Abstract
Nitric oxide (NO) regulates placental blood flow and actively participates in trophoblast invasion and placental development. Asymmetric dimethylarginine (ADMA) can inhibit NO synthase, which generates NO. ADMA has been associated with uterine artery flow disturbances such as preeclampsia. Substantial experimental evidence has reliably supported the hypothesis that an adverse in utero environment plays a role in postnatal physiological and pathophysiological programming. Growing evidence suggests that the placental nitrergic system is involved in epigenetic fetal programming. In this review, we discuss the roles of NO and ADMA in normal and compromised pregnancies as well as the link between placental insufficiency and epigenetic fetal programming.
1. Introduction
Nitric oxide (NO), the main vasodilator in the placenta, is involved in the regulation of feto-placental vascular reactivity, placental bed vascular resistance, trophoblast invasion and apoptosis, and platelet adhesion and aggregation in the intervillous space [1–3]. Placental vascular development is a crucial process required for adequate fetal development [1,4]. Placenta insufficiencies such as in gestational diabetes mellitus (GDM), intrauterine growth restriction (IUGR), and preeclampsia are related to vascular dysfunction of the placenta [5].
Plasma asymmetric dimethylarginine (ADMA) is recognized as a biomarker of endothelial disorders, cardiovascular disorders, hypercholesterolemia, and stroke [6–8]. Maternal plasma ADMA levels are reduced in a normal pregnancy but increase as the gestational age increases [9,10] and is increased in compromised pregnancies such as those with preeclampsia [9,10].
The placenta plays a central role in fetal programming by directly regulating blood flow, transporter activity, fetal nutrient supply and fetal growth [11,12]. Epigenetic mechanisms play a critical role during placental maturation and development [13]. NO is a known epigenetic molecule that plays a role in epigenetic fetal programming [14,15].
It has been suggested that therapeutic agents, which target placental blood flow and vascular development, ameliorate fetal growth restriction [2,5]. Thus, manipulation of the ADMA-NO pathways may have a therapeutic potential to rescue placental blood flow and improve long-term outcomes in patients with placental insufficiencies.
2. Metabolisms of NO and ADMA
NO is synthesized in every cell type by nitric oxide synthases (NOSs), namely nitric oxide synthase 1 (NOS1 or nNOS), nitric oxide synthase 2 (NOS2 or iNOS), and nitric oxide synthase 3 (NOS3 or eNOS). NOS1 and NOS3 are considered constitutive NOS. NO is formed from its precursor, l-arginine, by a family of NOSs in the presence of oxygen and the cofactor tetrahydrobiopterin, with the production of l-citrulline. NO produced in endothelial cells relaxes vascular smooth muscle via the cyclic guanosine monophosphate-dependent pathway and also has a potent vasodilatation effect. The exchange of amino acids between the intracellular environment and the plasma is facilitated by specific transporter systems. NOS activities depend on the ability of endothelial cells to take up their specific substrate l-arginine via a variety of membrane transport systems. In human endothelial cells, these membrane transport systems include y+, y+ L, b0,+, and B0,+[7,16].
ADMA is formed when arginine residues in proteins are methylated by the action of types I and II protein arginine methyltransferases (PRMT). ADMA is a competitive inhibitor of l-arginine for all 3 NOS isoforms. Approximately 20% of ADMA is excreted by the kidneys, whereas the other 80% is metabolized by two dimethylarginine dimethylaminohydrolases (DDAH-1 and -2) to l-citrulline and dimethylamine. System y+ transporters mediate ADMA uptake by neighboring cells and conform a family of proteins known as cationic amino acid transporters (CATs) that includes CAT-1, CAT-2A, CAT-2B, CAT-3, and CAT-4 isoforms [7]. The CATs are the main determinant of the ADMA distribution between the cytosol and the extracellular fluid. Intracellular ADMA can inhibit CAT, block NOS activity, and limit the cellular uptake of l-arginine, thereby contributing to oxidative stress and further NO biogenesis inhibition. The NO and ADMA synthesis and metabolism pathways are presented in Figure 1.
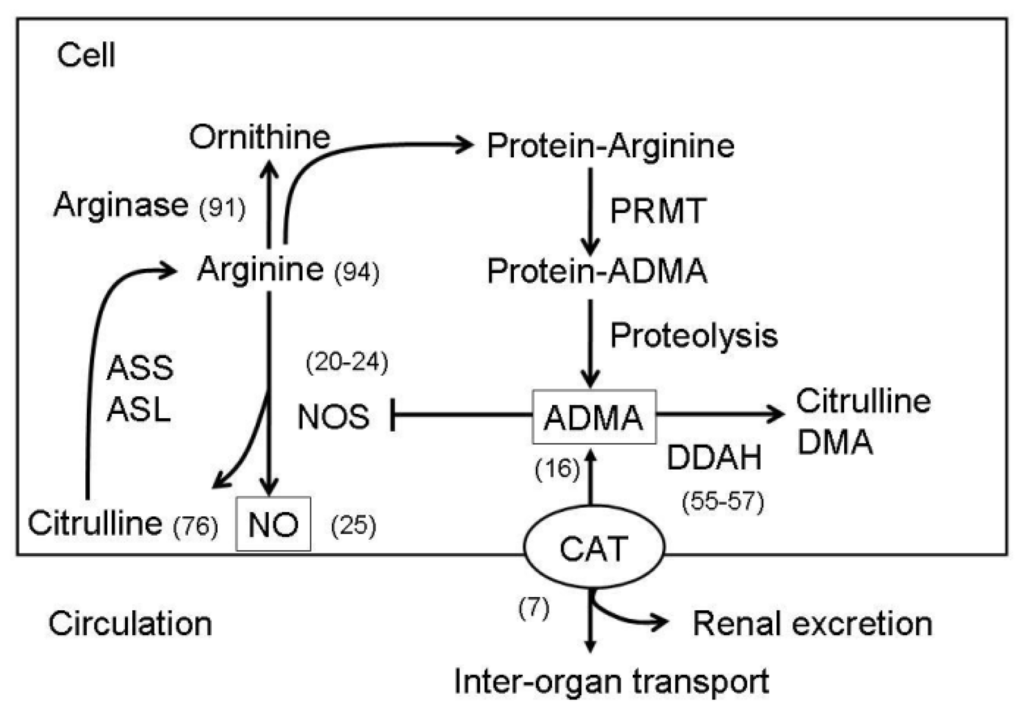
Figure 1.
