Mutations Associated with Functional Disorder of Xanthine Oxidoreductase and Hereditary Xanthinuria in Humans


Abstract
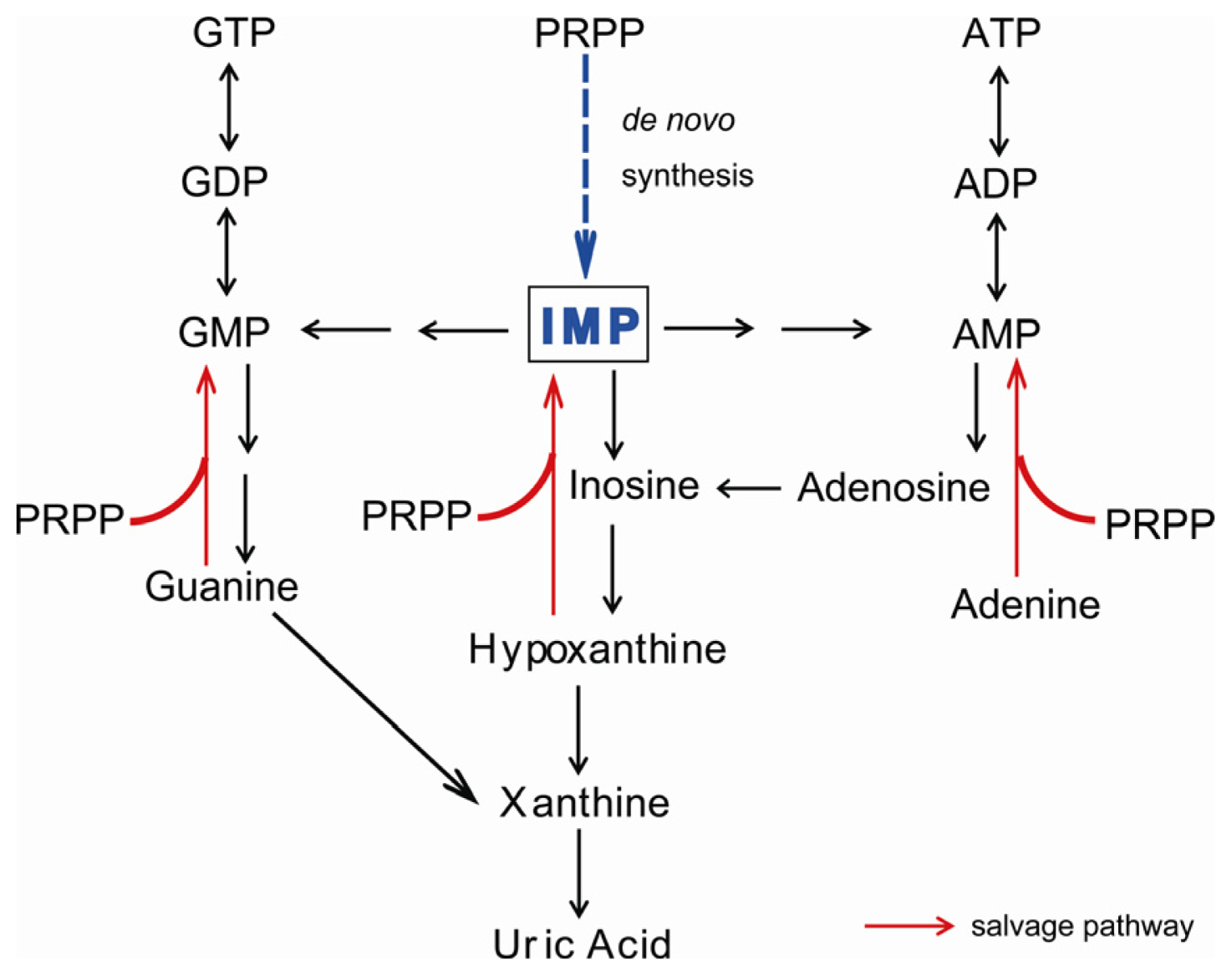
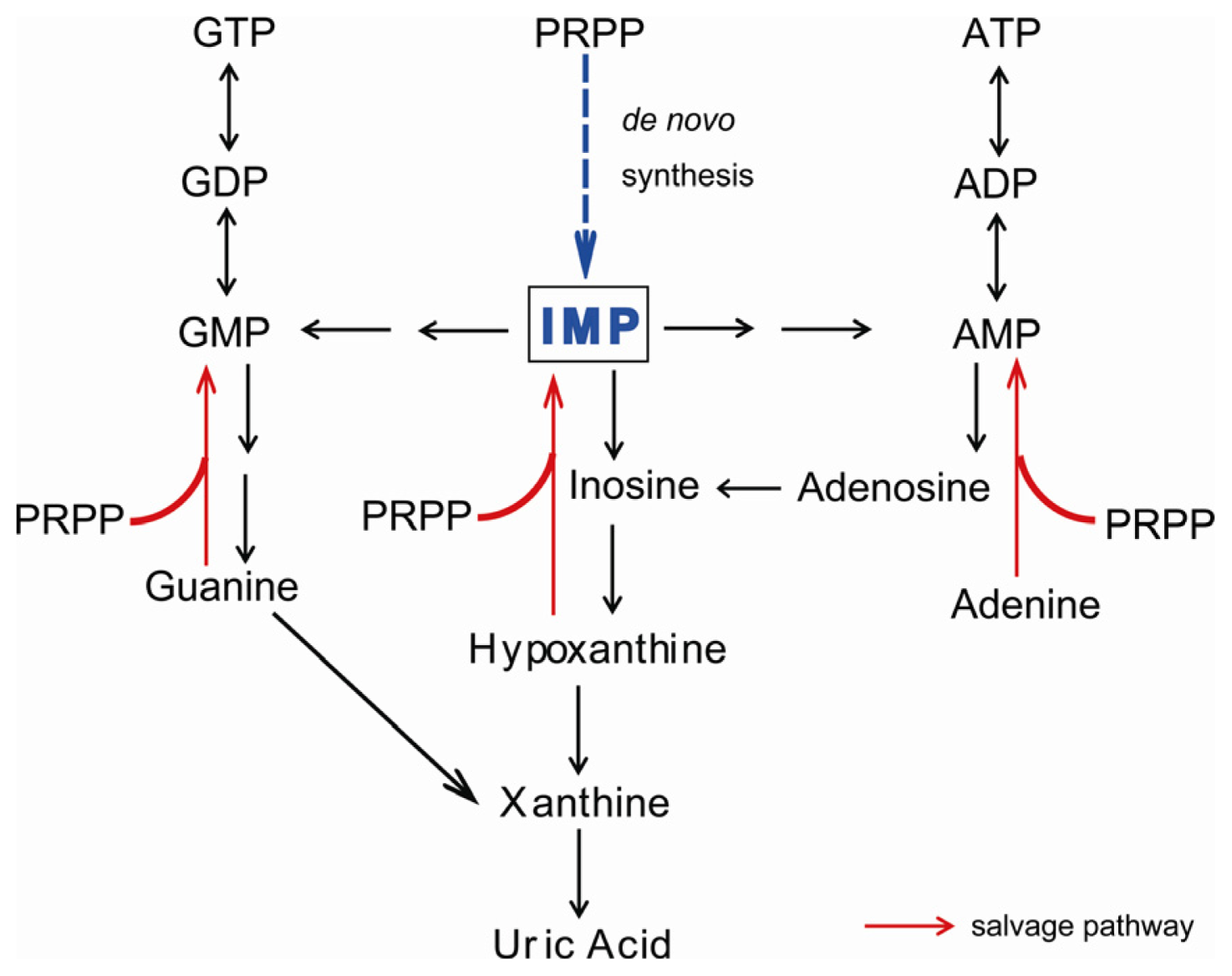
:1. Introduction
2. Symptoms of XOR Deficiency and Differential Diagnosis
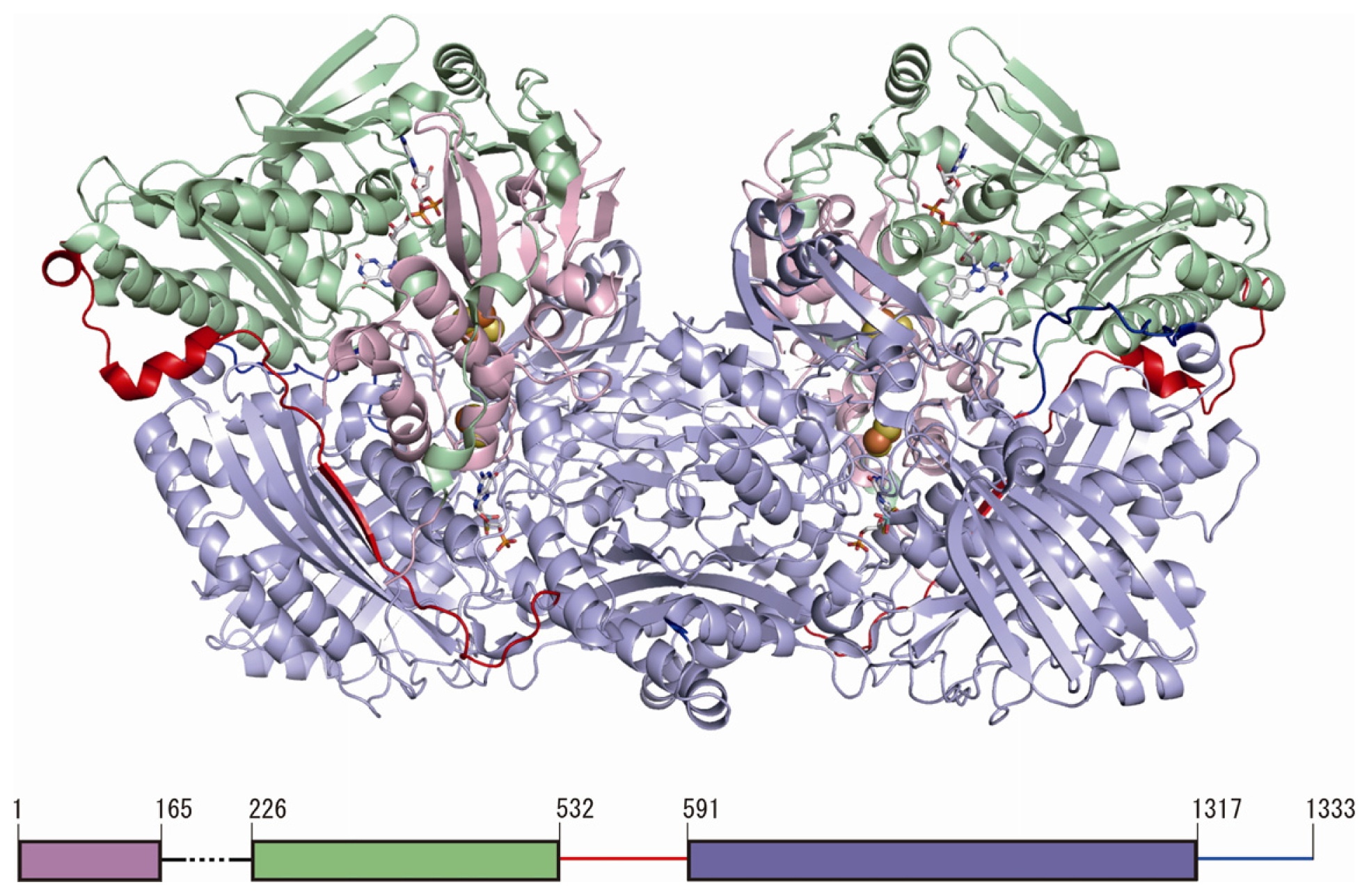
3. Overall Structure of Human Xanthine Oxidoreductase (XOR)
4. Residues Crucial for Enzyme Function: Experimental Studies
4.1. The N-Terminal Fe/S Domain
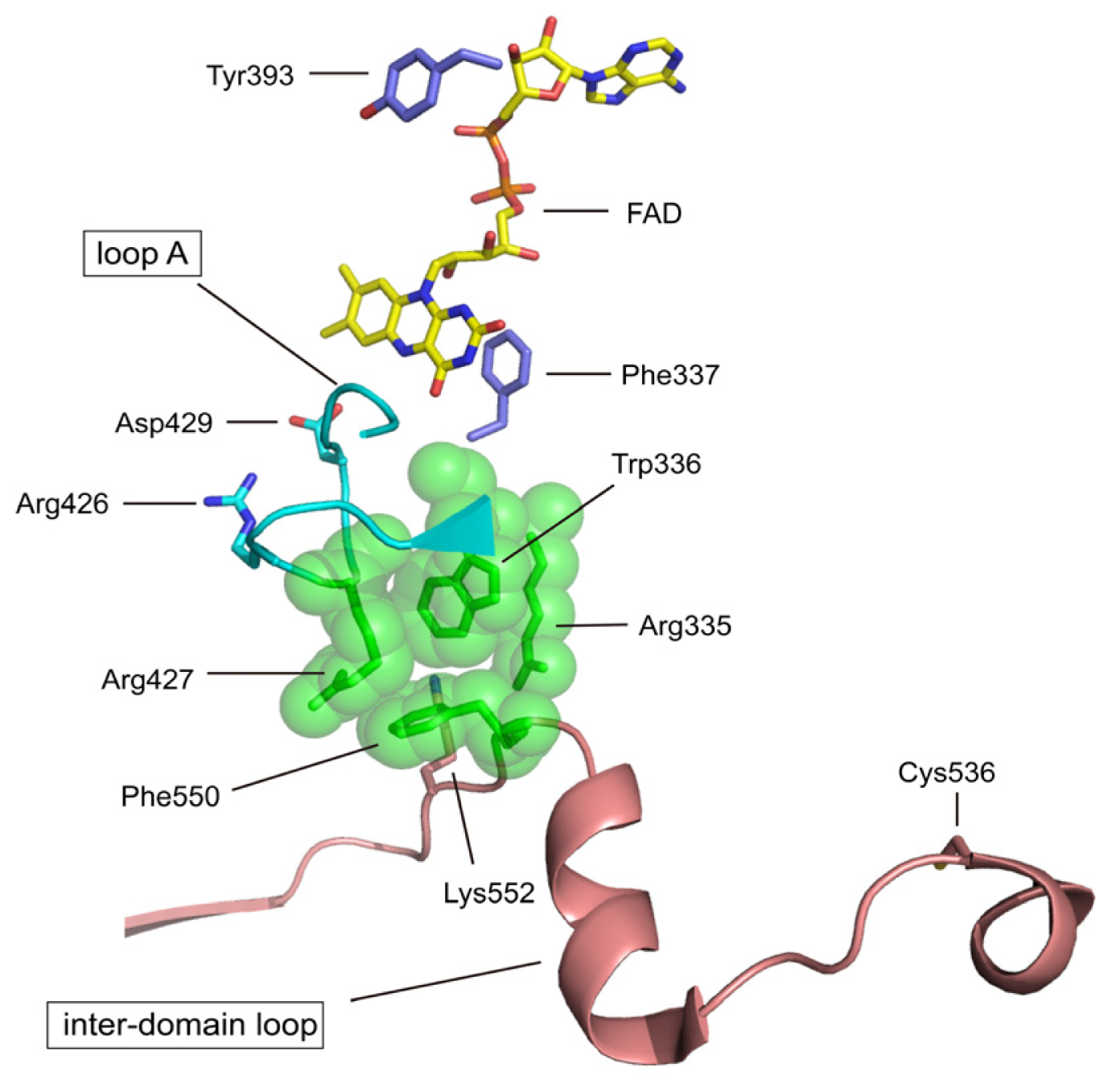
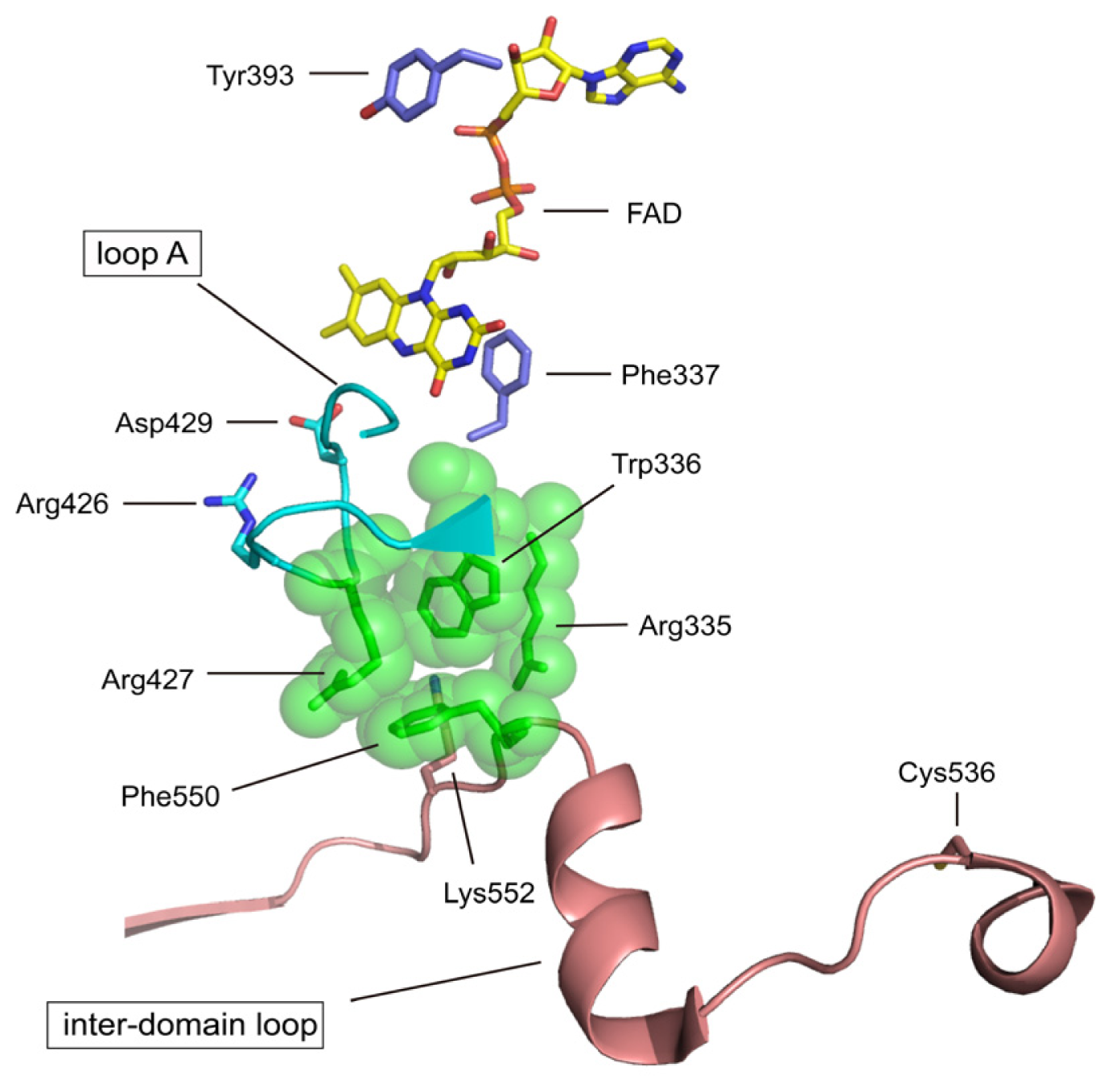
4.2. The Intermediate FAD Domain
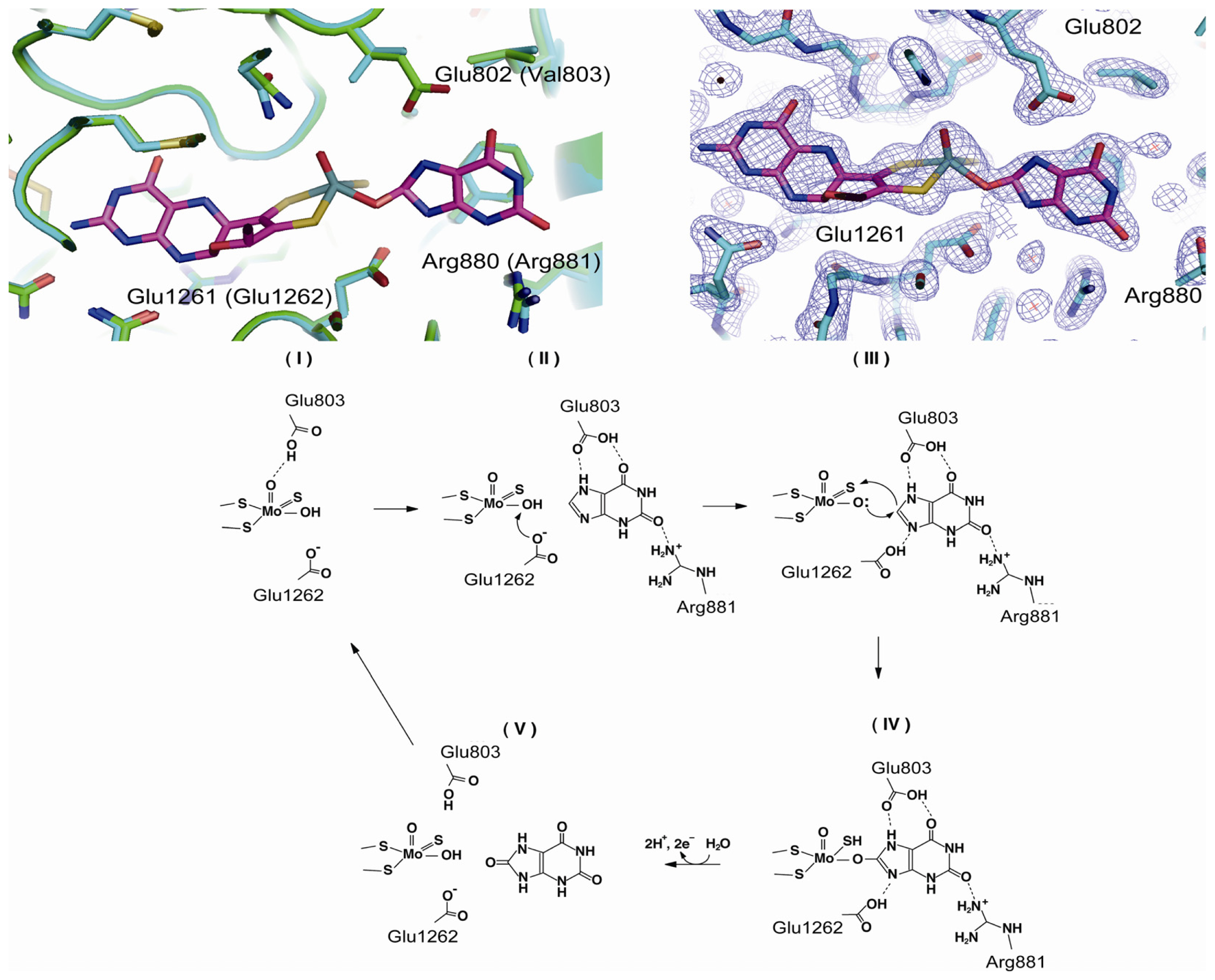
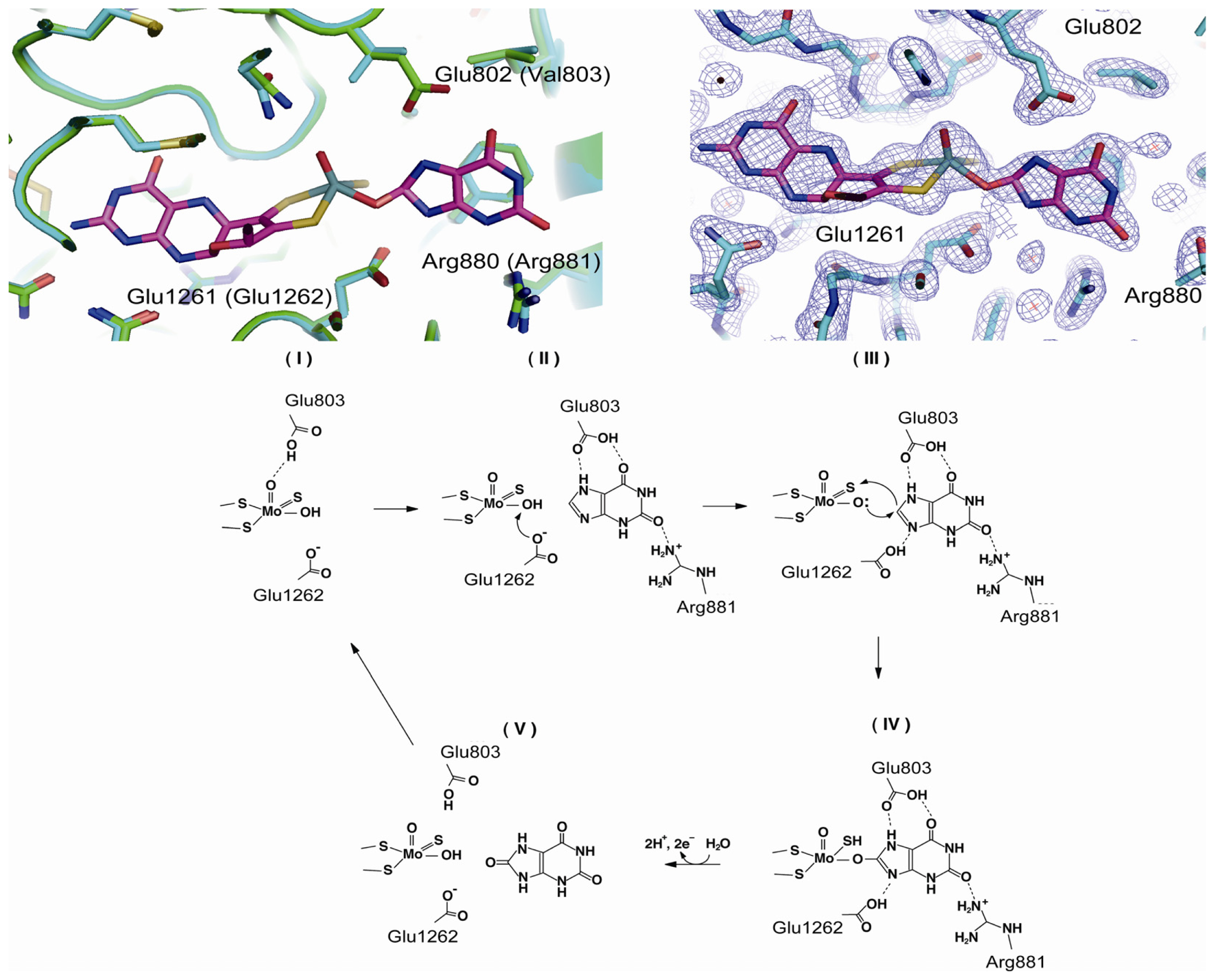
4.3. The C-Terminal Moco Domain
5. Mutations Causing Type I Xanthinuria
6. Type II Xanthinuria Is the Consequence of Mutation of Human Moco Sulfurase Gene
Acknowledgments
- Conflict of InterestAuthors declare no conflict of interests.
References
- Nishino, T. The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J. Biochem 1994, 116, 1–6. [Google Scholar]
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. Faseb J 1995, 9, 995–1003. [Google Scholar]
- Hille, R. The Mononuclear Molybdenum Enzymes. Chem. Rev 1996, 96, 2757–2816. [Google Scholar]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J 2008, 275, 3278–3289. [Google Scholar]
- Hille, R.; Nishino, T.; Bittner, F. Molybdenum enzymes in higher organisms. Coord. Chem. Rev 2011, 255, 1179–1205. [Google Scholar]
- Elion, G.B. Enzymatic and metabolic studies with allopurinol. Ann. Rheum. Dis 1966, 25, 608–614. [Google Scholar]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol 2004, 555, 589–606. [Google Scholar]
- Hellsten-Westing, Y. Immunohistochemical localization of xanthine oxidase in human cardiac and skeletal muscle. Histochemistry 1993, 100, 215–222. [Google Scholar]
- Linder, N.; Rapola, J.; Raivio, K.O. Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab Investig 1999, 79, 967–974. [Google Scholar]
- Angermuller, S.; Bruder, G.; Volkl, A.; Wesch, H.; Fahimi, H.D. Localization of xanthine oxidase in crystalline cores of peroxisomes. A cytochemical and biochemical study. Eur. J. Cell. Biol 1987, 45, 137–144. [Google Scholar]
- Ichikawa, M.; Nishino, T.; Nishino, T.; Ichikawa, A. Subcellular localization of xanthine oxidase in rat hepatocytes: High-resolution immunoelectron microscopic study combined with biochemical analysis. J. Histochem. Cytochem 1992, 40, 1097–1103. [Google Scholar]
- Elion, G.B.; Kovensky, A.; Hitchings, G.H. Metabolic studies of allopurinol, an inhibitor of xanthine oxidase. Biochem. Pharmacol 1966, 15, 863–880. [Google Scholar]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol. Rev 2006, 58, 87–114. [Google Scholar]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem 2003, 278, 1848–1855. [Google Scholar]
- Okamoto, K.; Matsumoto, K.; Hille, R.; Eger, B.T.; Pai, E.F.; Nishino, T. The crystal structure of xanthine oxidoreductase during catalysis: Implications for reaction mechanism and enzyme inhibition. Proc. Natl. Acad. Sci. USA 2004, 101, 7931–7936. [Google Scholar]
- Terkeltaub, R. Update on gout: New therapeutic strategies and options. Nat. Rev. Rheumatol 2010, 6, 30–38. [Google Scholar]
- Becker, M.A.; Schumacher, H.R., Jr; Wortmann, R.L.; MacDonald, P.A.; Eustace, D.; Palo, W.A.; Streit, J.; Joseph-Ridge, N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N. Engl. J. Med. 2005, 353, 2450–2461. [Google Scholar]
- Kamatani, N.; Fujimori, S.; Hada, T.; Hosoya, T.; Kohri, K.; Nakamura, T.; Ueda, T.; Yamamoto, T.; Yamanaka, H.; Matsuzawa, Y. An allopurinol-controlled, randomized, double-dummy, double-blind, parallel between-group, comparative study of febuxostat (TMX-67), a non-purine-selective inhibitor of xanthine oxidase, in patients with hyperuricemia including those with gout in Japan: phase 3 clinical study. J. Clin. Rheumatol 2011, 17, S13–S18. [Google Scholar]
