Isolation and Structural Characterization of Lignin from Cotton Stalk Treated in an Ammonia Hydrothermal System




Abstract
:1. Introduction
2. Results and Discussion
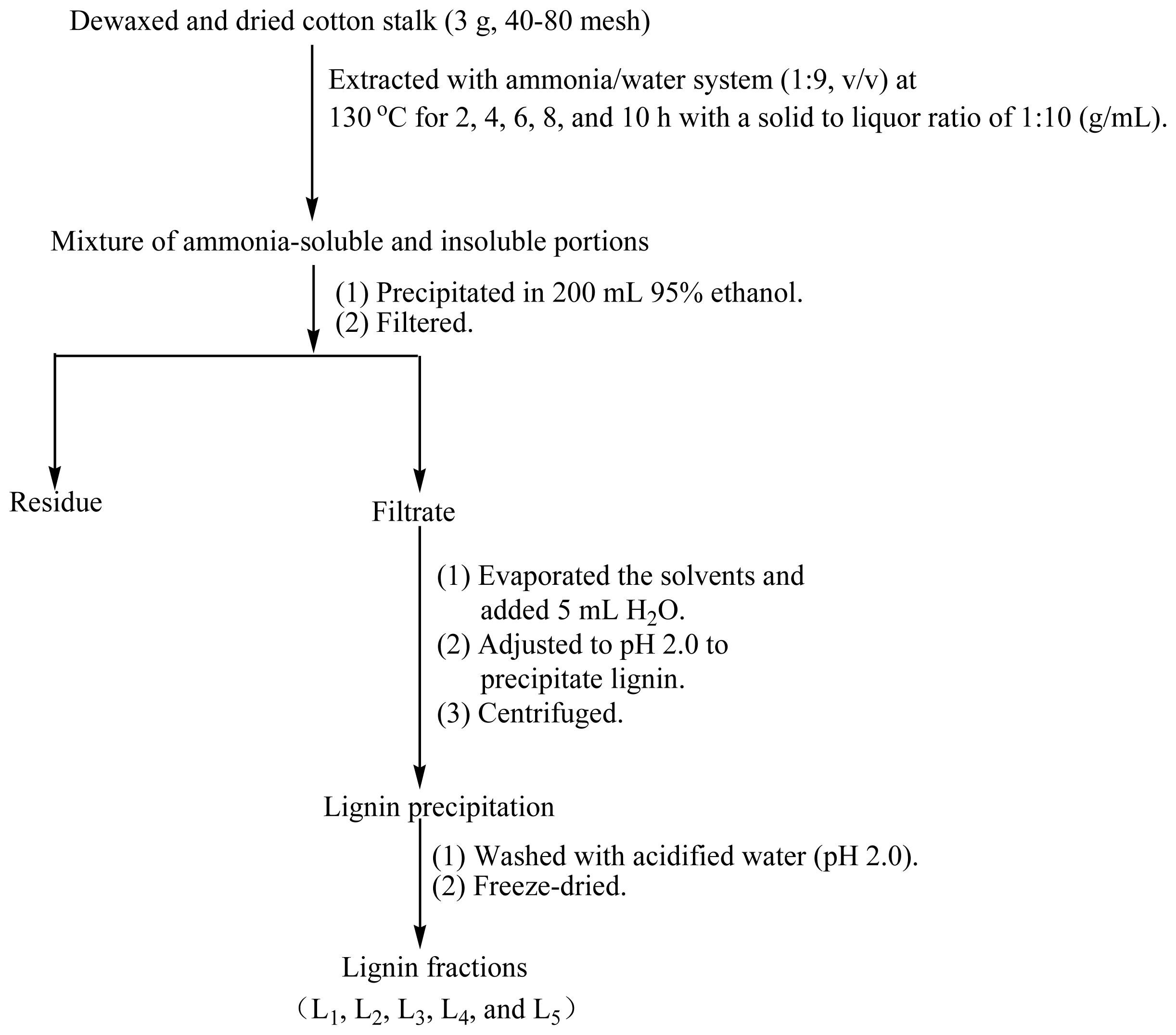
2.1. Purity of Lignin Fractions
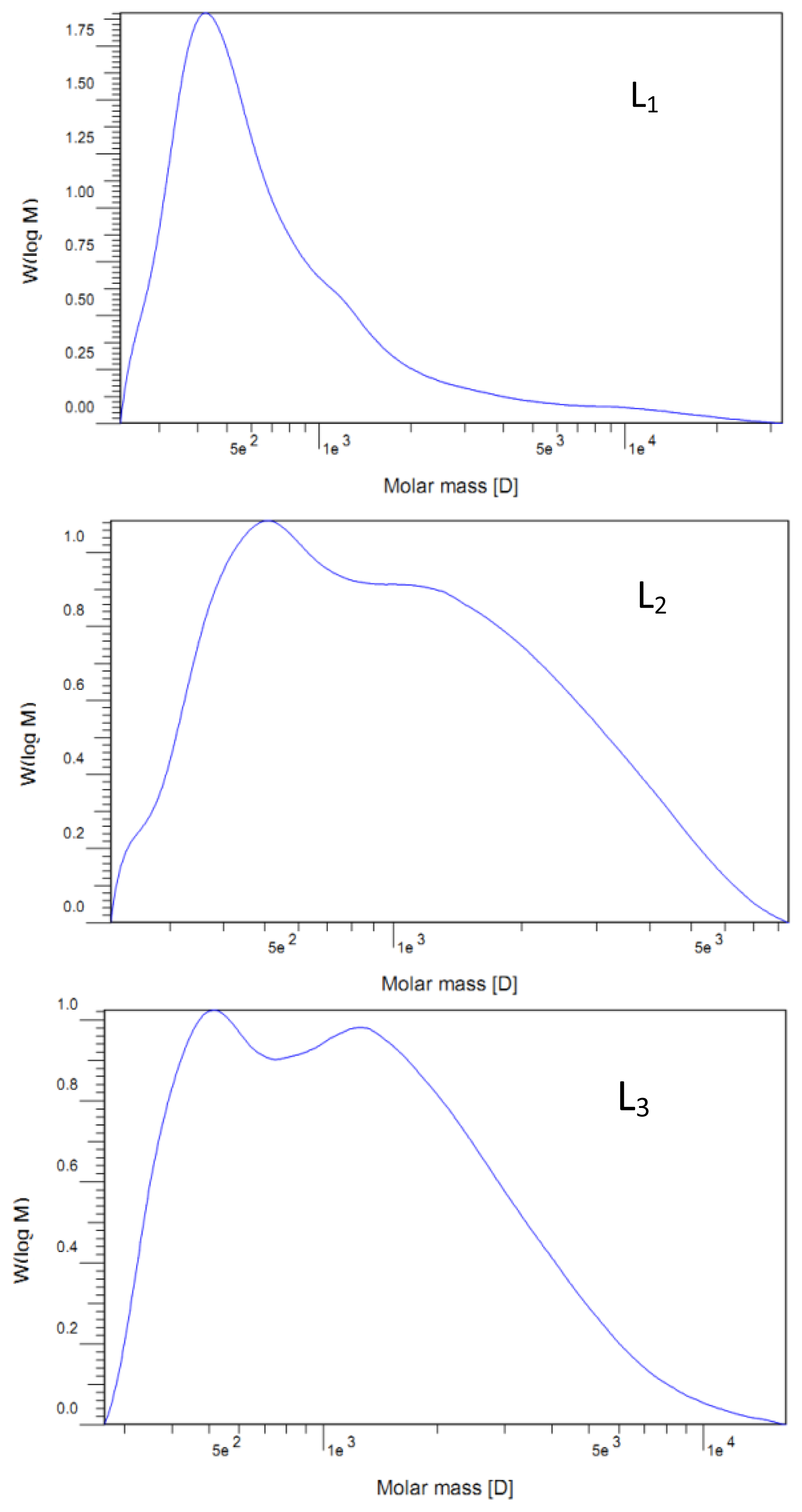
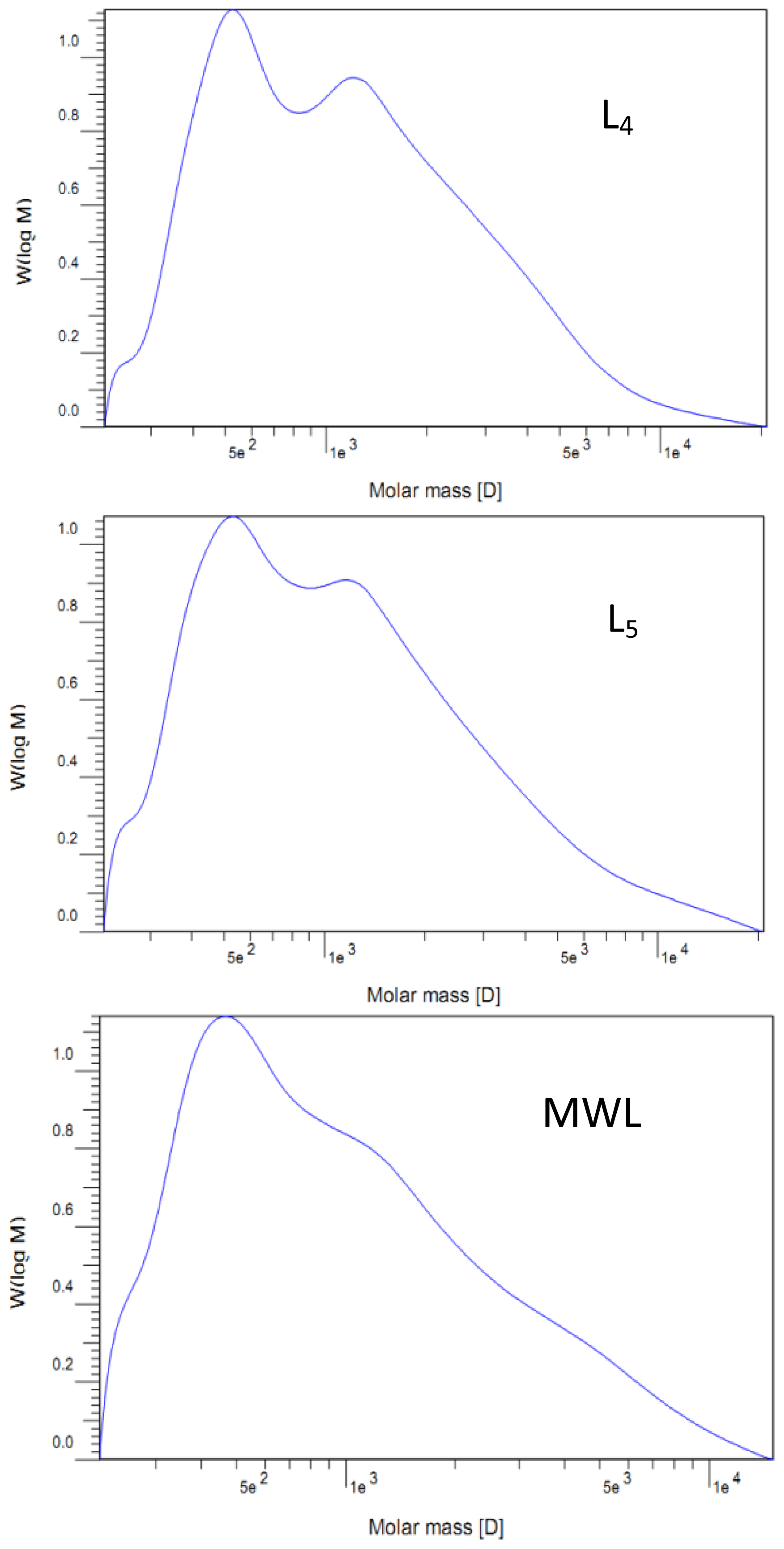
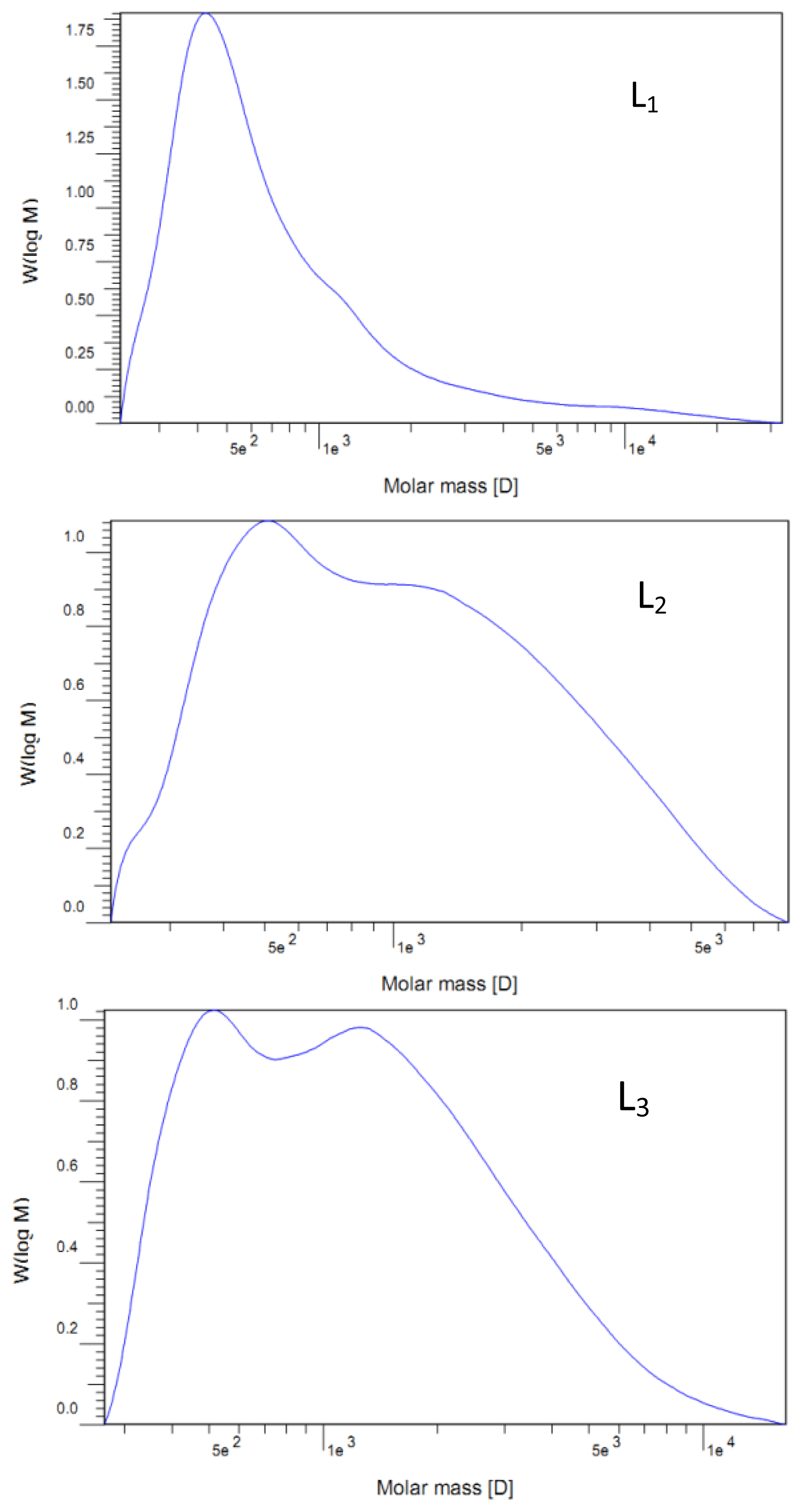
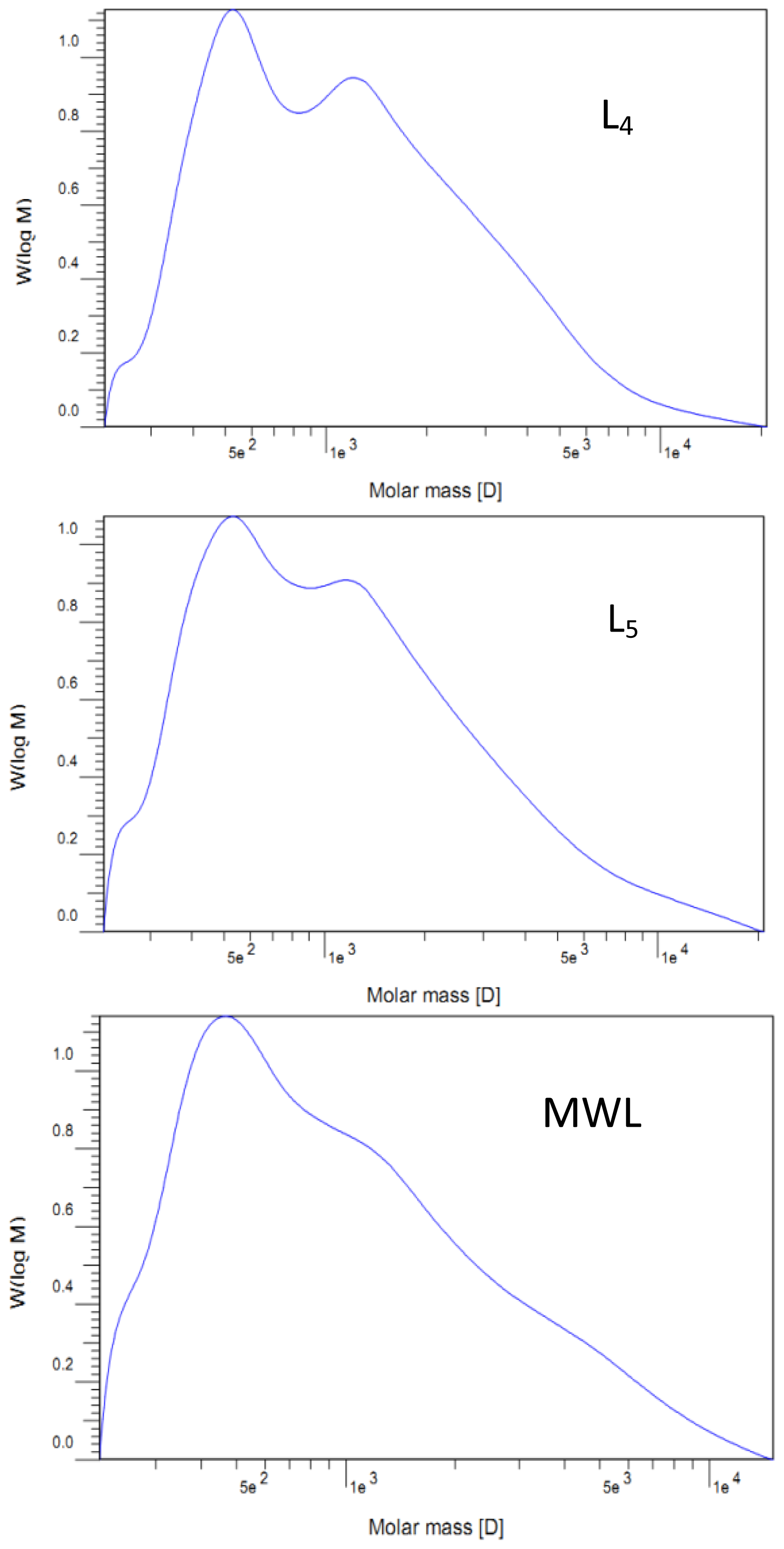
2.2. Molecular Weight Distribution
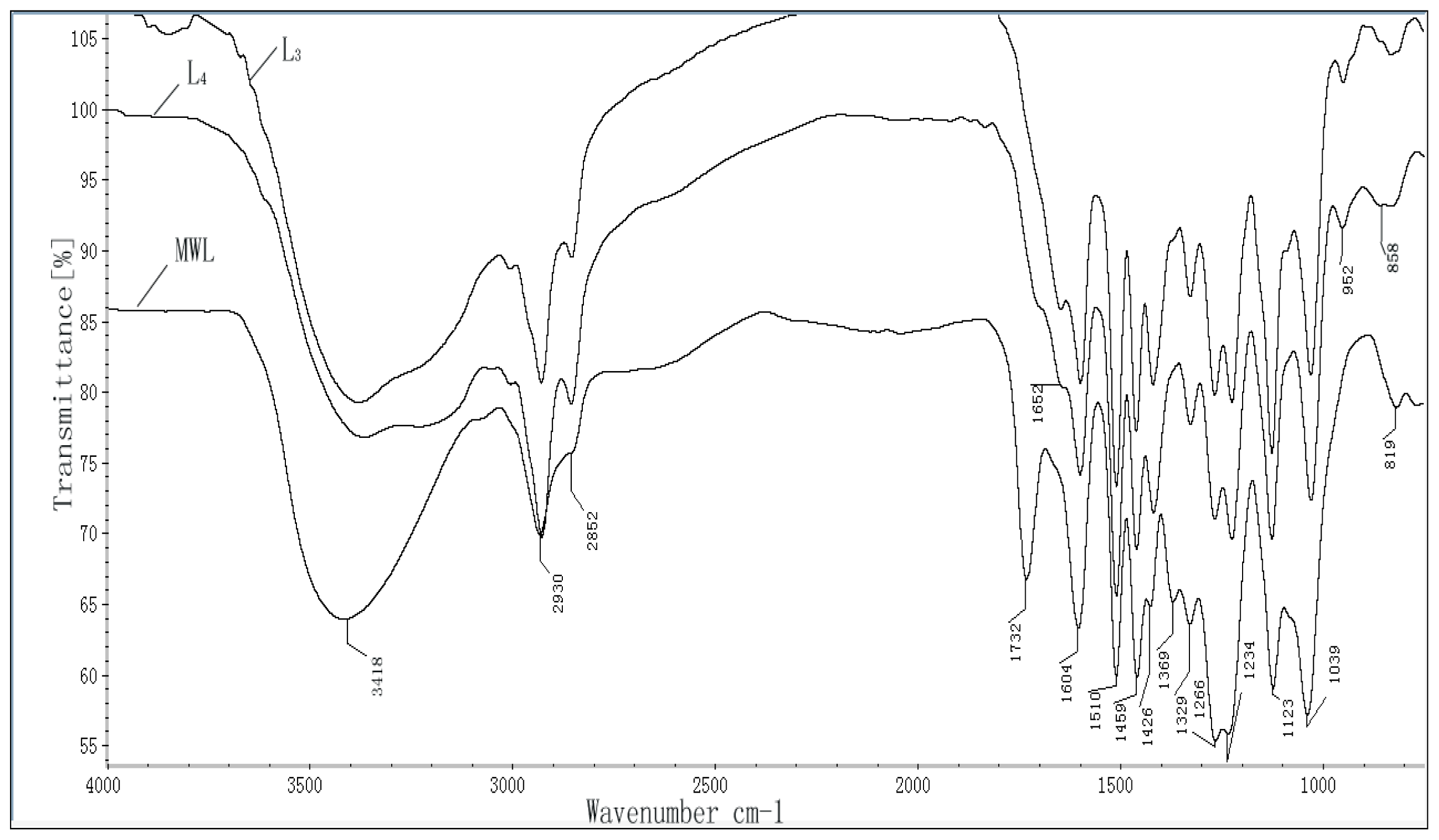
2.3. FT-IR Spectra
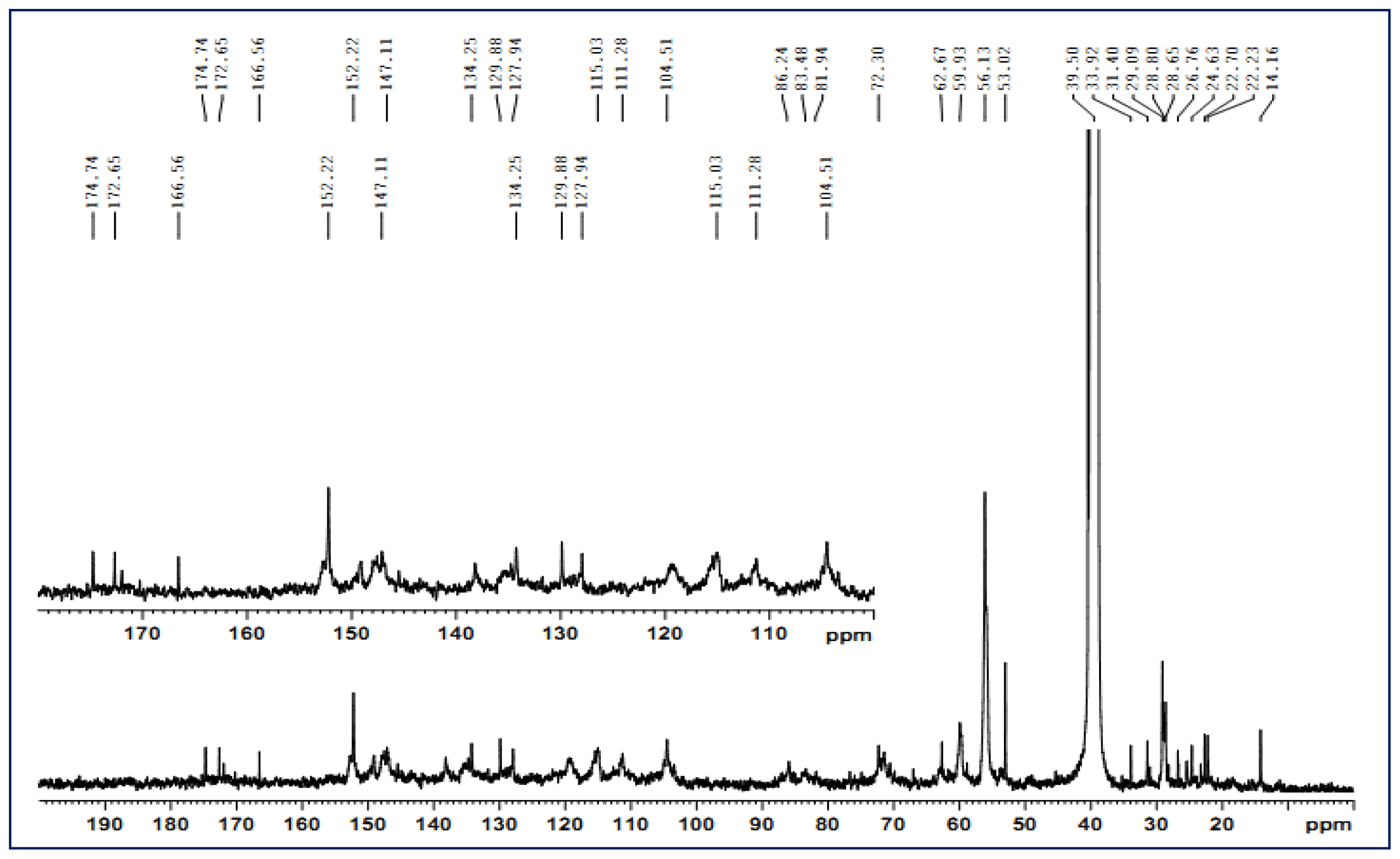
2.4. 13C-NMR Spectra
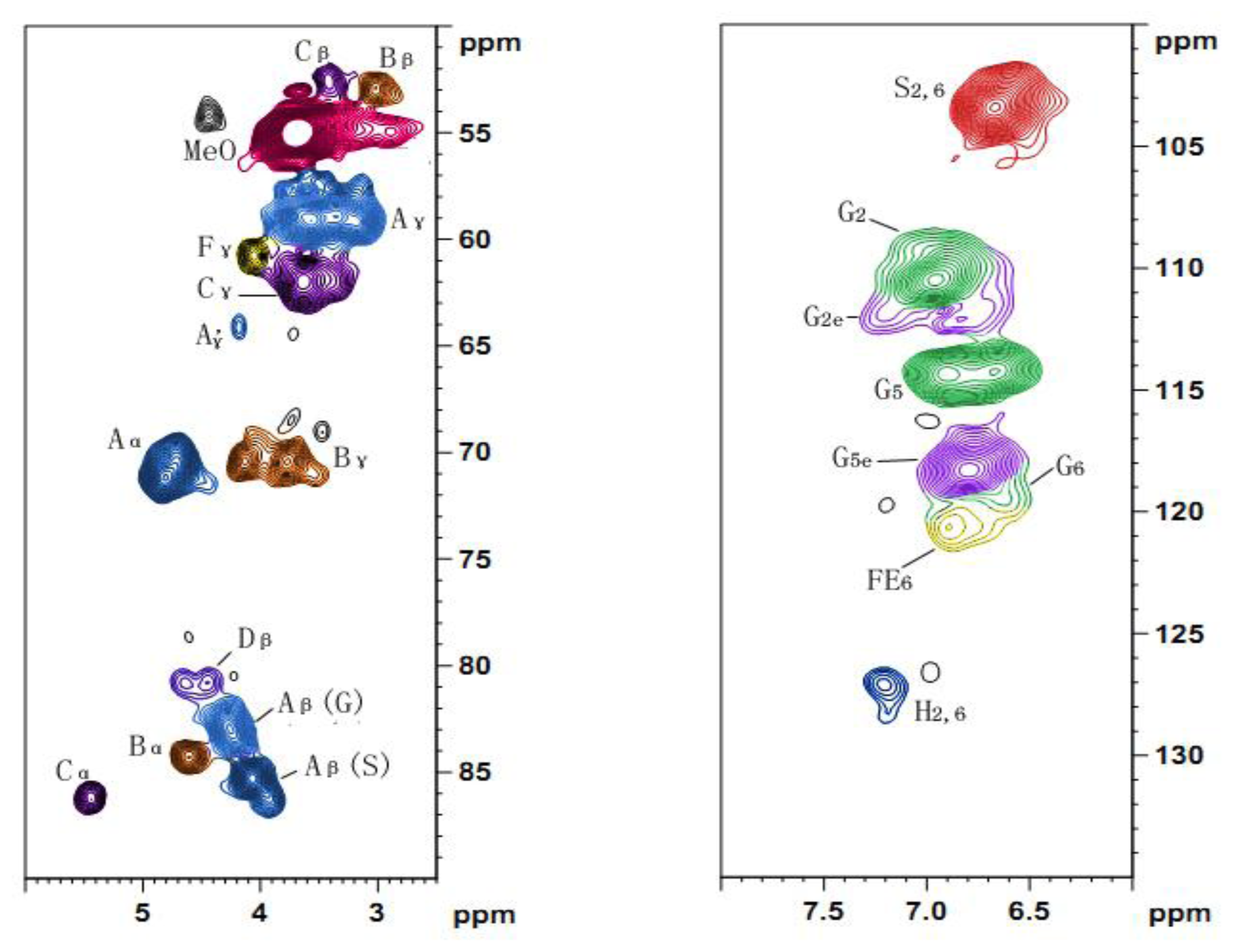
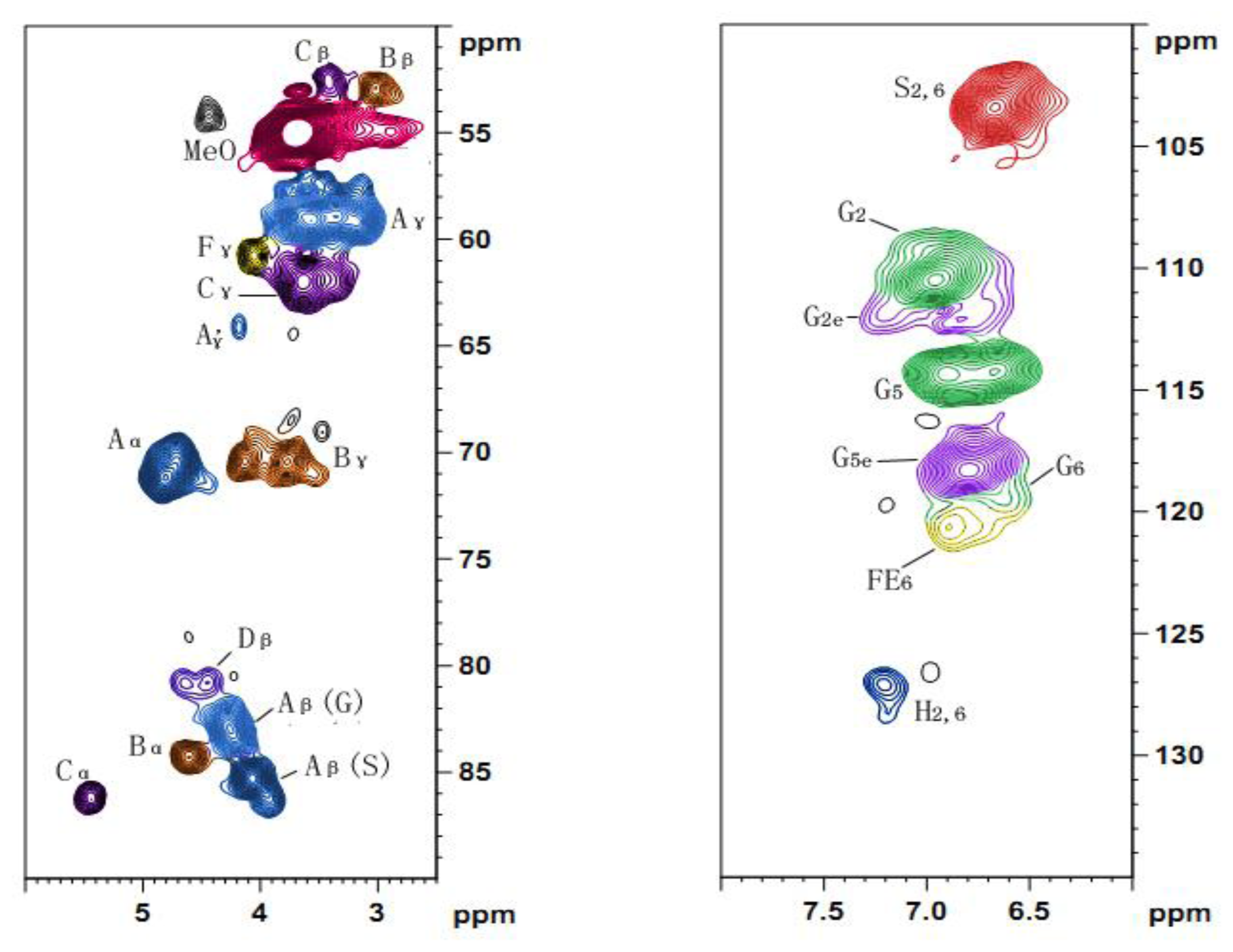
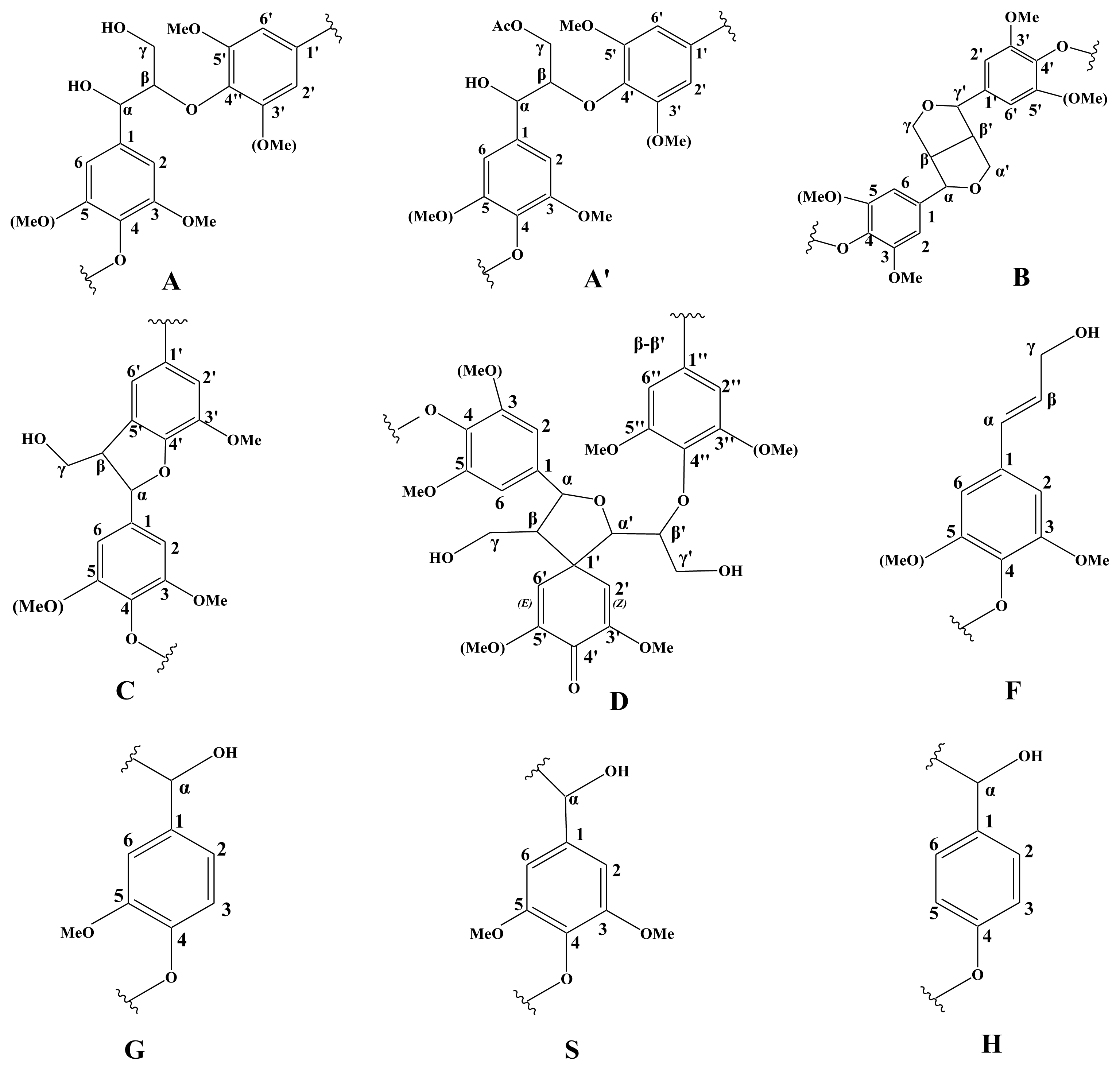
2.5. 2D HSQC NMR
3. Experimental Section
3.1. Materials
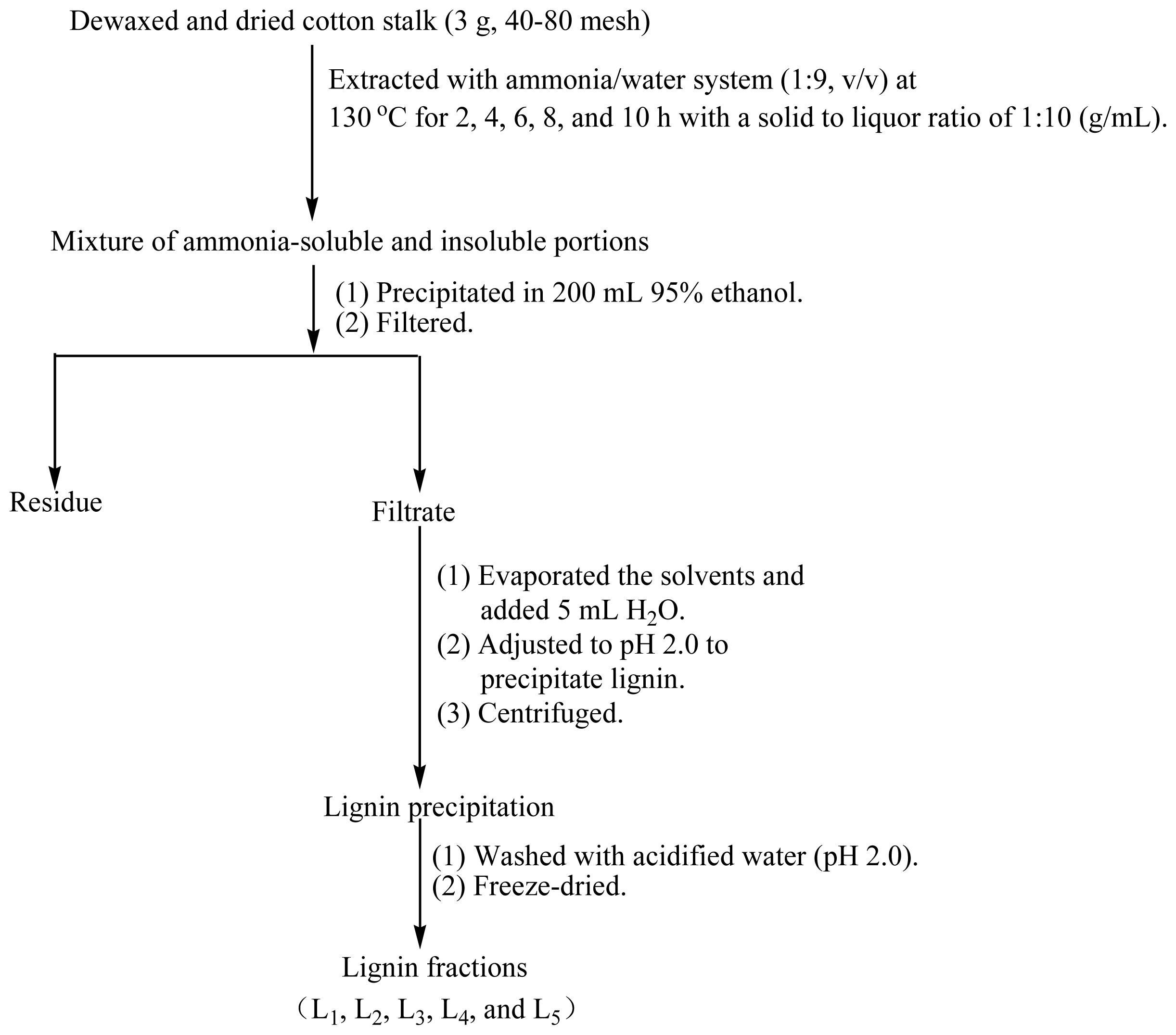
3.2. Isolation and Purification of Lignin Fractions
3.3. Sugar Analysis
3.4. Determination of Molecular Weight
3.5. FT-IR Spectral Characterization
3.6. Nuclear Magnetic Resonance Spectra
4. Conclusions
Acknowledgments
References
- Mohammadi-Rovshandeh, J.; Sereshti, H. The effect of extraction and prehydrolysis on the thermoplasticity and thermal stability of chemically modified rice straw. Iran. Polym. J 2005, 14, 855–862. [Google Scholar]
- Ikeda, T.; Holtman, K.; Kadla, J.F.; Chang, H.M.; Jameel, H. Studies on effect of ball milling on lignin structure using a modified DFRC method. J. Agric. Food Chem 2002, 50, 129–135. [Google Scholar]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol 2003, 54, 519–546. [Google Scholar]
- Rencoret, J.; Gutiérrez, A.; Nieto, L.; Jiménez-Barbero, J.; Faulds, C.B.; Kim, H.; Ralph, J.; Martínez, Á.T.; del Río, J.C. Lignin composition and structure in young versus adult Eucalyptus globulus plants. Plant Physiol. 2011, 155, 667–682. [Google Scholar]