A schema showing the synthesis and metabolisms of ADMA-NO pathways. Protein-incorporated ADMA is formed by PRMTs. Free ADMA is then released after protein degradation. Free ADMA can be transported by CAT to move in or out of the cells. ADMA can be transported to major organs for ADMA degradation or excreted by the kidneys. ADMA is metabolized by DDAH to generate citrulline and DMA. Free ADMA can compete with arginine for NOS to generate NO and citrulline. Citrulline can be converted to arginine by ASS and ASL. In addition to NOS, arginine is the substrate for other metabolic pathways, such as arginase and protein synthesis. ASS, argininosuccinate synthetase; ASL, argininosuccinate lyase; CAT, cationic amino acid transporter; DMA, dimethylamine; NOS, nitric oxide synthese; ADMA, assymetric dimethylarginine; PRMT, protein arginine methyltransferase; DDAH, dimethylarginine dimethylaminohydrolase. Numbers in parentheses indicate the representative references.
In preterm infants, plasma ADMA levels remain relatively constant during the first week of life but increase from 0.66 μM to 0.95 μM by the fourth week [17]. During childhood, ADMA levels slowly decrease from birth until around 25 years of age, with a mean decrease of 15 nM per year [18]. In adults, plasma ADMA concentrations tend to increase with age, with the mean concentration in the range of 0.4–0.6 μM [19].
3. Roles of NO and ADMA in Normal Pregnancies
During pregnancy, the mother and fetus have a complex anatomical and functional interaction. The placenta acts as the interface between the mother and the fetus and maintains fetal homoeostasis. The placenta originates from trophoblasts that differentiate into cytotrophoblasts and syncytiotrophoblasts, which form the primary villi. Villous cytotrophoblasts are specialized epithelial cells that anchor the fetus to the mother and establish blood flow to the placenta. The syncytiotrophoblasts, which comprise the transporting epithelium of the placenta, contain many transport proteins in the plasma membranes of both the mother and the fetus. These transport systems deliver nutrients to the fetus and provide a substrate for syncytiotrophoblast metabolism. Placental nutrient transport is dependent on vascular development; therefore, NO plays a critical role.
As pregnancy continues, NOS3 expression increases, primarily in the syncytiotrophoblasts [20]. In addition, NOS3 activity is present in the intermediate trophoblastic cells in the cell columns of the anchoring villi and in the trophoblastic cells at the implantation site [21]. On the other hand, NOS2 activity increases throughout pregnancy that peaks around mid-gestation [22]. Suzuki et al. proposed that NO plays a supportive role in promoting embryo survival [23]. Purcell et al. suggested that after implantation, both NOS3 and NOS2 might play a role in tissue remodeling, immunosuppression, and vasoregulation [24].
Plasma nitrates/nitrites have also been reported to be elevated in human pregnancy [25]. A study involving sheep showed that the pregnancy-associated increase in endothelium-dependent relaxation of the uterine arteries is predominantly regulated by the upregulation of NO release that results in decreased intracellular free calcium in the smooth muscle [26]. The human feto-placental vasculature lacks autonomic innervation, and therefore, NO confers autocrine and/or paracrine effects, playing an important role in the regulation of feto-placental blood flow. NO is the main vasodilator in the placenta and is involved feto-placental vascular reactivity regulation, placental bed vascular resistance, trophoblast invasion and apoptosis, and platelet adhesion and aggregation in the intervillous space [1].
Adequate morphology and function of the vascular tree are dependent on environmental cues such as blood flow, nutrient supply, and oxygen levels. Placenta vasculogenesis is the process of vessel formation from mesenchymal-derived hemangioblasts that differentiate into endothelial cells [27,28]. Differentiation of embryonic stem cells toward the endothelial lineage is an important step in vasculogenesis, and vascular endothelial growth factor (VEGF) is an important factor implicated in this process. In general, the initiation of vasculogenesis requires VEGF expression, which are effects mediated by NO [29]. The yolk sac plays a crucial role in embryo and placental vascular development. A study of mice yolk sacs showed that vasculogenesis is initiated by NO [30]. Furthermore, the spatio-temporal expression patterns of NOS2 and NOS3 were related to vasculogenesis in the yolk sac [30].
Angiogenesis, the formation of functional capillaries from pre-existing vasculature, helps drive villous development and differentiation [27]. In this process, single vessels are formed because endothelial precursor cells differentiate into endothelial cells. NO is a critical downstream mediator of potent angiogenic agents such as VEGF, basic fibroblast growth factor (FGF), and angiopoietin-1 [31]. The role of NO is more established in angiogenesis than in vasculogenesis [32].
The critical role of NO in angiogenesis has been shown in NOS3 knockout mice [33]. Likewise, NOS inhibition is accompanied by angiogenesis deficiencies as is exemplified by deficient vascular sprouting [34]. Interestingly, NO may also act upstream of angiogenic growth factors [35] because the effect of NO on VEGF production may be mediated through hypoxia-inducible factor-1.