- Massey, V.; Komai, H.; Palmer, G.; Elion, G.B. On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo [3,4-d]pyrimidines. J. Biol. Chem 1970, 245, 2837–2844. [Google Scholar]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Pai, E.F.; Nishino, T. Mechanism of inhibition of xanthine oxidoreductase by allopurinol: Crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleos. Nucleot. Nucleic Acids 2008, 27, 888–893. [Google Scholar]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar]
- Glantzounis, G.K.; Tsimoyiannis, E.C.; Kappas, A.M.; Galaris, D.A. Uric acid and oxidative stress. Curr. Pharm. Des 2005, 11, 4145–4151. [Google Scholar]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar]
- Jones, S.P.; Bolli, R. The ubiquitous role of nitric oxide in cardioprotection. J. Mol. Cell Cardiol 2006, 40, 16–23. [Google Scholar]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov 2008, 7, 156–167. [Google Scholar]
- Kobayashi, K.; Miki, M.; Tagawa, S. Pulse-radiolysis study of the reaction of nitric oxide with superoxide. J. Chem. Soc. Dalton. Trans. 1995, 2885–2889. [Google Scholar] [CrossRef]
- Nishino, T.; Nakanishi, S.; Okamoto, K.; Mizushima, J.; Hori, H.; Iwasaki, T.; Nishino, T.; Ichimori, K.; Nakazawa, H. Conversion of xanthine dehydrogenase into oxidase and its role in reperfusion injury. Biochem. Soc. Trans 1997, 25, 783–786. [Google Scholar]
- Okamoto, K.; Kusano, T.; Nishino, T. Chemical Nature and Reaction Mechanisms of the Molybdenum Cofactor of Xanthine Oxidoreductase. Curr. Pharmaceut. Des. 2012, in press. [Google Scholar]
- Li, H.; Samouilov, A.; Liu, X.; Zweier, J.L. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide generation in anoxic tissues. J. Biol. Chem 2001, 276, 24482–24489. [Google Scholar]
- Massey, V.; Brumby, P.E.; Komai, H. Studies on milk xanthine oxidase. Some spectral and kinetic properties. J. Biol. Chem 1969, 244, 1682–1691. [Google Scholar]
- Saito, T.; Nishino, T. Differences in redox and kinetic properties between NAD-dependent and O2-dependent types of rat liver xanthine dehydrogenase. J. Biol. Chem 1989, 264, 10015–10022. [Google Scholar]
- Dent, C.E.; Philpot, G.R. Xanthinuria, an inborn error (or deviation) of metabolism. Lancet 1954, 266, 182–185. [Google Scholar]
- Kojima, T.; Nishina, T.; Kitamura, M.; Hosoya, T.; Nishioka, K. Biochemical studies on the purine metabolism of four cases with hereditary xanthinuria. Clin. Chim. Acta 1984, 137, 189–198. [Google Scholar]
- Simmonds, H.A.; Reiter, S.; Nishino, T. Hereditary xanthinuria. In The Metabolic and Molecular Bases of Inherited Disease, 7th ed; Scriver, C.R., Ed.; McGraw-Hill Health Professions Division: New York, NY, USA, 1995; pp. 1781–1797. [Google Scholar]
- Mateos, F.A.; Puig, J.G.; Jimenez, M.L.; Fox, I.H. Hereditary xanthinuria. Evidence for enhanced hypoxanthine salvage. J. Clin. Invest 1987, 79, 847–852. [Google Scholar]
- Ichida, K.; Amaya, Y.; Kamatani, N.; Nishino, T.; Hosoya, T.; Sakai, O. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical type I xanthinuria. J. Clin. Invest 1997, 99, 2391–2397. [Google Scholar]
- Ichida, K.; Matsumura, T.; Sakuma, R.; Hosoya, T.; Nishino, T. Mutation of human molybdenum cofactor sulfurase gene is responsible for classical xanthinuria type II. Biochem. Biophys. Res. Commun 2001, 282, 1194–1200. [Google Scholar]
- Yamamoto, T.; Higashino, K.; Kono, N.; Kawachi, M.; Nanahoshi, M.; Takahashi, S.; Suda, M.; Hada, T. Metabolism of pyrazinamide and allopurinol in hereditary xanthine oxidase deficiency. Clin. Chim. Acta 1989, 180, 169–175. [Google Scholar]