- Gellerstedt, G.; Henriksson, G. Lignins: Major sources, structure and properties. In Monomers, Polymers and Composites from Renewable Resources; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume Chapter 9, pp. 201–204. [Google Scholar]
- Ross, K.; Mazza, G. Characteristics of lignin from flax shives as affected by extraction conditions. Int. J. Mol. Sci 2010, 11, 4035–4050. [Google Scholar]
- Campbell, M.M.; Sederoff, R.R. Variation in lignin content and composition. mechanisms of control and implications for the genetic improvement of plants. Plant Physiol 1996, 110, 3–13. [Google Scholar]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol 2010, 153, 895–905. [Google Scholar]
- Sticklen, M.B. Plant genetic engineering for biofuel production: towards affordable cellulosic ethanol. Nat. Rev. Genet 2008, 9, 433–443. [Google Scholar]
- Weng, J.K.; Li, X.; Bonawitz, N.D.; Chapple, C. Emerging strategies of lignin engineering and degradation for cellulosic biofuel production. Curr. Opin. Biotechnol 2008, 19, 166–172. [Google Scholar]
- Mansfield, S.D. Solutions for dissolution-engineering cell walls for deconstruction. Curr Opin. Biotechnol 2009, 20, 286–294. [Google Scholar]
- Xu, F.; Sun, R.C.; Zhai, M.Z.; Sun, J.X.; Jiang, J.X.; Zhao, G.J. Comparative study of three lignin fractions isolated from mild ball-milled Tamarix austromogoliac and Caragana sepium. J. Appl. Polym. Sci 2008, 108, 1158–1168. [Google Scholar]
- Kondo, R.; Sako, T.; Limori, T.; Imamura, H. Formation of glycosidic lignin-carbohydrate complex in the dehydrogenative polymerization of coniferyl alcohol. Mokuzai Gakkaishi 1990, 36, 332–338. [Google Scholar]
- Guerra, A.; Filpponen, I.; Lucia, L.A.; Argyropoulos, D.S. Comparative evaluation of three lignin isolation protocols for various wood species. J. Agric. Food Chem 2006, 54, 9696–9705. [Google Scholar]
- Pye, E.K. Industrial lignin production and applications. In Biorefineries-Industrial Processes and Products; Kamm, B., Gruber, P.R., Kamm, M., Eds.; Wiley-VCH: Weinheim, Germany, 2008; Volume Chapter 5, pp. 165–200. [Google Scholar]