Maternal plasma ADMA levels are reduced in a normal pregnancy but increase as the gestational age increases [9,10]. Holden et al. reported that the mean plasma ADMA concentratio in 20 nonpregnant women was 0.82 μmol/L [10]. The mean plasma ADMA concentration was 0.40 μmmol/L in 33 first-trimester pregnancies, 0.52 μmol/L in 50 second-trimester pregnancies, and 0.56 μmmol/L in 44 third- trimester pregnancies [10].
In early pregnancy, the reduction in ADMA and concomitant increase in NO may lead to hemodynamic adaptation, a higher need of organ perfusion in pregnancy, and uterine relaxation to allow for undisturbed intrauterine growth of the fetus. In late pregnancy, physiologically increased ADMA levels thus help prepare the uterine muscle fibers for the higher contractile activity that is necessary for successful delivery by antagonizing NO-induced uterine relaxation. This is reflected by the higher ADMA level after cesarean birth compared with vaginal birth [36]. After birth, ADMA levels fall, which may contribute to decreased NO production and bioavailability in neonatal vascular beds [36].
4. Roles of NO and ADMA in Compromised Pregnancies
NO regulates feto-placental vascular reactivity and placental blood flow. It has been suggested that placental angiogenesis abnormalities and angiogenic factor expression are associated with a variety of compromised pregnancies such as IUGR, GDM, and preeclampsia [37,38].
In an ewe model, circulating NO and its metabolites were elevated in pregnancies with multiple fetuses compared with singletons [39]. NOS3 can interact with the VEGF system and can enhance its production and function in placental tissues during early pregnancy [40]. In this context, placental expression of NOS3 is reduced in compromised pregnancies, including those of humans [41].
A normal pregnancy is accompanied by increasing metabolic activity of the placental mitochondria throughout gestation. The placenta thus generates reactive oxygen species (ROS) and reactive nitrogen species (RNS) in normal pregnancies and increased levels of the same in compromised pregnancies. In parallel, altered expressions of amino acid transporters in the placenta are also found in compromised pregnancies [1]. In this regard, Khullar et al. showed that NO−- and superoxide (O2 −)-derived free radicals impair placental syncytiotrophoblast amino acid uptake and increase Na+ permeability in vitro[42].
Due to their abundance in the placenta, superoxide and NO can interact to generate peroxynitrite (ONOO−), a potent pro-oxidant. Peroxynitrite formation will alter the placental NO level, which in turn will affect physiological function. The presence of nitrative stress was associated with diminished vascular reactivity of the fetal placenta, a situation that could be recapitulated in vitro by peroxynitrite treatment [43]. Roberts et al. found many nitrated proteins in the placenta [44]. Nitration of the placental proteins is found in normal pregnancies but at increased levels in pathologic pregnancies [45]. For instance, p38 mitogen-activated protein kinase could be nitrated with peroxynitrite at the tyrosine residues and could be associated with the loss of catalytic activity [45].
Recent studies have suggested that the plasma concentrations of ADMA may serve as a risk biomarker for endothelial dysfunction, hypercholesterolemia, preeclampsia, stroke, and cardiovascular disease [6]. Declining ADMA levels is observed in a normal pregnancy with increasing gestational age [9]. In compromised pregnancy such as preeclampsia, ADMA levels rise to levels greater than that seen in normal pregnancy [10]. Holden et al. showed that the mean plasma ADMA concentration of the 18 third-trimester preeclamptic subjects was 1.17 μmol/L, significantly higher than both the normotensive gestational age-matched and the nonpregnant control groups [10]. The association of abnormal uterine artery Doppler waveforms with high ADMA concentrations [46] in pregnancy supports the role of endogenous NOS inhibitors adversely affecting maternal vasodilation and blood pressure. Consistent with these findings, increased vasoconstriction of human stem villous feto-placental arteries [47] and increased ovine umbilical vascular resistance [48] are caused by an NOS inhibitor, NG-monomethyl-l-arginine. Suzuki et al. showed that NG-nitro-l-argininemethyl ester, an NOS inhibitor, caused apoptosis in the decidua, suggesting that NO in the decidua is essential to cell survival and the maintenance of uterine formation [49].
4.1. Preeclampsia
Preeclampsia is a major cause of fetal growth restriction, premature delivery, and maternal death worldwide. The underlying pathology of preeclampsia is presumably because of a relatively hypoxic or ischemic placenta. Preeclampsia is a disorder unique to pregnancy characterized by maternal hypertension, proteinuria, and edema.
In preeclampsia, the cytotrophoblast fails to adopt a vascular adhesion phenotype, resulting in compromised blood flow to the maternal-fetal interface. Through its unique angiogenic/vasculogenic properties, NO may be critical for cytotrophoblast endovascular invasion, an essential feature of normal placentation. Current evidence supports altered NO production in the feto-placental unit in preeclampsia [50]. In this regard, a reduced NO formation has been hypothesized to account for the abnormal placental perfusion in preeclampsia [51].
Increased ROS/RNS level contributes to endothelial dysfunction in the placenta and in the maternal vasculature and has been implicated as the pathophysiological feature of preeclampsia. An arginine deficiency documented in the preeclampsia placenta caused decreased NO and increased superoxide formation, leading to NO degradation and excess peroxynitrite formation [52]. Similarly, peroxynitrite formation is capable of attenuating vascular responses in the preeclampsia placenta [43]. In parallel, Myatt et al. showed elevated NOS3 expression in the placental vascular endothelium of preeclamptic women that was proposed to be a compensatory mechanism [53].