- Ichida, K.; Yoshida, M.; Sakuma, R.; Hosoya, T. Two siblings with classical xanthinuria type 1: Significance of allopurinol loading test. Intern. Med 1998, 37, 77–82. [Google Scholar]
- Pryde, D.C.; Dalvie, D.; Hu, Q.; Jones, P.; Obach, R.S.; Tran, T.D. Aldehyde oxidase: An enzyme of emerging importance in drug discovery. J. Med. Chem 2010, 53, 8441–8460. [Google Scholar]
- Garattini, E.; Terao, M. The role of aldehyde oxidase in drug metabolism. Expert Opin. Drug Metab. Toxicol 2012, 8, 487–503. [Google Scholar]
- Kucera, J.; Bulkova, T.; Rychla, R.; Jahn, P. Bilateral xanthine nephrolithiasis in a dog. J. Small Anim. Pract 1997, 38, 302–305. [Google Scholar]
- Van Zuilen, C.D.; Nickel, R.F.; van Dijk, T.H.; Reijngoud, D.J. Xanthinuria in a family of Cavalier King Charles spaniels. Vet. Q 1997, 19, 172–174. [Google Scholar]
- Tsuchida, S.; Kagi, A.; Koyama, H.; Tagawa, M. Xanthine urolithiasis in a cat: A case report and evaluation of a candidate gene for xanthine dehydrogenase. J. Feline Med. Surg 2007, 9, 503–508. [Google Scholar]
- Miranda, M.; Rigueira, L.; Suarez, M.L.; Carbajales, P.; Moure, P.; Fidalgo, L.E.; Failde, D.; Vazquez, S. Xanthine nephrolithiasis in a galician blond beef calf. J. Vet. Med. Sci 2010, 72, 921–923. [Google Scholar]
- Oda, M.; Satta, Y.; Takenaka, O.; Takahata, N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol. Biol. Evol 2002, 19, 640–653. [Google Scholar]
- Bradbury, M.G.; Henderson, M.; Brocklebank, J.T.; Simmonds, H.A. Acute renal failure due to xanthine stones. Pediatr. Nephrol 1995, 9, 476–477. [Google Scholar]
- Thomas, N.; Stephen, D.C.; Abraham, B.; Kekre, N.; Seshadri, M.S. Xanthinuria—An unusual cause for renal stone disease. J. Assoc. Physicians. India 1996, 44, 203–204. [Google Scholar]
- Kiss, A.; Barenyi, M.; Csontai, A. Xanthine stone in the urinary bladder of a male child. Urol. Int 1999, 63, 242–244. [Google Scholar]
- Al-Eisa, A.A.; Al-Hunayyan, A.; Gupta, R. Pediatric urolithiasis in Kuwait. Int. Urol. Nephrol 2002, 33, 3–6. [Google Scholar]
- Arikyants, N.; Sarkissian, A.; Hesse, A.; Eggermann, T.; Leumann, E.; Steinmann, B. Xanthinuria type I: A rare cause of urolithiasis. Pediatr. Nephrol 2007, 22, 310–314. [Google Scholar]
- Gargah, T.; Essid, A.; Labassi, A.; Hamzaoui, M.; Lakhoua, M.R. Xanthine urolithiasis. Saudi J. Kidney Dis. Transpl 2010, 21, 328–331. [Google Scholar]
- Martin, H.M.; Hancock, J.T.; Salisbury, V.; Harrison, R. Role of xanthine oxidoreductase as an antimicrobial agent. Infect. Immun 2004, 72, 4933–4939. [Google Scholar]
- Vorbach, C.; Scriven, A.; Capecchi, M.R. The housekeeping gene xanthine oxidoreductase is necessary for milk fat droplet enveloping and secretion: Gene sharing in the lactating mammary gland. Gene Dev 2002, 16, 3223–3235. [Google Scholar]
- McManaman, J.L.; Palmer, C.A.; Wright, R.M.; Neville, M.C. Functional regulation of xanthine oxidoreductase expression and localization in the mouse mammary gland: Evidence of a role in lipid secretion. J. Physiol 2002, 545, 567–579. [Google Scholar]
- Nakazono, K.; Watanabe, N.; Matsuno, K.; Sasaki, J.; Sato, T.; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. USA 1991, 88, 10045–10048. [Google Scholar]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar]
- Elion, G.B. The purine path to chemotherapy. Science 1989, 244, 41–47. [Google Scholar]
- Johnson, J.L.; Duran, M. Molybdenum Cofactor Deficiency and Isolated Sulfite Oxidase Deficiency. In The Metabolic & Molecular Bases of Inherited Disease, 8th ed; Scriver, C.R., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3163–3177. [Google Scholar]