- Kim, H.S.; Cho, D.H.; Won, K.; Kim, Y.H. Inactivation of Coprinus cinereus peroxidase during the oxidation of various phenolic compounds originated from lignin. Enzyme Microb. Tech 2009, 45, 150–155. [Google Scholar]
- Wang, K.; Xu, F.; Sun, R.C. Molecular Characteristics of kraft-AQ pulping lignin fractionated by sequential organic solvent extraction. Int. J. Mol. Sci 2010, 11, 2988–3001. [Google Scholar]
- Zhang, A.P.; Lu, F.C.; Liu, C.F.; Sun, R.C. Isolation and characterization of lignins from Eucalyptus tereticornis (12ABL). J. Agric. Food Chem 2010, 58, 11287–11293. [Google Scholar]
- Björkman, A. Studies on finely divided wood. Part V. the effect of milling. Svensk Papperst 1957, 60, 329–335. [Google Scholar]
- Bardet, M.; Robert, D.R. On the reactions and degradation of the lignin during steam hydrolysis of aspen wood. Svensk Papperst 1985, 88, 61–67. [Google Scholar]
- Ramos, L.P. The chemistry involved in the steam treatment of lignocellulosic materials. Quim. Nova 2003, 26, 863–871. [Google Scholar]
- Li, J.; Henriksson, G.; Gellerstedt, G. Lignin depolymerization/repolymerization and its critical role for delignification of aspen wood by stream explosion. Bioresour. Technol 2007, 98, 3061–3068. [Google Scholar]
- Sannigrahi, P.; Kim, D.H.; Jung, S.; Ragauskas, A.J. Pseudo lignin and pretreatment chemistry. Energy Environ. Sci 2011, 4, 1306–1310. [Google Scholar]
- Klinke, H.B.; Thomsen, A.B.; Ahring, B.K. Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl. Microbiol. Biotechnol 2004, 66, 10–26. [Google Scholar]
- Björkman, A. Isolation of lignin from finely divided wood with neutral solvents. Nature 1954, 174, 1057–1058. [Google Scholar]
- Xiao, L.P.; Shi, Z.J.; Xu, F.; Sun, R.C. Characterization of MWLs from Tamarix ramosissima isolated before and after hydrothermal treatment by spectroscopical and wet chemical methods. Holzforschung 2011, 66, 341–348. [Google Scholar]