ADMA levels have been shown to increase even before the development of preeclampsia, suggesting that increased ADMA may be linked with the occurrence of preeclampsia in high-risk women [46]. In the study of Savvidou et al., women who developed preeclampsia at a later stage had both bilateral uterine artery notches and impaired brachial artery flow-mediated dilation at 23–25 weeks’ gestation [46]. Since brachial artery flow-mediated dilation depends mainly on NO release, the increased ADMA levels and resultant endothelial dysfunction are thought to be attributable to the occurrence of preeclampsia. Placenta expresses high levels of DDAH-2 [54]. Anderssohn et al. showed decreased DDAH activity in the placenta from preeclamptic patients [55]. However, the association between DDAH polymorphisms and preeclampsia susceptibility remains inconclusive [56,57]. Whether impaired DDAH activity results in elevated ADMA levels that impair NO release and contributes to placental vascular dysfunction in preeclampsia awaits further elucidation.
4.2. Gestational Diabetes Mellitus
GDM is a syndrome characterized by glucose intolerance leading to maternal hyperglycemia, endothelial dysfunction, and abnormal regulation of vascular tone [58]. Placentas from GDM pregnancies are larger than normal [59] and show decreased formation of terminal villi and increased numbers of intermediate villi compared to those from normal pregnancies [60]. These vascular changes are likely to affect placental vascular resistance and vascular volume, leading to metabolic changes at the feto-placental microvascular and macrovascular endothelium [61].
Increased NO synthesis has also been reported in human placental veins and arteries from pregnancies with GDM [62]. GDM is associated with endothelial dysfunction characterized by an altered endothelial l-arginine/NO signaling pathway. In primary cultures of human umbilical cord endothelial cells (HUVECs) isolated from pregnancies with GDM, synthesis of NO [63,64] and l-arginine transport [64] and its intracellular concentration [61] are increased. Thus, altered placental vascular activity is characteristic of GDM and may be a consequence of a functional dissociation between NO synthesis and l-arginine uptake and/or bioavailability to the vascular endothelium and smooth muscle in the human placental circulation.
In addition, increased growth and vascularization in the GDM rat placenta are associated with higher levels of matrix metalloproteinase-2 (MMP-2) and MMP-9 [65], the activities of which are positively regulated by NO [66].
Akturk et al. investigated ADMA concentration in women with GDM and normal glucose tolerance during late pregnancy [67]. They found that ADMA concentration was elevated in women with GDM during late pregnancy and was positively correlated with the glucose levels in glucose challenge test [67].
4.3. Prenatal Malnutrition
Nutrient transfer across the placenta is essential for fetal growth and development. Prenatal malnutrition has important consequences in fetal growth and intrauterine programming. The placenta may act as a nutrient sensor, modifying nutrient and hormone availability to feto-placental tissues in relation to environmental challenges.
Placental NOS activities are 40%–45% lower in protein-deficient pigs than in protein-adequate pigs [68]. Maternal undernutrition in sheep decreases concentrations of arginine, citrulline, and polyamines in the maternal plasma, fetal plasma, and allantoic fluid [69]. In line with these findings, maternal undernutrition impairs NO-dependent vasodilation and increases arterial blood pressure in the ovine fetus [70].
The expression of angiogenic growth factors such as VEGF and basic FGF-2 as well as vascularity are altered in prenatally malnourished ewes [71]. Both VEGF [72] and FGF-2 [73] have been implicated in stimulating NO production in endothelium. On the other hand, NO can regulate both VEGF and FGF-2 expressions [31].
The labyrinthine layer, the site of feto-maternal interaction in the mouse placenta, contains both fetal and maternal blood vessels circulating independently of each other. Rutland et al. reported a reduction in labyrinthine blood vessel length and decreased expression of vascular endothelial adhesion molecules in the murine placenta in response to gestational protein malnutrition, suggesting that maternal nutrition alterations change placental vascular function [74].
We found increased plasma ADMA levels in the adult offspring from a 50% maternal caloric restriction rat model [75]. Since caloric restriction causes long-term somatic effects, we propose that increased plasma ADMA plays an important role in fetal programming.
4.4. Prenatal Glucocorticoid and Stress Exposure
Prenatal glucocorticoid therapy is used to prophylactically impede morbid symptoms associated with preterm delivery, such as respiratory distress syndrome and intraventricular hemorrhage. A recent medical practice survey showed that the proportion of women receiving antenatal corticosteroids had increased consistently over a seven-year period for those deliveries at 24–35 weeks and for those deliveries after 34 weeks [76]. It is well known that prenatal glucocorticoid and stress exposure lead to programming of hypothalamic-pituitary-adrenal function and behavior and have long-term effects on the offspring [77,78]. The effects of prenatal stress on fetal outcome are mediated in part by elevated fetal glucocorticoid exposure.
Altered placental glucocorticoid metabolism may influence placental efficiency by changing placental morphology, hormone synthesis, and transport physiology. VEGF is an important factor in the vasculogenesis process. Hewitt et al. first showed that prenatal glucocorticoid exposure induced fetal and placental growth restrictions by inhibiting placental VEGF expression and reducing placental vascularization [79]. VEGF-induced angiogenesis requires NO formation [80] derived from NOS3 activity [81]. In endothelial cells, VEGF induces NO synthesis via NOS3 through the activation of VEGFR-1 [82] and VEGFR-2 [83].
5. Placental Insufficiency and Developmental Programming
5.1. Role of the Placental Nitrergic System in Epigenetic Fetal Programming
Epigenetic modifications refer to stable and heritable gene expression changes that are not mediated by the alterations of the DNA sequence. Epigenetic mechanisms play a critical role during placental maturation and development [13]. Growing evidence indicates that NO exerts control over epigenetic mechanisms, including the function of histone deacetylase (HDAC) [14,15]. NO exerts its regulatory function on chromatin and gene expression via two chemical reactions: S-nitrosylation and tyrosine nitration [14]. Endothelial specialization and blood vessel formation are controlled by epigenetic mechanisms in the first stages of vascular development [84]. A study using HUVECs showed that the HDAC inhibitor, trichostatin A, reduced angiogenesis partially via regulation of NOS3-derived NO bioavalability [85]. Using a mouse cell line, Zeng et al. detected HDAC 3 in blood vessels during embryogenesis [86]. Together, NO might play a role in epigenetic fetal programming.
