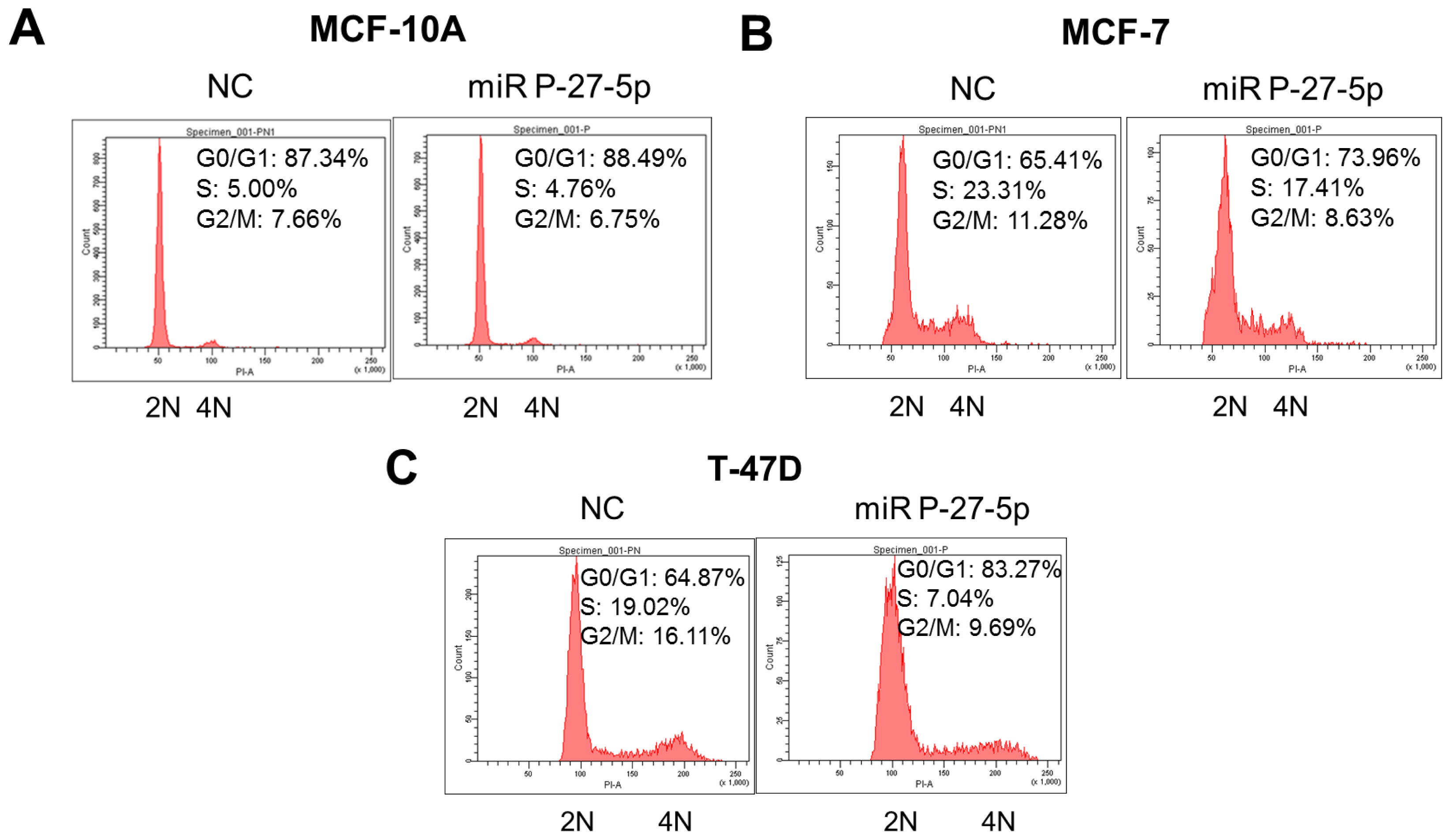
2.3. miR P-27-5p Induces G1 Arrest in Breast Cancer Cells
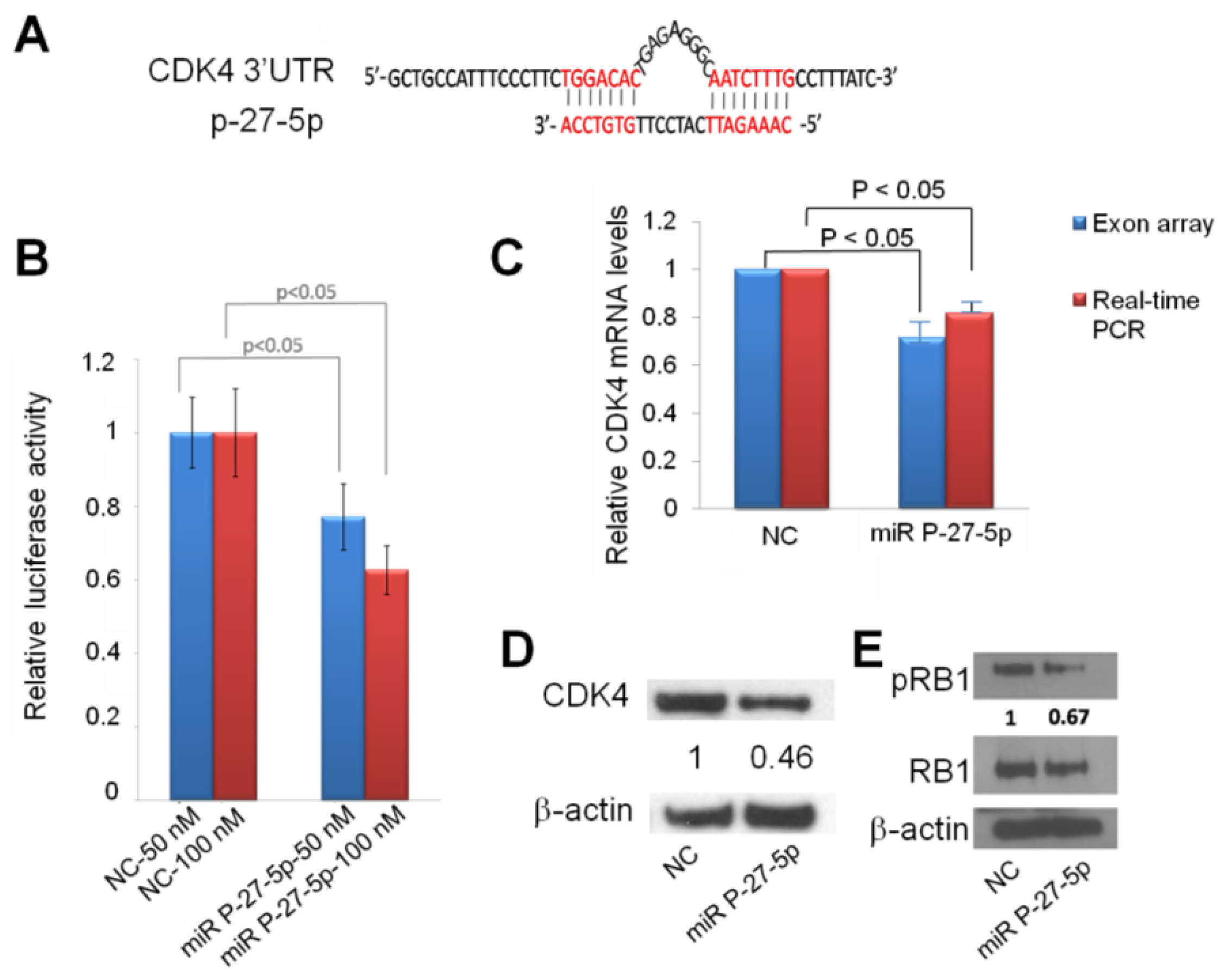
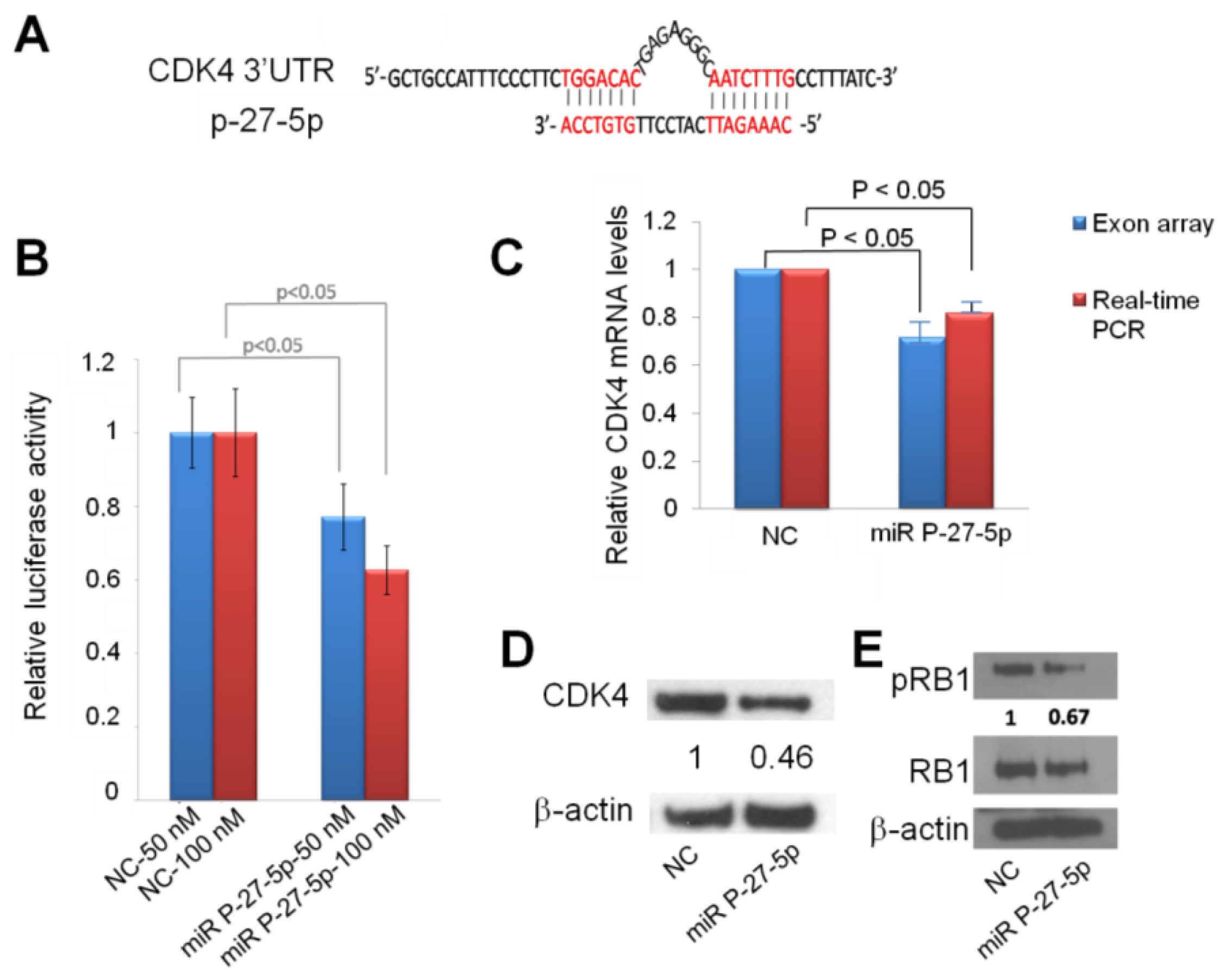
2.4. Down-Regulation of CDK4 by miR P-27-5p at mRNA and Protein Levels via Direct Targeting 3′-UTR
2.5. miR P-27-5p Inhibits RB1 Phosphorylation
2.6. Discussion
3. Experimental Section
3.1. Cell Culture and Transfection
3.2. Exon Arrays and Data Analysis
3.3. Protein Interaction Network and Functional Enrichment
3.4. Cell Viability Assay
3.5. Cell Cycle Analysis
3.6. Construction of the CDK4 3′-UTR Report Plasmids and Luciferase Assay
3.7. Quantitative Real-Time PCR
3.8. Western Blot
3.9. Statistical Analysis
4. Conclusions
Supplementary Material
ijms-13-06352-s001.xlsAcknowledgments
References
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar]
- Ventura, A.; Jacks, T. MicroRNAs and cancer: Short RNAs go a long way. Cell 2009, 136, 586–591. [Google Scholar]
- Mattie, M.D.; Benz, C.C.; Bowers, J.; Sensinger, K.; Wong, L.; Scott, G.K.; Fedele, V.; Ginzinger, D.; Getts, R.; Haqq, C. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol. Cancer 2006, 5. [Google Scholar] [CrossRef]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar]
- Lieberman, J. Micromanaging cancer. N. Engl. J. Med 2009, 361, 1500–1501. [Google Scholar]
- Chang, Y.M.; Juan, H.F.; Lee, T.Y.; Chang, Y.Y.; Yeh, Y.M.; Li, W.H.; Shih, A.C. Prediction of human miRNAs using tissue-selective motifs in 3′ UTRs. Proc. Natl. Acad. Sci. USA 2008, 105, 17061–17066. [Google Scholar]
- Yue, H.; Eastman, P.S.; Wang, B.B.; Minor, J.; Doctolero, M.H.; Nuttall, R.L.; Stack, R.; Becker, J.W.; Montgomery, J.R.; Vainer, M.; et al. An evaluation of the performance of cDNA microarrays for detecting changes in global mRNA expression. Nucleic Acids Res 2001, 29. [Google Scholar] [CrossRef]