- Zhang, B.; Huang, H.J.; Ramaswamy, S. Reaction kinetics of the hydrothermal treatment of lignin. Appl. Biochem. Biotechnol 2008, 147, 119–131. [Google Scholar]
- Wyman, C.E.; Dale, B.E.; Elander, R.T.; Holtzapple, M.; Ladisch, M.R.; Lee, Y.Y. Coordinated development of leading biomass pretreatment technologies. Bioresour. Technol 2005, 96, 1959–1966. [Google Scholar]
- Kim, T.H.; Kim, J.S.; Sunwoo, C.S.; Lee, Y.Y. Pretreatment of corn stover by aqueous ammonia. Bioresour. Technol 2003, 90, 39–47. [Google Scholar]
- Deng, H.; Lu, J.J.; Li, G.X.; Zhang, G.L.; Wang, X.G. Adsorption of methylene blue on adsorbent materials produced from cotton stalk. Chem. Eng. J 2011, 172, 326–334. [Google Scholar]
- Rencoret, J.; Marques, G.; Gutierrez, A.; Nieto, L.; Jimenez-Barbero, J.; Martinez, A.T.; del Rio, J.C. Isolation and structural characterization of the milled-wood lignin from Paulownia fortunei wood. Ind. Crop. Prod 2009, 30, 137–143. [Google Scholar]
- Fasching, M.; Schröder, P.; Wollboldt, R.P.; Weber, H.K.; Sixta, H. A new and facile method for isolation of lignin from wood based on complete wood dissolution. Holzforschung 2008, 62, 15–23. [Google Scholar]
- Scalbert, A.; Monties, B. Comparison of wheat straw lignin preparations II, Straw lignin solubilisation in alkali. Holzforschung 1986, 40, 249–254. [Google Scholar]
- Sun, R.C.; Lawther, J.M.; Banks, W.B. Effects of extraction time and different alkalis on the composition of alkali soluble wheat straw lignins. J. Agric. Food Chem 1996, 44, 3965–3970. [Google Scholar]
- Sun, R.C.; Sun, X.F.; Fowler, P.; Tomkinson, J. Structural and physico-chemical characterization of lignins solubilized during alkaline peroxide treatment of barley straw. Eur. Polym. J 2002, 38, 1399–1407. [Google Scholar]