ADMA-related enzymes are also involved in compromised pregnancies and programming. Free ADMA levels are controlled by two counterbalancing pathways: PRMT I-related and DDAH-related. Several PRMTs have been associated with epigenetic regulation in which PRMTs act as histone methyltransferases or secondary co-regulators of transcription or facilitate mRNA splicing and stability [87]. At the organism level, several PRMTs seem to be important for development and may play an important role in organogenesis. Likewise, PRMT 5 was known to be a maternal detrimental factor of embyogenesis in fish [88]. However, the relationship between PRMTs and fetal programming remains poorly understood.
We and others have found that ADMA is involved in the fetal development of chronic kidney diseases and hypertension [7,89,90]. ADMA is implicated in the hypertension seen in spontaneous hypertensive rat [89]. Similarly, high prenatal salt intake in dams cause persistent hypertension in male offspring [90]. The observations that salt intake in dams cause adult male offspring increased serum ADMA concentrations and hypertension are in agreement with the fetal programming hypothesis [90]. In the study of Savvidou et al.[46], elevated plasma ADMA levels were seen in women who later on developed preeclampsia, suggesting the role of ADMA in fetal programming. However, the exact role of ADMA in fetal programming remains unclear and needs further studies.
5.2. Manipulations of the ADMA-NO Pathway to Prevent Compromised Pregnancies and Fetal Programming
Treatment of compromised pregnancy and fetal programming is currently limited to the management of complications. As stated above, the high morbidity and mortality of compromised pregnancy and the resulting fetal programming is likely due to an impaired ADMA-NO pathway. Thus, to preserve NO bioavailability, several therapeutic strategies have been used to either increase NO synthesis or decrease its breakdown. We recently reviewed that treatment with metformin, oral contraceptives, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, fenofibrate, folic acid, α-lipoic acid, melatonin, vitamin E, and N-acetylcysteine can lower plasma ADMA levels [16,91,92]; however, a specific ADMA-lowering agent remains unavailable.
l-arginine administration has been tested in many human diseases and experimental animals as a way to improve NO bioavailability. Some favorable effects of supplemental l-arginine on blood pressure and pregnancy outcomes were reported in patients with preeclampsia [93]. However, the data from human trials remain inconclusive. Since oral l-arginine treatment is hampered by pre-systemic elimination (e.g., hepatic and intestinal arginase) and l-citrulline can be converted to l-arginine, oral l-citrulline supplementation is a potential way to raise plasma l-arginine concentration and augment NO [94]. This thought is supported by our recent study that showed that maternal supplementation with l-citrulline increased renal NO bioavailability in the offspring of calorically restricted mothers [75]. In addition, l-arginine availability can be impaired because of consumption via other metabolic pathways (i.e., arginase), reduced de novo synthesis, and impaired transport. For example, arginase inhibitor may be a potentially therapeutic approach to restoring l-arginine-NO to protect compromised pregnancies [95].
The impact of oxidative stress in pregnancy-related disorders and the use of antioxidants in perinatal medicine have been well reviewed elsewhere [96]. Importantly, the use of oxidative stress in interpreting developmental programming is an emerging hypothesis [97]. Even though some antioxidants have detrimental effects in experimental preeclampsia, antioxidant therapy to counter oxidative stress in human trials has failed to prevent preeclampsia [98]. It is clear that identification of the source of ROS and control of its production is a better strategy than non-selective use of antioxidants to prevent compromised pregnancy and fetal programming.
6. Conclusions
Placental nutrient transport is dependent on vascular development, which determines blood flow to the placenta. NO regulates placental blood flow. ADMA has been associated with uterine artery flow disturbances. Substantial experimental evidence has reliably supported the roles of NO and ADMA in normal pregnancy and placenta insufficiency as well as the link between placenta insufficiency and epigenetic fetal programming. With expanded knowledge on the mechanisms of the impaired ADMA-NO pathway, additional targets for therapeutic intervention will be identified. A multifaceted approach to restoring NO bioavailability will be required to prevent compromised pregnancies and fetal programming.
Acknowledgements
The study was supported in part by grants from Chang Gung Memorial Hospital, Kaohsiung, Taiwan, CMRPG8B0111 to Li-Tung Huang, and grant CMRPG8B0171 and grant NSC 98-2314-B-182A-006-MY3 from National Science Council, Taiwan to You-Lin Tain. We thank the center for Translation Research in Biomedical Sciences and center for Gene Proteomics Research, Kaohsiung Chang Gung Memorial hospital for supports.
- Conflict of InterestThe authors declare no conflict of interest.