- Ideker, T.; Thorsson, V.; Ranish, J.A.; Christmas, R.; Buhler, J.; Eng, J.K.; Bumgarner, R.; Goodlett, D.R.; Aebersold, R.; Hood, L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 2001, 292, 929–934. [Google Scholar]
- Hughes, T.R.; Marton, M.J.; Jones, A.R.; Roberts, C.J.; Stoughton, R.; Armour, C.D.; Bennett, H.A.; Coffey, E.; Dai, H.; He, Y.D.; et al. Functional discovery via a compendium of expression profiles. Cell 2000, 102, 109–126. [Google Scholar]
- Abdueva, D.; Wing, M.R.; Schaub, B.; Triche, T.J. Experimental comparison and evaluation of the Affymetrix exon and U133Plus2 GeneChip arrays. PLoS One 2007, 2. [Google Scholar] [CrossRef]
- Seo, J.; Kim, M.; Kim, J. Identification of novel genes differentially expressed in PMA-induced HL-60 cells using cDNA microarrays. Mol. Cells 2000, 10, 733–739. [Google Scholar]
- Le Naour, F.; Hohenkirk, L.; Grolleau, A.; Misek, D.E.; Lescure, P.; Geiger, J.D.; Hanash, S.; Beretta, L. Profiling changes in gene expression during differentiation and maturation of monocyte-derived dendritic cells using both oligonucleotide microarrays and proteomics. J. Biol. Chem 2001, 276, 17920–17931. [Google Scholar]
- Dong, Y.; Sui, L.; Sugimoto, K.; Tai, Y.; Tokuda, M. Cyclin D1-CDK4 complex, a possible critical factor for cell proliferation and prognosis in laryngeal squamous cell carcinomas. Int. J. Cancer 2001, 95, 209–215. [Google Scholar]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar]
- Sun, Y.; Li, Y.X.; Wu, H.J.; Wu, S.H.; Wang, Y.A.; Luo, D.Z.; Liao, D.J. Effects of an indolocarbazole-derived CDK4 inhibitor on breast cancer cells. J. Cancer 2011, 2, 36–51. [Google Scholar]
- Lin, S.L.; Chang, D.C.; Ying, S.Y.; Leu, D.; Wu, D.T. MicroRNA miR-302 inhibits the tumorigenecity of human pluripotent stem cells by coordinate suppression of the CDK2 and CDK4/6 cell cycle pathways. Cancer Res 2010, 70, 9473–9482. [Google Scholar]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar]