- Ichida, K.; Amaya, Y.; Noda, K.; Minoshima, S.; Hosoya, T.; Sakai, O.; Shimizu, N.; Nishino, T. Cloning of the cDNA encoding human xanthine dehydrogenase (oxidase): Structural analysis of the protein and chromosomal location of the gene. Gene 1993, 133, 279–284. [Google Scholar]
- Amaya, Y.; Yamazaki, K.; Sato, M.; Noda, K.; Nishino, T.; Nishino, T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J. Biol. Chem 1990, 265, 14170–14175. [Google Scholar]
- Minoshima, S.; Wang, Y.; Ichida, K.; Nishino, T.; Shimizu, N. Mapping of the gene for human xanthine dehydrogenase (oxidase) (XDH) to band p23 of chromosome 2. Cytogenet. Cell Genet 1995, 68, 52–53. [Google Scholar]
- Xu, P.; Huecksteadt, T.P.; Hoidal, J.R. Molecular cloning and characterization of the human xanthine dehydrogenase gene (XDH). Genomics 1996, 34, 173–180. [Google Scholar]
- Wright, R.M.; Vaitaitis, G.M.; Wilson, C.M.; Repine, T.B.; Terada, L.S.; Repine, J.E. cDNA cloning, characterization, and tissue-specific expression of human xanthine dehydrogenase/xanthine oxidase. Proc. Natl. Acad. Sci. USA 1993, 90, 10690–10694. [Google Scholar]
- Saksela, M.; Raivio, K.O. Cloning and expression in vitro of human xanthine dehydrogenase/oxidase. Biochem. J 1996, 315, 235–239. [Google Scholar]
- Garattini, E.; Fratelli, M.; Terao, M. Mammalian aldehyde oxidases: Genetics, evolution and biochemistry. Cell Mol. Life Sci 2008, 65, 1019–1048. [Google Scholar]
- Boer, D.R.; Thapper, A.; Brondino, C.D.; Romao, M.J.; Moura, J.J. X-ray crystal structure and EPR spectra of “arsenite-inhibited” Desulfovibriogigas aldehyde dehydrogenase: A member of the xanthine oxidase family. J. Am. Chem. Soc 2004, 126, 8614–8615. [Google Scholar]
- Yamaguchi, Y.; Matsumura, T.; Ichida, K.; Okamoto, K.; Nishino, T. Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: roles of active site residues in binding and activation of purine substrate. J. Biochem 2007, 141, 513–524. [Google Scholar]
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Hori, H.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Mechanism of the conversion of xanthine dehydrogenase to xanthine oxidase: Identification of the two cysteine disulfide bonds and crystal structure of a non-convertible rat liver xanthine dehydrogenase mutant. J. Biol. Chem 2005, 280, 24888–24894. [Google Scholar]
- Iwasaki, T.; Okamoto, K.; Nishino, T.; Mizushima, J.; Hori, H. Sequence motif-specific assignment of two [2Fe-2S] clusters in rat xanthine oxidoreductase studied by site-directed mutagenesis. J. Biochem 2000, 127, 771–778. [Google Scholar]
- Kuwabara, Y.; Nishino, T.; Okamoto, K.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proc. Natl. Acad. Sci. USA 2003, 100, 8170–8175. [Google Scholar]
- Nishino, T. The nicotinamide adenine dinucleotide-binding site of chicken liver xanthine dehydrogenase. Evidence for alteration of the redox potential of the flavin by NAD binding or modification of the NAD-binding site and isolation of a modified peptide. J. Biol. Chem 1989, 264, 5468–5473. [Google Scholar]
- Nishino, T. The conversion from the dehydrogenase type to the oxidase type of rat liver xanthine dehydrogenase by modification of cysteine residues with fluorodinitrobenzene. J. Biol. Chem 1997, 272, 29859–29864. [Google Scholar]
- Nishino, T.; Tsushima, K.; Hille, R.; Massey, V. Inhibition of milk xanthine oxidase by fluorodinitrobenzene. J. Biol. Chem 1982, 257, 7348–7353. [Google Scholar]
- Huber, R.; Hof, P.; Duarte, R.O.; Moura, J.J.; Moura, I.; Liu, M.Y.; LeGall, J.; Hille, R.; Archer, M.; Romao, M.J. A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proc. Natl. Acad. Sci. USA 1996, 93, 8846–8851. [Google Scholar]