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| CAT | Cationic amino acid transporters |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| FGF | Fibroblast growth factor |
| GDM | Gestational diabetes mellitus |
| HDAC | Histone deacetylase |
| HUVEC | Human umbilical cord endothelial cells |
| IUGR | Intrauterine growth restriction |
| MMP | Matrix metalloproteinases |
| NO | Nitric oxide |
| NOS1 or nNOS | Nitric oxide synthase 1 |
| NOS2 or iNOS | Nitric oxide synthase 2 |
| NOS3 or eNOS | Nitric oxide synthase 3 |
| PRMT | Protein arginine methyltransferases |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| VEGF | Vascular endothelial growth factor |
References
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol 2006, 572, 25–30. [Google Scholar]
- Reynolds, L.P.; Borowicz, P.P.; Caton, J.S.; Vonnahme, K.A.; Luther, J.S.; Buchanan, D.S.; Hafez, S.A.; Grazul-Bilska, A.T.; Redmer, D.A. Uteroplacental vascular development and placental function: an update. Int. J. Dev. Biol 2010, 54, 355–366. [Google Scholar]
- Krause, B.J.; Hanson, M.A.; Casanello, P. Role of nitric oxide in placental vascular development and function. Placenta 2011, 32, 797–805. [Google Scholar]
- Godfrey, M. The role of the placenta in fetal programming—A review. Placenta 2002, 23, S20–S27. [Google Scholar]
- Reynolds, L.P.; Caton, J.S.; Redmer, D.A.; Grazul-Bilska, A.T.; Vonnahme, K.A.; Borowicz, P.P.; Luther, J.S.; Wallace, J.M.; Wu, G.; Spencer, T.E. Evidence for altered placental blood flow and vascularity in compromised pregnancies. J. Physiol 2006, 572, 51–58. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1023–1030. [Google Scholar]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res 2009, 60, 448–460. [Google Scholar]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev 2010, 6, 82–90. [Google Scholar]
- Fickling, S.A.; Williams, D.; Vallance, P.; Nussey, S.S.; Whitley, G.S. Plasma of endogenous inhibitor of nitric oxide synthesis in normal pregnancy and pre-eclampsia. Lancet 1993, 342, 242–243. [Google Scholar]
- Holden, D.P.; Fickling, S.A.; Whitley, G.S.; Nussey, S.S. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am. J. Obstet. Gynecol 1998, 178, 551–556. [Google Scholar]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. (Lond.) 2007, 113, 1–13. [Google Scholar]
- Reynolds, L.P.; Caton, J.S. Role of the pre- and post-natal environment in developmental programming of health and productivity. Mol. Cell Endocrinol 2012, 354, 54–59. [Google Scholar]
- Rugg-Gunn, P.J. Epigenetic features of the mouse trophoblast. Reprod. Biomed. Online 2012, 25, 21–30. [Google Scholar]
- Illi, B.; Colussi, C.; Grasselli, A.; Farsetti, A.; Capogrossi, M.C.; Gaetano, C. NO sparks off chromatin: Tales of a multifaceted epigenetic regulator. Pharmacol. Ther 2009, 123, 344–352. [Google Scholar]
- Nott, A.; Riccio, A. Nitric oxide-mediated epigenetic mechanisms in developing neurons. Cell Cycle 2009, 8, 725–730. [Google Scholar]
- Tain, Y.L.; Huang, L.T. Asymmetric dimethylarginine: Clinical applications in pediatric medicine. J. Formos. Med. Assoc 2011, 110, 70–77. [Google Scholar]
- Vida, G.; Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Boger, S.M. Plasma asymmetric dimethylarginine concentration during the perinatal period. Neonatology 2007, 92, 8–13. [Google Scholar]
- Lucke, T.; Kanzelmeyer, N.; Kemper, M.J.; Tsikas, D.; Das, A.M. Developmental changes in the l-arginine/nitric oxide pathway from infancy to adulthood: Plasma asymmetric dimethylarginine levels decrease with age. Clin. Chem. Lab. Med 2007, 45, 1525–1530. [Google Scholar]
- Horowitz, J.D.; Heresztyn, T. An overview of plasma concentrations of asymmetric dimethylarginine (ADMA) in health and disease and in clinical studies: Methodological considerations. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2007, 851, 42–50. [Google Scholar]
- Rossmanith, W.G.; Hoffmeister, U.; Wolfahrt, S.; Kleine, B.; McLean, M.; Jacobs, R.A.; Grossman, A.B. Expression and functional analysis of endothelial nitric oxide synthase (eNOS) in human placenta. Mol. Hum. Reprod 1999, 5, 487–494. [Google Scholar]
- Ariel, I.; Hochberg, A.; Shochina, M. Endothelial nitric oxide synthase immunoreactivity in early gestation and in trophoblastic disease. J. Clin. Pathol 1998, 51, 427–431. [Google Scholar]
- Suzuki, T.; Ikeda, Y.; Yoshikawa, H.; Tanaka, K.; Morita, H.; Yamamoto, M.; Takizawa, T. Gestational changes in production of NO and expression of NOS mRNA isoforms in the rat placenta. J. Vet. Med. Sci 2009, 71, 495–498. [Google Scholar]
- Suzuki, T.; Mori, C.; Yoshikawa, H.; Miyazaki, Y.; Kansaku, N.; Tanaka, K.; Morita, H.; Takizawa, T. Changes in nitric oxide production levels and expression of nitric oxide synthase isoforms in the rat uterus during pregnancy. Biosci. Biotechnol. Biochem 2009, 73, 2163–2166. [Google Scholar]