- Yoon, S.; Choi, Y.C.; Lee, S.; Jeong, Y.; Yoon, J.; Baek, K. Induction of growth arrest by miR-542–3p that targets survivin. FEBS Lett 2010, 584, 4048–4052. [Google Scholar]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar]
- Tseng, C.W.; Lin, C.C.; Chen, C.N.; Huang, H.C.; Juan, H.F. Integrative network analysis reveals active microRNAs and their functions in gastric cancer. BMC Syst. Biol 2011, 5. [Google Scholar] [CrossRef]
- Tseng, C.W.; Yang, J.C.; Chen, C.N.; Huang, H.C.; Chuang, K.N.; Lin, C.C.; Lai, H.S.; Lee, P.H.; Chang, K.J.; Juan, H.F. Identification of 14-3-3beta in human gastric cancer cells and its potency as a diagnostic and prognostic biomarker. Proteomics 2011, 11, 2423–2439. [Google Scholar]
- Mathivanan, S.; Periaswamy, B.; Gandhi, T.K.; Kandasamy, K.; Suresh, S.; Mohmood, R.; Ramachandra, Y.L.; Pandey, A. An evaluation of human protein-protein interaction data in the public domain. BMC Bioinform 2006, 7. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar]
- Sutherland, R.L.; Musgrove, E.A. CDK inhibitors as potential breast cancer therapeutics: New evidence for enhanced efficacy in ER+ disease. Breast Cancer Res 2009, 11. [Google Scholar] [CrossRef]
- Zelivianski, S.; Cooley, A.; Kall, R.; Jeruss, J.S. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol. Cancer Res 2010, 8, 1375–1387. [Google Scholar]
- Farazi, T.A.; Horlings, H.M.; ten Hoeve, J.J.; Mihailovic, A.; Halfwerk, H.; Morozov, P.; Brown, M.; Hafner, M.; Reyal, F.; van Kouwenhove, M.; et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res 2011, 71, 4443–4453. [Google Scholar]
- Takada, S.; Berezikov, E.; Yamashita, Y.; Lagos-Quintana, M.; Kloosterman, W.P.; Enomoto, M.; Hatanaka, H.; Fujiwara, S.; Watanabe, H.; Soda, M.; et al. Mouse microRNA profiles determined with a new and sensitive cloning method. Nucleic Acids Res 2006, 34. [Google Scholar] [CrossRef]