- Palmer, G.; Massey, V. Electron paramagnetic resonance and circular dichroism studies on milk xanthine oxidase. J. Biol. Chem 1969, 244, 2614–2620. [Google Scholar]
- Porras, A.G.; Palmer, G. The room temperature potentiometry of xanthine oxidase. Ph-dependent redox behavior of the flavin, molybdenum, and iron-sulfur centers. J. Biol. Chem 1982, 257, 11617–11626. [Google Scholar]
- Caldeira, J.; Belle, V.; Asso, M.; Guigliarelli, B.; Moura, I.; Moura, J.J.; Bertrand, P. Analysis of the electron paramagnetic resonance properties of the [2Fe-2S]1+ centers in molybdenum enzymes of the xanthine oxidase family: Assignment of signals I and II. Biochemistry 2000, 39, 2700–2707. [Google Scholar]
- Hille, R.; Hagen, W.R.; Dunham, W.R. Spectroscopic studies on the iron-sulfur centers of milk xanthine oxidase. J. Biol. Chem 1985, 260, 10569–10575. [Google Scholar]
- Ishikita, H.; Eger, B.T.; Okamoto, K.; Nishino, T.; Pai, E.F. Protein conformational gating of enzymatic activity in xanthine oxidoreductase. J. Am. Chem. Soc 2012, 134, 999–1009. [Google Scholar]
- Massey, V.; Schopfer, L.M.; Nishino, T.; Nishino, T. Differences in protein structure of xanthine dehydrogenase and xanthine oxidase revealed by reconstitution with flavin active site probes. J. Biol. Chem 1989, 264, 10567–10573. [Google Scholar]
- Saito, T.; Nishino, T.; Massey, V. Differences in environment of FAD between NAD-dependent and O2-dependent types of rat liver xanthine dehydrogenase shown by active site probe study. J. Biol. Chem 1989, 264, 15930–15935. [Google Scholar]
- Hunt, J.; Massey, V. Purification and properties of milk xanthine dehydrogenase. J. Biol. Chem 1992, 267, 21479–21485. [Google Scholar]
- Tsujii, A.; Nishino, T. Mechanism of transition from xanthine dehydrogenase to xanthine oxidase: Effect of guanidine-HCL or urea on the activity. Nucleos. Nucleot. Nucleic Acids 2008, 27, 881–887. [Google Scholar]
- Okamoto, K.; Kawaguchi, Y.; Eger, B.T.; Pai, E.F.; Nishino, T. Crystal Structures of Urate Bound Form of Xanthine Oxidoreductase: Substrate Orientation and Structure of the Key Reaction Intermediate. J. Am. Chem. Soc 2010, 132, 17080–17083. [Google Scholar]
- Leimkuhler, S.; Stockert, A.L.; Igarashi, K.; Nishino, T.; Hille, R. The role of active site glutamate residues in catalysis of Rhodobacter capsulatus xanthine dehydrogenase. J. Biol. Chem 2004, 279, 40437–40444. [Google Scholar]
- Bergmann, F.; Dikstein, S. Studies on uric acid and related compounds III. Observations on the specificity of mammalian xanthine oxidases. J. Biol. Chem 1956, 223, 765–780. [Google Scholar]
- Cao, H.; Pauff, J.M.; Hille, R. Substrate orientation and catalytic specificity in the action of xanthine oxidase: The sequential hydroxylation of hypoxanthine to uric acid. J. Biol. Chem 2010, 285, 28044–28053. [Google Scholar]
- Jezewska, M.M. Xanthine accumulation during hypoxanthine oxidation by milk xanthine oxidase. Eur. J. Biochem 1973, 36, 385–390. [Google Scholar]
- Metz, S.; Thiel, W. A combined QM/MM study on the reductive half-reaction of xanthine oxidase: substrate orientation and mechanism. J. Am. Chem. Soc 2009, 131, 14885–14902. [Google Scholar]
- Nakamura, M.; Yuichiro, Y.; Jorn Oliver, S.; Tomohiro, M.; Schwab, K.O.; Takeshi, N.; Tatsuo, H.; Ichida, K. Identification of a xanthinuria type I case with mutations of xanthine dehydrogenase in an Afghan child. Clin. Chim. Acta 2012, 414, 158–160. [Google Scholar]
- Sakamoto, N.; Yamamoto, T.; Moriwaki, Y.; Teranishi, T.; Toyoda, M.; Onishi, Y.; Kuroda, S.; Sakaguchi, K.; Fujisawa, T.; Maeda, M.; et al. Identification of a new point mutation in the human xanthine dehydrogenase gene responsible for a case of classical type I xanthinuria. Hum. Genet 2001, 108, 279–283. [Google Scholar]