- Purcell, T.L.; Given, R.; Chwalisz, K.; Garfield, R.E. Nitric oxide synthase distribution during implantation in the mouse. Mol. Hum. Reprod 1999, 5, 467–475. [Google Scholar]
- Seligman, S.P.; Buyon, J.P.; Clancy, R.M.; Young, B.K.; Abramson, S.B. The role of nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol 1994, 171, 944–948. [Google Scholar]
- Xiao, D.; Pearce, W.; Zhang, L. Pregnancy enhances endothelium-dependent relaxation of ovine uterine artery: Role of NO and intracellular Ca2+. Am. J. Physiol. Heart Circ. Physiol 2001, 281, H183–H190. [Google Scholar]
- Kaufmann, P.; Mayhew, T.M.; Charnock-Jones, D.S. Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 2004, 25, 114–126. [Google Scholar]
- Demir, R.; Kayisli, U.A.; Cayli, S.; Huppertz, B. Sequential steps during vasculogenesis and angiogenesis in the very early human placenta. Placenta 2006, 27, 535–539. [Google Scholar]
- Shizukuda, Y.; Tang, S.; Yokota, R.; Ware, J.A. Vascular endothelial growth factor-induced endothelial cell migration and proliferation depend on a nitric oxide-mediated decrease in protein kinase C activity. Circ. Res 1999, 85, 247–256. [Google Scholar]
- Nath, A.K.; Enciso, J.; Kuniyasu, M.; Hao, X.Y.; Madri, J.A.; Pinter, E. Nitric oxide modulates murine yolk sac vasculogenesis and rescues glucose induced vasculopathy. Development 2004, 131, 2485–2496. [Google Scholar]
- Frank, S.; Stallmeyer, B.; Kampfer, H.; Schaffner, C.; Pfeilschifter, J. Differential regulation of vascular endothelial growth factor and its receptor fms-like-tyrosine kinase is mediated by nitric oxide in rat renal mesangial cells. Biochem. J 1999, 338, 367–374. [Google Scholar]
- Han, R.N.; Stewart, D.J. Defective lung vascular development in endothelial nitric oxide synthasedeficient mice. Trends Cardiovasc. Med 2006, 16, 29–34. [Google Scholar]
- Zhao, X.; Lu, X.; Feng, Q. Deficiency in endothelial nitric oxide synthase impairs myocardial angiogenesis. Am. J. Physiol. Heart Circ. Physiol 2002, 283, H2371–H2378. [Google Scholar]
- Kon, K.; Fujii, S.; Kosaka, H.; Fujiwara, T. Nitric oxide synthase inhibition by N(G)-nitro-l-arginine methyl ester retards vascular sprouting in angiogenesis. Microvasc. Res 2003, 65, 2–8. [Google Scholar]
- Dulak, A.; Jozkowicz, J. Regulation of vascular endothelial growth factor synthesis by nitric oxide: Facts and controversies. Antioxid. Redox. Signal 2003, 5, 123–132. [Google Scholar]
- Vida, G.; Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Böger, S.M. Birth by cesarean section is associated with elevated neonatal plasma levels of dimethylarginines. Pediatr. Int 2012, 54, 476–479. [Google Scholar]
- Reynolds, L.P.; Borowicz, P.P.; Vonnahme, K.A.; Johnson, M.L.; Grazul-Bilska, A.T.; Wallace, J.M.; Caton, J.S.; Redmer, D.A. Animal models of placental angiogenesis. Placenta 2005, 26, 689–708. [Google Scholar]
- Myatt, L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta 2010, 24, S66–S69. [Google Scholar]
- Vonnahme, K.A.; Wilson, M.E.; Li, Y.; Rupnow, H.L.; Phernetton, T.M.; Ford, S.P.; Magness, R.R. Circulating levels of nitric oxide and vascular endothelial growth factor throughout ovine pregnancy. J. Physiol 2005, 565, 101–109. [Google Scholar]
- Reynolds, L.P.; Redmer, D.A. Angiogenesis in the placenta. Biol. Reprod 2001, 64, 1033–1040. [Google Scholar]
- Bird, I.M.; Zhang, L.; Magness, R.R. Possible mechanisms underlying pregnancy-induced changes in uterine artery endothelial function. Am. J. Physiol. Regul. Integr. Comp. Physiol 2003, 284, R245–R258. [Google Scholar]
- Khullar, S.; Greenwood, S.L.; McCord, N.; Glazier, J.D.; Ayuk, P.T. Nitric oxide and superoxide impair human placental amino acid uptake and increase Na+ permeability: Implications for fetal growth. Free Radic. Biol. Med 2004, 36, 271–277. [Google Scholar]
- Kossenjans, W.; Eis, A.; Sahay, R.; Brockman, D.; Myatt, L. Role of peroxynitrite in altered fetal-placental vascular reactivity in diabetes or preeclampsia. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1311–H1139. [Google Scholar]
- Roberts, V.H.; Webster, R.P.; Brockman, D.E.; Pitzer, B.A.; Myatt, L. Post-translational modifications of the P2X(4) purinergic receptor subtype in the human placenta are altered in preeclampsia. Placenta 2007, 28, 270–277. [Google Scholar]
- Webster, R.P.; Macha, S.; Brockman, D.; Myatt, L. Peroxynitrite treatment in vitro disables catalytic activity of recombinant p38 MAPK. Proteomics 2006, 6, 4838–4844. [Google Scholar]
- Savvidou, M.D.; Hingorani, A.D.; Tsikas, D.; Frolich, J.C.; Vallance, P.; Nicolaides, K.H. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet 2003, 361, 1511–1517. [Google Scholar]
- McCarthy, A.L.; Woolfson, R.G.; Evans, B.J.; Davies, D.R.; Raju, S.K.; Poston, L. Functional characteristics of small placental arteries. Am. J. Obstet. Gynecol 1994, 170, 945–951. [Google Scholar]
- Chang, J.K.; Roman, C.; Heymann, M.A. Effect of endothelium-derived relaxing factor inhibition on the umbilical-placental circulation in fetal lambs in utero. Am. J. Obstet. Gynecol 1992, 166, 727–734. [Google Scholar]