- Faix, O. Classification of lignins from different botanical origins by FT-IR spectroscopy. Holzforschung 1991, 45, 21–27. [Google Scholar]
- Tai, D.S.; Terazawa, M.; Chen, C.L.; Chang, H.M. Lignin biodegradation products from birch wood by phanerochaete chrysosporium. part I. fractionation of methanol-extractive and characterization of ether-insoluble low-molecular-weight fraction. Holzforschung 1990, 44, 185–190. [Google Scholar]
- Li, M.F.; Fan, Y.M.; Sun, R.C.; Xu, F. Characterization of extracted lignin of bamboo (Neosinocalamus affinis) pretreated with sodium hydroxide/urea solution at low temperature. Bioresources 2010, 3, 1762–1778. [Google Scholar]
- Del, R.; Jose, C.; Rencoret, J.; Marques, G.; Li, J.B.; Gellerstedt, G.; Jiménez-Barbero, J.; Martínez, A.T.; Gutiérrez, A. Structural characterization of the lignin from jute (Corchorus capsularis) fibers. J. Agric. Food Chem 2009, 57, 10271–10281. [Google Scholar]
- Zhang, X.M.; Yuan, T.Q.; Peng, F.; Xu, F.; Sun, R.C. Separation and structural characterization of lignin from hybrid poplar based on complete dissolution in DMSO/LiCl. Separ. Sci. Technol 2010, 45, 2497–2506. [Google Scholar]
- Wen, J.L.; Sun, Z.J.; Sun, Y.C.; Sun, S.N.; Xu, F.; Sun, R.C. Structural characterization of alkali-extractable lignin fractions from bamboo. J. Biobased Mater. Bioenergy 2010, 4, 1–18. [Google Scholar]
- Lu, H.F.; Hu, R.F.; Ward, A.; Amidon, T.E.; Liang, B.; Liu, S.J. Hot-water extraction and its effect on soda pulping of aspen woodchips. Biomass Bioenergy 2012, 39, 5–13. [Google Scholar]
- Nguyen, T.A.D.; Kim, K.R.; Han, S.J.; Cho, H.Y.; Kim, J.W.; Park, S.M.; Park, J.C.; Sim, S.J. Pretreatment of rice straw with ammonia and ionic liquid for lignocellulose conversion to fermentable sugars. Bioresour. Technol 2010, 101, 7432–7438. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sugars (%) | Lignin fractions a | |||||