- Jurecka, A.; Stiburkova, B.; Krijt, J.; Gradowska, W.; Tylki-Szymanska, A. Xanthine dehydrogenase deficiency with novel sequence variations presenting as rheumatoid arthritis in a 78-year-old patient. J. Inherit. Metab. Dis. 2010. [Google Scholar] [CrossRef]
- Stiburkova, B.; Krijt, J.; Vyletal, P.; Bartl, J.; Gerhatova, E.; Korinek, M.; Sebesta, I. Novel mutations in xanthine dehydrogenase/oxidase cause severe hypouricemia: Biochemical and molecular genetic analysis in two Czech families with xanthinuria type I. Clin. Chim. Acta 2012, 413, 93–99. [Google Scholar]
- Levartovsky, D.; Lagziel, A.; Sperling, O.; Liberman, U.; Yaron, M.; Hosoya, T.; Ichida, K.; Peretz, H. XDH gene mutation is the underlying cause of classical xanthinuria: A second report. Kidney Int 2000, 57, 2215–2220. [Google Scholar]
- Kudo, M.; Moteki, T.; Sasaki, T.; Konno, Y.; Ujiie, S.; Onose, A.; Mizugaki, M.; Ishikawa, M.; Hiratsuka, M. Functional characterization of human xanthine oxidase allelic variants. Pharmacogenetics Genom 2008, 18, 243–251. [Google Scholar]
- Gok, F.; Ichida, K.; Topaloglu, R. Mutational analysis of the xanthine dehydrogenase gene in a Turkish family with autosomal recessive classical xanthinuria. Nephrol. Dial. Transplant 2003, 18, 2278–2283. [Google Scholar]
- Fujiwara, Y.; Kawakami, Y.; Shinohara, Y.; Ichida, K. A case of hereditary xanthinuria type 1 accompanied by bilateral renal calculi. Intern. Med 2012, 51, 1879–1884. [Google Scholar]
- Kikuchi, H.; Fujisaki, H.; Furuta, T.; Okamoto, K.; Leimkuhler, S.; Nishino, T. Different inhibitory potency of febuxostat towards mammalian and bacterial xanthine oxidoreductases: Insight from molecular dynamics. Sci. Rep 2012, 2, 331. [Google Scholar]
- Massey, V.; Edmondson, D. On the mechanism of inactivation of xanthine oxidase by cyanide. J. Biol. Chem 1970, 245, 6595–6598. [Google Scholar]
- Wahl, R.C.; Rajagopalan, K.V. Evidence for the inorganic nature of the cyanolyzable sulfur of molybdenum hydroxylases. J. Biol. Chem 1982, 257, 1354–1359. [Google Scholar]
- Nishino, T.; Nishino, T.; Tsushima, K. Purification of highly active milk xanthine oxidase by affinity chromatography on Sepharose 4B/folate gel. FEBS Lett 1981, 131, 369–372. [Google Scholar]
- Ikegami, T.; Nishino, T. The presence of desulfo xanthine dehydrogenase in purified and crude enzyme preparations from rat liver. Arch. Biochem. Biophys 1986, 247, 254–260. [Google Scholar]
- Nishino, T. Purification of hepatic xanthine dehydrogenase from chicken fed a high-protein diet. Biochim. Biophys. Acta 1974, 341, 93–98. [Google Scholar]
- Itoh, R.; Nishino, T.; Usami, C.; Tsushima, K. An immunochemical study of the changes in chicken liver xanthine dehydrogenase activity during dietary adaptation. J. Biochem 1978, 84, 19–26. [Google Scholar]
- Wahl, R.C.; Warner, C.K.; Finnerty, V.; Rajagopalan, K.V. Drosophila melanogaster ma-l mutants are defective in the sulfuration of desulfo Mo hydroxylases. J. Biol. Chem 1982, 257, 3958–3962. [Google Scholar]
- Reiter, S.; Simmonds, H.A.; Zollner, N.; Braun, S.L.; Knedel, M. Demonstration of a combined deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxipurinol. Clin. Chim. Acta 1990, 187, 221–234. [Google Scholar]
- Amrani, L.; Primus, J.; Glatigny, A.; Arcangeli, L.; Scazzocchio, C.; Finnerty, V. Comparison of the sequences of the Aspergillus nidulans hxB and Drosophila melanogaster ma-l genes with nifS from Azotobacter vinelandii suggests a mechanism for the insertion of the terminal sulphur atom in the molybdopterin cofactor. Mol. Microbiol 2000, 38, 114–125. [Google Scholar]