- Kuhn, D.E.; Nuovo, G.J.; Martin, M.M.; Malana, G.E.; Pleister, A.P.; Jiang, J.; Schmittgen, T.D.; Terry, A.V., Jr; Gardiner, K.; Head, E.; et al. Human chromosome 21-derived miRNAs are overexpressed in down syndrome brains and hearts. Biochem. Biophys. Res. Commun 2008, 370, 473–477. [Google Scholar]
- Kwon, O.H.; Park, J.L.; Kim, M.; Kim, J.H.; Lee, H.C.; Kim, H.J.; Noh, S.M.; Song, K.S.; Yoo, H.S.; Paik, S.G.; et al. Aberrant up-regulation of LAMB3 and LAMC2 by promoter demethylation in gastric cancer. Biochem. Biophys. Res. Commun 2011, 406, 539–545. [Google Scholar]
- Kita, Y.; Mimori, K.; Tanaka, F.; Matsumoto, T.; Haraguchi, N.; Ishikawa, K.; Matsuzaki, S.; Fukuyoshi, Y.; Inoue, H.; Natsugoe, S.; et al. Clinical significance of LAMB3 and COL7A1 mRNA in esophageal squamous cell carcinoma. Eur. J. Surg. Oncol 2009, 35, 52–58. [Google Scholar]
- Calaluce, R.; Bearss, D.J.; Barrera, J.; Zhao, Y.; Han, H.; Beck, S.K.; McDaniel, K.; Nagle, R.B. Laminin-5 beta3A expression in LNCaP human prostate carcinoma cells increases cell migration and tumorigenicity. Neoplasia 2004, 6, 468–479. [Google Scholar]
- De Guire, V.; Caron, M.; Scott, N.; Menard, C.; Gaumont-Leclerc, M.F.; Chartrand, P.; Major, F.; Ferbeyre, G. Designing small multiple-target artificial RNAs. Nucleic Acids Res 2010, 38. [Google Scholar] [CrossRef]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet 2009, 10, 578–585. [Google Scholar]
- Idogawa, M.; Sasaki, Y.; Suzuki, H.; Mita, H.; Imai, K.; Shinomura, Y.; Tokino, T. A single recombinant adenovirus expressing p53 and p21-targeting artificial microRNAs efficiently induces apoptosis in human cancer cells. Clin. Cancer Res 2009, 15, 3725–3732. [Google Scholar]
- Prud’homme, G.J.; Glinka, Y.; Toulina, A.; Ace, O.; Subramaniam, V.; Jothy, S. Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Zhong, Z.; Yeow, W.S.; Zou, C.; Wassell, R.; Wang, C.; Pestell, R.G.; Quong, J.N.; Quong, A.A. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res 2010, 70, 2105–2114. [Google Scholar]