|---|---|---|---|---|---|---|
| L1 | L2 | L3 | L4 | L5 | MWL | |
| Rhamnose | ND b | ND | 0.02 | 0.01 | 0.03 | 0.09 |
| Arabinose | ND | ND | 0.02 | 0.01 | 0.05 | 0.09 |
| Galactose | 0.03 | 0.01 | 0.07 | 0.04 | 0.05 | 0.09 |
| Glucose | 1.18 | 1.21 | 0.70 | 0.36 | 0.83 | 2.04 |
| Xylose | 0.05 | 0.03 | 0.12 | 0.01 | 0.09 | 4.43 |
| Mannose | 0.03 | ND | 0.05 | ND | ND | ND |
| Total | 1.29 | 1.25 | 0.98 | 0.43 | 1.06 | 6.74 |
| Lignin fractions a | ||||||
|---|---|---|---|---|---|---|
| L1 | L2 | L3 | L4 | L5 | MWL | |
| Mw | 1250 | 1390 | 1700 | 1690 | 1740 | 1520 |
| Mn | 560 | 760 | 890 | 830 | 790 | 700 |
| Mw/Mn | 2.23 | 1.83 | 1.91 | 2.04 | 2.20 | 2.17 |
| Absorption band (cm−1) | Assignment |
|---|---|
| 3418 | O–H stretching vibration in aromatic and aliphatic OH groups |
| 2930 | C–H stretching vibrations in OCH3 |
| 2930, 2852 | C–H asymmetric and symmetrical vibrations in saturated CH2 |
| 1732 | Unconjugated carbonyl groups |
| 1652 | Carbonyl stretching in para-substituted ketones or aryl aldehydes |
| 1604 | Aromatic ring vibrations and C=O stretching (S > G) |
| 1510 | Aromatic skeletal vibrations (G > S) |
| 1459 | Asymmetric C–H deformations (in CH3 and –CH2–) |
| 1426 | Aromatic skeletal vibrations combined with C–H in plane deform |
| 1360 | COO-asymmetric and symmetrical vibrations in carboxylate groups |
| 1329 | Syringyl units |
| 1266 | Guaiacyl units |
| 1234 | C–C, C–O, and C=O stretch (G condensed > G etherified) |
| 1123 | Aromatic in-plane C–H bending (typical for S units) |
| 1039 | Aromatic C–H in-plane deformation (G > S units) |
| 952 | –HC=CH-out-of-plane deform. (trans) |
| 858 | Aromatic C–H out of bending |
| 819 | C–H out-of-plane in position 2 and 6 of S units, and in all positions of H units |
| PPM | Intensity | Assignment | PPM | Intensity | Assignment |
|---|---|---|---|---|---|
| 174.7 | M | Aliphatic carboxyl carbon | 72.3 | W | C-α, G and S units |