- Watanabe, T.; Ihara, N.; Itoh, T.; Fujita, T.; Sugimoto, Y. Deletion mutation in Drosophila ma-l homologous, putative molybdopterin cofactor sulfurase gene is associated with bovine xanthinuria type II. J. Biol. Chem 2000, 275, 21789–21792. [Google Scholar]
- Yamamoto, T.; Moriwaki, Y.; Takahashi, S.; Tsutsumi, Z.; Tuneyoshi, K.; Matsui, K.; Cheng, J.; Hada, T. Identification of a new point mutation in the human molybdenum cofactor sulferase gene that is responsible for xanthinuria type II. Metabolism 2003, 52, 1501–1504. [Google Scholar]
- Peretz, H.; Naamati, M.S.; Levartovsky, D.; Lagziel, A.; Shani, E.; Horn, I.; Shalev, H.; Landau, D. Identification and characterization of the first mutation (Arg776Cys) in the C-terminal domain of the Human Molybdenum Cofactor Sulfurase (HMCS) associated with type II classical xanthinuria. Mol. Genet. Metabol 2007, 91, 23–29. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inherited disorders of purine metabolism |
| Genetic defects in the molybdoflavoprotein enzymes: |
| Xanthinuria type I (xanthine oxidoreductase deficiency) |
| Xanthinuria type II (molybdenum cofactor sulfurase deficiency: combined xanthine oxidoreductase and aldehyde oxidase deficiencies) |
| Molybdenum cofactor deficiency |
| Purine nucleoside phosphorylase deficiency |
| Phosphoribosylpyrophosphate synthetase deficiency |
| Secondary reduction in uric acid biosynthesis |
| Hepatic failure |
| Inherited renal hypouricemia (isolated renal tubule reabsorption defect) |
| Renal hypouricemia-1 [URAT1 (SLC22A12) deficiency] |
| Renal hypouricemia-2 [URAT9 (SLC22A9) deficiency] |
| Inherited causes of the Fanconi renotubular syndrome and its variants (the syndrome of multiple renal tubule reabsorption defects) |
| Fanconi renotubular syndrome 1 |
| Cystinosis (accumulation of intralysosomal cystine) |
| Galactosemia (galactose-1-phosphate uridylyltransferase deficiency) |
| Hereditary fructose intolerance (fructose 1-phosphate aldolase B deficiency) |
| Glycogen storage disease type 1 (glucose-6-phosphate deficiency) |
| Wilson’s disease [ATPase, Cu2+ transporting, beta polypeptide (ATP7B) deficiency] |
| Mitochondrial complex IV deficiency (cytochrome c oxidase deficiency) |
| Acquired causes of the Fanconi renotubular syndrome and its variants |
| Metal poisoning (e.g., Cd, Zn, Cu, Pb, Hg) |
| Multiple myeloma |
| Nephrotic syndrome |
| Malignant disease |
| Autoimmune disease (e.g., Sjogren’s syndrome) |
| Thermal burns |
| Primary hyperparathyroidism |
| Acute renal tubular necrosis |
| Renal transplant rejection |
| Drugs |
| Xanthine oxidoreductase inhibitor (e.g., allopurinol, febuxostat) |
| Drugs used either as uricosuric agents or to block other aspects of renal tubule excretion (e.g., sulfinpyrazone, probenecid, benzbromarone) |
| Non-steroidal anti-inflammatory drugs with uricosuric properties (e.g., phenylbutazone, azapropazone, high dose of aspirin) |
| Coumarin anticoagulants (e.g., warfarin) |
| Outdated tetracycline (5 alpha-6-anhydro-4-epitetracycline) |
| Nutritional deficiencies |
| Vitamines B12, C, D |
| Kwashiorkor |
| Corresponding human residue No. | Residue in experimental animal | Function | Experiments |
|---|---|---|---|
| The Fe/S domain | |||
| Cys43 | rat Cys43 | Fe/S II ligand | mutation to Ser [71] |
| Cys51 | rat Cys51 | Fe/S II ligand | mutation to Ser or Ala [71] |
| Cys116 | rat Cys115 | Fe/S I ligand | mutation to Ser [71] |
| Lys185 | rat Lys184 | interdomain | Trypsin [62] |
| The FAD domain | |||
| Arg427 | bovine Arg427 | A member of the cluster XDH/XO conversion | mutation to Gln [72] |
| Arg335 | bovine Arg335 | A member of the cluster XDH/XO conversion | mutation to Ala [72] |
| Trp336 | bovine Trp336 & rat Trp335 | A member of the cluster XDH/XO conversion | mutation to Ala [72] |