- Ye, X.; Liu, Z.; Hemida, M.G.; Yang, D. Targeted delivery of mutant tolerant anti-coxsackievirus artificial microRNAs using folate conjugated bacteriophage Phi29 pRNA. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- dChip Software: Analysis and visualization of gene expression and SNP microarrays. Available online: http://biosun1.harvard.edu/complab/dchip accessed on 3 January 2012.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Gene List | p-Value | Adj. p-Value |
|---|---|---|---|
| protein modification process (GO:0006464) | STK36 STYX USE1 EIF5A CASK RPS6KB2 AURKB MAP3K6 MAP3K5 ERO1LB ST3GAL3 TRAK1 IFNG CLK4 LOX INSR SYK EGFR DBNL BCR TNIK PTPRF DTL WNK1 PTPRS PIM2 HDAC11 SUV420H2 PPP1CA HUWE1 MAPK3 AMFR UBA52 TNFAIP1 GGCX FGFR4 ERBB3 STK11 ERBB2 MAPKAPK3 NEDD8 ANAPC11 EPHB3 ACP1 SRC EPHB6 UBE2D2 PTK6 PRKRA GNMT TRAF7 MGAT4B B4GALT1 P4HB MAP2K2 TKT HDAC5 CDC42BPG HDAC3 PLK1 DOHH GRK6 SPTBN1 EAF2 MERTK HDAC6 BCKDK FASTK UCHL1 CAD AKT1 PAK7 PIGM PRMT2 ASH2L HSF1 PAK4 TGFA RAB6A KLHL20 CTBP1 PIGX DDB1 PIGU PIGT CDK6 UBE2I CDK4 PRKCE CDK2 SRPK1 G6PD BTG1 DDB2 DHPS SERP1 PRKCZ PPP4R2 RAB3D UBE3A USP5 GPAA1 ABI2 STUB1 IGF1R STT3A CDC2L2 PER1 C19ORF62 PRKAA2 PPP3CA THBS1 YES1 QSOX1 KAT2A PDK2 LRSAM1 FLT1 FLT4 SIRT2 RPS6KA1 MAPK13 CSNK1E RSRC1 MAPK15 ARAF JAK1 DPM2 DPM3 IRF4 RBM14 RNF40 | 7.98 × 10−8 | 6.34 × 10−6 |
| protein amino acid phosphorylation (GO:0006468) | BCKDK FASTK STK36 CASK RPS6KB2 CAD AURKB AKT1 PAK7 MAP3K6 MAP3K5 HSF1 PAK4 IFNG CLK4 TGFA INSR SYK EGFR DBNL CTBP1 BCR TNIK WNK1 CDK6 PIM2 CDK4 PRKCE CDK2 SRPK1 MAPK3 PRKCZ FGFR4 ERBB3 STK11 ERBB2 MAPKAPK3 ABI2 EPHB3 SRC IGF1R EPHB6 CDC2L2 PTK6 PRKRA PRKAA2 THBS1 YES1 PDK2 FLT1 MAP2K2 FLT4 TKT CDC42BPG RPS6KA1 MAPK13 CSNK1E PLK1 RSRC1 MAPK15 ARAF GRK6 JAK1 SPTBN1 MERTK | 3.24 × 10−6 | 1.43 × 10−4 |