| 172.7 | M | As above | 62.7 | M | C-5, xylose unit |
| 166.6 | W | C-α, carboxylic carbon | 59.9 | S | C-γ, G and S units |
| 152.2 | M | C-3/C-5, S units | 56.1 | S | OCH3, G and S units |
| 134.3 | W | C-1, S units etherified; C-1, G units etherified | 53.02 | VW | C-β, β-5′ units |
| 129.9 | W | C-1, G units | 33.9 | W | CH3 in ketones or in aliphatic side chain |
| 127.9 | W | C-2/C-6, H units | 31.4 | W | As above |
| 115.0 | M | C-5, G units | 29.1 | M | CH2 in aliphatic side chain |
| 111.3 | M | C-2, G units | 25.3 | W | CH3 or CH2 group in side chains |
| 104.5 | M | C-2/C-6, S units | 24.6 | M | As above |
| 86.2 | W | C-β, β-O-4′ | 22.7 | W | As above |
| 83.5 | W | C-α, β-β′ | 14.2 | W | γ-CH3 in n-propyl side chain |
| Lables | δC/δH | Assignment |
|---|---|---|
| Cβ | 52.6/3.41 | Cβ–Hβ in phenylcoumaran substructures (C) |
| Bβ | 52.9/3.02 | Cβ–Hβ in β-β′ (resinol) substructures (B) |
| MeO | 54.9/3.70 | C–H in methoxyls |
| Aγ | 59.1/3.26 and 3.60 | Cγ–Hγ in β-O-4′ substructures (A) |
| Fγ | 60.7/4.06 | Cγ–Hγ in p-hydroxycinnamyl alcohol end groups (F) |
| Cγ | 62.0/3.64 | Cγ–Hγ in phenylcoumaran substructures (C) |
| A′γ | 64.1/4.18 | Cγ–Hγ in γ-acylated β-O-4′ substructures (A′ and A″) |
| Bγ | 70.4/3.76 and 4.13 | Cγ–Hγ in β-β′ resinol substructures (B) |
| Aα | 71.1/4.76 | Cα–Hα in β-O-4′ substructures linked to an S units (A) |
| Dβ′ | 80.9/4.45 | Cβ′–Hβ′ in spirodienone substructures (D) |
| Aβ(G) | 83.2/4.23 | Cβ–Hβ in β-O-4′ substructures linked to G and H units (A) |
| Bα | 84.3/4.61 | Cα–Hα in β-β′ (resinol) substructures (B) |
| Aβ(S) | 85.5/4.07 | Cβ–Hβ in β-O-4′ substructures linked to S units (A) |
| Aβ(S) | 86.1/3.91 | Cβ–Hβ in β-O-4′ substructures linked to S units (A) |
| Cα | 86.2/5.44 | Cα–Hα in phenylcoumaran substructures (C) |
| S2,6 | 103.3/6.66 | C2,6–H2,6 in etherified S units (S) |