| Phe337 | rat Phe336 | redox potential of FAD | mutation to Leu (to be published) |
| Tyr393 | chicken Tyr419 | NAD+ binding | chemical modification with FSBA [73] |
| Asp429 | rat Asp428 | redox potential of FAD | mutation (to be published) |
| Cys536 | rat Cys535 | disulfide formation with Cys992 XDH/XO conversion | mutation to Ala [70] & chemical modification with FDNB [74] |
| Lys552 | rat Lys551 | Interdomain trypsin XDH/XO | Trypsin [62] |
| The Moco domain | |||
| Lys755 | bovine Lys754 | kcat slower | chemical modification with FDNB [74,75] |
| Lys772 | bovine Lys771 | kcat slower | chemical modification with FDNB [74,75] |
| Glu803 | human | purine binding | mutation to Val [69] |
| Arg881 | human | purine binding | mutation to Met [69] |
| Cys993 | rat Cys992 | disulfide with Cys535 XDH/XO conversion | mutation to Arg [70] & chemical modification with FDNB [74] |
| Glu1262 | human | mutation to Ala [69,76] | |
| Cys1318 | rat Cys1316 | disulfie with Cys1324? | mutation to Ser [70] |
| Cys1326 | rat Cys1324 | disulfide with Cys1316? | mutation to Ser [70] & chemical modification with FDNB [74] |
| Codon change | Amino acid change | Codon number | Phenotype | Reference |
|---|---|---|---|---|
| c. 140_141insG (c. 140dupG) | p.Cys48LeufsX12 | 47 | Xanthinuria, type 1 | [92] |
| c. 445C > T | p.Arg149Cys | 149 | Xanthinuria, type 1 | [93] |
| c. 641delC | p.Pro214GlnfsX4 | 214 | Xanthinuria, type 1 | [94,95] |
| c. 682C > T | p.Arg228X | 228 | Xanthinuria, type 1 | [37] |
| c. 1664_1665insC (c.1664dupC) | p.Ala556SerfsX15 | 555 | Xanthinuria, type 1 | [96] |
| c. 1663C > T | p.Pro555Ser | 555 | Decreased activity | [97] |
| c. 1820G > A | p.Arg607Gln | 607 | Decreased activity | [97] |
| c. 1868C > T | p.Thr623Ile | 623 | Decreased activity | [97] |
| c. 2107A > G | p.Ile703Val | 703 | Increased activity | [97] |
| c. 2164A > T | p.Lys722X | 722 | Xanthinuria, type 1 | [98] |
| c. 2473C > T | p.Arg825X | 825 | Xanthinuria, type 1 | [95] |
| c. 2567delC | p.Thr856LysfsX73 | 856 | Xanthinuria, type 1 | [37,96] |
| c. 2641C > T | p.Arg881X | 881 | Xanthinuria, type 1 | [95] |
| c. 2727C > A | p.Asn909Lys | 909 | Decreased activity | [97] |
| c. 2729C > A | p.Thr910Lys | 910 | XDH deficiency | [97] |
| c. 2729C > T | p.Thr910Met | 910 | Xanthinuria, type 1 | [52,92] |
| c. 3449C > G | p.Pro1150Arg | 1150 | Decreased activity | [97] |
| c. 3662A > G | p.His1221Arg | 1221 | Increased activity | [97] |
| c. 3953G > A | p.Cys1318Tyr | 1318 | Decreased activity | [97] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ichida, K.; Amaya, Y.; Okamoto, K.; Nishino, T. Mutations Associated with Functional Disorder of Xanthine Oxidoreductase and Hereditary Xanthinuria in Humans. Int. J. Mol. Sci. 2012, 13, 15475-15495. https://doi.org/10.3390/ijms131115475
Ichida K, Amaya Y, Okamoto K, Nishino T. Mutations Associated with Functional Disorder of Xanthine Oxidoreductase and Hereditary Xanthinuria in Humans. International Journal of Molecular Sciences. 2012; 13(11):15475-15495. https://doi.org/10.3390/ijms131115475
Chicago/Turabian StyleIchida, Kimiyoshi, Yoshihiro Amaya, Ken Okamoto, and Takeshi Nishino. 2012. "Mutations Associated with Functional Disorder of Xanthine Oxidoreductase and Hereditary Xanthinuria in Humans" International Journal of Molecular Sciences 13, no. 11: 15475-15495. https://doi.org/10.3390/ijms131115475
APA StyleIchida, K., Amaya, Y., Okamoto, K., & Nishino, T. (2012). Mutations Associated with Functional Disorder of Xanthine Oxidoreductase and Hereditary Xanthinuria in Humans. International Journal of Molecular Sciences, 13(11), 15475-15495. https://doi.org/10.3390/ijms131115475