| phosphorylation (GO:0016310) | BCKDK UQCRC1 FASTK STK36 SNCA CASK RPS6KB2 CAD AURKB AKT1 PAK7 MAP3K6 MAP3K5 HSF1 PAK4 IFNG CLK4 TGFA INSR SYK EGFR DBNL CTBP1 BCR TNIK WNK1 CDK6 PIM2 CDK4 PRKCE CDK2 SRPK1 ATP6V1A MAPK3 PRKCZ FGFR4 NDUFB8 ERBB3 STK11 ERBB2 MAPKAPK3 ABI2 ATP6V1B1 EPHB3 SRC IGF1R EPHB6 CDC2L2 PTK6 PRKRA PRKAA2 THBS1 YES1 PDK2 ATP5J2 FLT1 MAP2K2 FLT4 TKT CDC42BPG RPS6KA1 MAPK13 PLK1 CSNK1E RSRC1 MAPK15 ARAF GRK6 JAK1 SPTBN1 MERTK | 2.66 × 10−5 | 9.24 × 10−4 |
| regulation of cell proliferation (GO:0042127) | FGF18 EIF5A GJA1 STRN SSR1 MEN1 MAGED1 PGR BNIPL CDKN2A ASH2L HSF1 APOE CDKN2D HEY2 IFNG TGFA RARB DLG5 ODZ1 NR2F2 INSR LTA SYK EGFR CTBP1 CAPNS1 PTPRF DTL RXFP2 HTR4 ESR1 CDK6 ESR2 RB1 CDK4 RPS4X CDK2 RBBP9 MXD4 NME2 BTG1 NME1 DHPS ATPIF1 SMARCA2 CAV2 PRKCZ DERL2 FGFR4 MVD TBC1D8 ERBB3 STK11 FOXM1 ERBB2 BCL2L1 MIF IGF1R PRKRA ADRA2A RNF10 BCL6 THBS1 TINF2 EMD B4GALT1 FLT1 NF2 CREB3 TAF6 FLT4 CSNK2B TKT FZD7 CAPN1 TSC2 GLMN PBX1 ADRA1D IGFBP5 | 8.40 × 10−7 | 4.35 × 10−5 |
| post-translational protein modification (GO:0043687) | STK36 STYX USE1 CASK RPS6KB2 AURKB MAP3K6 MAP3K5 ERO1LB IFNG CLK4 INSR SYK EGFR DBNL BCR TNIK PTPRF DTL WNK1 PTPRS HDAC11 PIM2 SUV420H2 PPP1CA HUWE1 MAPK3 AMFR TNFAIP1 GGCX FGFR4 ERBB3 STK11 ERBB2 MAPKAPK3 NEDD8 ANAPC11 EPHB3 ACP1 SRC EPHB6 UBE2D2 PTK6 PRKRA TRAF7 MAP2K2 TKT HDAC5 CDC42BPG HDAC3 PLK1 GRK6 SPTBN1 EAF2 MERTK HDAC6 BCKDK FASTK UCHL1 CAD AKT1 PAK7 PRMT2 HSF1 ASH2L PAK4 TGFA RAB6A KLHL20 CTBP1 DDB1 PIGU PIGT UBE2I CDK6 PRKCE CDK4 SRPK1 CDK2 BTG1 DDB2 PRKCZ RAB3D UBE3A USP5 GPAA1 ABI2 STUB1 IGF1R CDC2L2 C19ORF62 PRKAA2 PPP3CA THBS1 YES1 QSOX1 KAT2A PDK2 LRSAM1 FLT1 FLT4 SIRT2 RPS6KA1 MAPK13 CSNK1E RSRC1 MAPK15 ARAF JAK1 IRF4 RBM14 RNF40 | 2.96 × 10−7 | 1.96 × 10−5 |
| cellular protein metabolic process (GO:0044267) | USE1 RPLP2 AURKB CCT3 MAP3K6 MAP3K5 ERO1LB RPLP0 RPLP1 IFNG CLK4 TRAK1 FAU RPL10 PSENEN SYK DBNL BCR TNIK PTPRF WNK1 PTPRS PIM2 SUV420H2 PPP1CA RPS19 HUWE1 BACE2 BACE1 MAPK3 AMFR GGCX ERBB3 RAD23A ERBB2 EPHB3 ACP1 EPHB6 RPS28 MGAT4B RPSA MAP2K2 TKT