| G2 | 110.5/6.94 | C2–H2 in G units (G) |
| G2e | 112.1/7.21 | C2–H2 in etherified G units (G) |
| G5 | 114.3/6.67 and 6.90 | C5–H5 in G units (G) |
| G5e | 118.3/6.79 | C5–H5 in etherified G units (G) |
| G6 | 119.5/6.59 | C6–H6, G units (G) |
| H2,6 | 127.2/7.20 | C2,6–H2,6 in H units (H) |
| Linkage relative abundance (% of total side chains involved) | Relative proportion (%) |
|---|---|
| Lignin inter-unit linkages | |
| β-Aryl-ether units (β-O-4′, A/A′) | 75.6 |
| Resinol substructures (β-β′, B) | 12.2 |
| Phenylcoumaran substructure (β-5′, C) | 7.4 |
| p-Hydroxycinnamyl alcohol end groups (F) | 4.9 |
| Percentage of γ-acetylation | 0.8 |
| Lignin aromatic units | |
| H (%) | 1 |
| S (%) | 40 |
| G (%) | 59 |
| S/G ratio | 0.7 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kang, S.; Xiao, L.; Meng, L.; Zhang, X.; Sun, R. Isolation and Structural Characterization of Lignin from Cotton Stalk Treated in an Ammonia Hydrothermal System. Int. J. Mol. Sci. 2012, 13, 15209-15226. https://doi.org/10.3390/ijms131115209
Kang S, Xiao L, Meng L, Zhang X, Sun R. Isolation and Structural Characterization of Lignin from Cotton Stalk Treated in an Ammonia Hydrothermal System. International Journal of Molecular Sciences. 2012; 13(11):15209-15226. https://doi.org/10.3390/ijms131115209
Chicago/Turabian StyleKang, Sumin, Lingping Xiao, Lingyan Meng, Xueming Zhang, and Runcang Sun. 2012. "Isolation and Structural Characterization of Lignin from Cotton Stalk Treated in an Ammonia Hydrothermal System" International Journal of Molecular Sciences 13, no. 11: 15209-15226. https://doi.org/10.3390/ijms131115209
APA StyleKang, S., Xiao, L., Meng, L., Zhang, X., & Sun, R. (2012). Isolation and Structural Characterization of Lignin from Cotton Stalk Treated in an Ammonia Hydrothermal System. International Journal of Molecular Sciences, 13(11), 15209-15226. https://doi.org/10.3390/ijms131115209