RPS6 RPS7 CCT7 CDC42BPG RPL18A GRK6 BCKDK NARS FASTK QARS CANX HSF1 ASH2L TGFA RAB6A CTBP1 SRPK1 G6PD EEF1G EEF1D SERP1 TUFM RAB3D USP5 GPAA1 ABI2 RPL36 STT3A MRPL12 PER1 C19ORF62 PRKAA2 PPP3CA THBS1 QSOX1 PDK2 LRSAM1 EIF2B1 RPL28 RPL29 RPS6KA1 CSNK1E RPL22 PSMD11 MAPK13 ARAF MAPK15 DPM2 DPM3 RBM14 STK36 STYX EIF5A CASK RPS6KB2 ST3GAL3 LOX INSR RPS27A EGFR DTL EEF2 HDAC11 UBA52 TNFAIP1 FGFR4 DERL2 EEF1B2 STK11 MAPKAPK3 NEDD8 ANAPC11 DERL3 SRC EIF3D UBE2D2 PSMB6 EIF3H RPL6 PTK6 PSMB3 EIF3F PRKRA RPL8 EIF3K EIF3L GNMT TRAF7 B4GALT1 P4HB EEF1A1 SEC11A EIF4B HDAC5 ATF6 HDAC3 PPIA DOHH PLK1 PSMC3 SPTBN1 DNAJB1 EAF2 MERTK HDAC6 COPA UCHL1 CAD AKT1 PAK7 PRMT2 PIGM PAK4 PSMD2 KLHL20 PIGX DDB1 PIGU PIGT UBE2I CDK6 PRKCE RPS4X CDK4 CDK2 BTG1 TBCD DDB2 DHPS PRKCZ PPP4R2 FKBP8 APH1A UBE3A FKBP4 APH1B STUB1 IGF1R CDC2L2 LARS YES1 KAT2A FLT1 FLT4 SIRT2 NCSTN RPL13A RSRC1 JAK1 IRF4 RNF40 | 4.47 × 10−13 | 1.78 × 10−10 |
| positive regulation of cellular process (GO:0048522) | MMS19 XRCC5 FGF18 THRA SNCA UTRN RPS6KB2 CASK EIF5A ITSN1 SSR1 MAGED1 PGR MAP3K5 CDKN2A HTRA2 CD44 PACSIN3 APOE IFNG PSENEN RARB INSR SYK EGFR PLD2 CAPNS1 PTPRF PCBD1 DFFA DTL HTR4 MED12 PIM2 TBR1 NCAM1 SPAG9 NME2 RPS19 NME3 PIAS3 NME1 VAMP3 SMARCA2 SMARCA4 CAV2 DERL2 FGFR4 MVD ERBB3 PFKFB2 GLUD1 ERBB2 RRM2B ANAPC11 BCL2L1 TUBB PSMB6 ARPC2 SMARCB1 PSMB3 P2RY2 NUMB PRKRA ADRA2A RNF10 TRAF7 TRAF4 B4GALT1 DVL2 TP53BP1 TEAD1 TKT TPD52L1 RPS6 SREBF2 ATF6 TRAF3IP2 HDAC5 ATF4 PSMC3 PLK1 IKBKG GLMN RFX3 PDZK1 PDCD6 ADRA1D PC HDAC6 E2F1 RTN4 LZTS2 FASTK GJA1 PPOX RFXANK MEN1 AKT1 BNIPL FOS CCNE1 ASH2L MAPT HEY2 PSMD2 TGFA RHOC NR2F2 CASP2 LTA ZFP36 RXFP2 ESR1 CDK6 ESR2 RB1 PFKM CDK4 RPS4X PRKCE HMGA1 CDK2 FLNA PRPF6 SCAP NCOA3 BTG1 IGF2R SORT1 DHPS EEF1D C6ORF108 BID MBL2 PRKCZ APH1A TBC1D8 UBE3A USP5 FOXM1 APH1B TRAIP STUB1 CALCOCO1 MIF IGF1R MRPL12 SOS2 C19ORF62 BCL6 THBS1 TINF2 TERF1 PHLDA1 FLT1 NF2 CREB3 PSAP FLT4 MAP1B BIRC5 DPYSL2 FZD7 CAPN1 NCSTN PKNOX1 PSMD11 CSNK1E MAPK15 BRE PBX1 IRF4 RBM14 DNM2 | 1.41 × 10−10 | 2.52 × 10−8 |
| interphase (GO:0051325) | E2F1 EGFR TPD52L1 BIRC5 CDK6 ZNF655 RB1 CDK4 RPS6 CDK2 AKT1 CCNE1 SIN3A CDKN2A CDKN2D PPP3CA TERF1 DNM2 | 1.90 × 10−5 | 6.87 × 10−4 |
| regulation of cell cycle (GO:0051726) | E2F1 E2F2 CDT1 MEN1 AKT1 GSS CDKN2A CLP1 CDKN2D IFNG TGFA FANCG NR2F2 INSR EGFR BCR DTL DDB1 CDK6 RB1 TACC3 CDK4 CDK2 RAD1 CHMP1A SGSM3 PRNP MAD2L2 DST SMARCA4 CAV2 STK11 FOXM1 ZNF655 CDC37 MIF BCL6 C19ORF62 THBS1 TERF1 TAF6 CREB3 BIRC5 RPS6 SIRT2 HDAC3 PLK1 TSC2 CKS2 BRE DGKZ | 6.92 × 10−7 | 3.82 × 10−5 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tseng, C.-W.; Huang, H.-C.; Shih, A.C.-C.; Chang, Y.-Y.; Hsu, C.-C.; Chang, J.-Y.; Li, W.-H.; Juan, H.-F. Revealing the Anti-Tumor Effect of Artificial miRNA p-27-5p on Human Breast Carcinoma Cell Line T-47D. Int. J. Mol. Sci. 2012, 13, 6352-6369. https://doi.org/10.3390/ijms13056352
Tseng C-W, Huang H-C, Shih AC-C, Chang Y-Y, Hsu C-C, Chang J-Y, Li W-H, Juan H-F. Revealing the Anti-Tumor Effect of Artificial miRNA p-27-5p on Human Breast Carcinoma Cell Line T-47D. International Journal of Molecular Sciences. 2012; 13(5):6352-6369. https://doi.org/10.3390/ijms13056352
Chicago/Turabian StyleTseng, Chien-Wei, Hsuan-Cheng Huang, Arthur Chun-Chieh Shih, Ya-Ya Chang, Chung-Cheng Hsu, Jen-Yun Chang, Wen-Hsiung Li, and Hsueh-Fen Juan. 2012. "Revealing the Anti-Tumor Effect of Artificial miRNA p-27-5p on Human Breast Carcinoma Cell Line T-47D" International Journal of Molecular Sciences 13, no. 5: 6352-6369. https://doi.org/10.3390/ijms13056352
APA StyleTseng, C.-W., Huang, H.-C., Shih, A. C.-C., Chang, Y.-Y., Hsu, C.-C., Chang, J.-Y., Li, W.-H., & Juan, H.-F. (2012). Revealing the Anti-Tumor Effect of Artificial miRNA p-27-5p on Human Breast Carcinoma Cell Line T-47D. International Journal of Molecular Sciences, 13(5), 6352-6369. https://doi.org/10.3390/ijms13056352