- Suzuki, T.; Nagamatsu, C.; Kushima, T.; Miyakoshi, R.; Tanaka, K.; Morita, H.; Sakaue, M.; Takizawa, T. Apoptosis caused by an inhibitor of NO production in the decidua of rat from mid-gestation. Exp. Biol. Med. (Maywood) 2010, 235, 455–462. [Google Scholar]
- Lowe, D.T. Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric Oxide 2000, 4, 441–458. [Google Scholar]
- Baylis, C.; Beinder, E.; Suto, T.; August, P. Recent insights into the roles of nitric oxide and renin-angiotensin in the pathophysiology of preeclamptic pregnancy. Semin. Nephrol 1998, 18, 208–230. [Google Scholar]
- Noris, M.; Todeschini, M.; Cassis, P.; Pasta, F.; Cappellini, A.; Bonazzola, S.; Macconi, D.; Maucci, R.; Porrati, F.; Benigni, A.; et al. l-arginine depletion in preeclampsia orients nitric oxide synthase toward oxidant species. Hypertension 2004, 43, 614–622. [Google Scholar]
- Myatt, L.; Eis, A.L.; Brockman, D.E.; Kossenjans, W.; Greer, I.A.; Lyall, F. Endothelial nitric oxide in placental villous tissue from normal, pre-eclamptic and intrauterine restricted pregnancies. Hum. Reprod 1997, 12, 714–718. [Google Scholar]
- Leiper, J.; MacAllister, R.; Whitley, G.; Santa Maria, J.; Chubb, A.; Charles, I.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology to microbial arginine deiminases. Biochem. J 1999, 343, 209–214. [Google Scholar]
- Anderssohn, M.; Maass, L.M.; Diemert, A.; Lüneburg, N.; Atzler, D.; Hecher, K.; Böger, R.H. Severely decreased activity of placental dimethylarginine dimethylaminohydrolase in pre-eclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol 2012, 161, 152–156. [Google Scholar]
- Akbar, F.; Heinonen, S.; Pirskanen, M.; Uimari, P.; Tuomainen, T.P.; Salonen, J.T. Haplotypic association of DDAH1 with susceptibility to pre-eclampsia. Mol. Hum. Reprod 2005, 11, 73–77. [Google Scholar]
- Kim, Y.J.; Park, B.H.; Park, H.; Jung, S.C.; Pang, M.G.; Ryu, H.M.; Lee, K.S.; Eom, S.M.; Park, H.Y. No association of the genetic polymorphisms of endothelial nitric oxide synthase, dimethylarginine dimethylaminohydrolase, and vascular endothelial growth factor with preeclampsia in Korean populations. Twin Res. Hum. Genet 2008, 11, 77–83. [Google Scholar]
- Sobrevia, L.; Mann, G.E. Dysfunction of the endothelial nitric oxide signalling pathway in diabetes and hyperglycaemia. Exp. Physiol 1997, 82, 423–452. [Google Scholar]
- Kucuk, M.; Doymaz, F. Placental weight and placental weight-to-birth weight ratio are increased in diet- and exercise-treated gestational diabetes mellitus subjects but not in subjects with one abnormal value on 100-g oral glucose tolerance test. J. Diabetes Complicat 2009, 23, 25–31. [Google Scholar]
- Daskalakis, G.; Marinopoulos, S.; Krielesi, V.; Papapanagiotou, A.; Papantoniou, N.; Mesogitis, S.; Antsaklis, A. Placental pathology in women with gestational diabetes. Acta. Obstet. Gynecol. Scand 2008, 87, 403–407. [Google Scholar]
- Sobrevia, L.; Abarzúa, F.; Nien, J.K.; Salomón, C.; Westermeier, F.; Puebla, C.; Cifuentes, F.; Guzmán-Gutiérrez, E.; Leiva, A.; Casanello, P. Review: Differential placental macrovascular and microvascular endothelial dysfunction in gestational diabetes. Placenta 2011, 25, S159–S164. [Google Scholar]
- Figueroa, R.; Martinez, E.; Fayngersh, R.P.; Tejani, N.; Mohazzab, H.K.M.; Wolin, M.S. Alterations in relaxation to lactate and H2O2 in human placental vessels from gestational diabetic pregnancies. Am. J. Physiol 2000, 278, H706–H713. [Google Scholar]
- Farías, M.; Puebla, C.; Westermeier, F.; Jo, M.J.; Pastor-Anglada, M.; Casanello, P.; Sobrevia, L. Nitric oxide reduces SLC29A1 promoter activity and adenosine transport involving transcription factor complex hCHOP-C/EBPα in human umbilical vein endothelial cells from gestational diabetes. Cardiovas. Res 2010, 86, 45–54. [Google Scholar]
- Vásquez, G.; Sanhueza, F.; Vásquez, R.; González, M.; San Martín, R.; Casanello, P.; Sobrevia, L. Role of adenosine transport in gestational diabetes-induced l-arginine transport and nitric oxide synthesis in human umbilical vein endothelium. J. Physiol 2004, 560, 111–122. [Google Scholar]
- Stojanovic, N.; Lewandowski, K.; Salata, I.; Bienkiewicz, M.; Tuck, S.; Prelevic, G.; Press, M. Serum levels of matrix metalloproteinases MMP-2 and MMP-9 and their inhibitors in women with glucose intolerance in pregnancy and normal controls. Gynecol. Endocrinol 2010, 26, 201–207. [Google Scholar]
- Novaro, V.; Colman-Lerner, A.; Ortega, F.V.; Jawerbaum, A.; Paz, D.; Lo Nostro, F.; Pustovrh, C.; Gimeno, M.F.; González, E. Regulation of metalloproteinases by nitric oxide in human trophoblast cells in culture. Reprod. Fertil. Dev 2001, 13, 411–420. [Google Scholar]
- Akturk, M.; Altinova, A.; Mert, I.; Dincel, A.; Sargin, A.; Buyukkagnici, U.; Arslan, M.; Danisman, N. Asymmetric dimethylarginine concentrations are elevated in women with gestational diabetes. Endocrine 2010, 38, 134–141. [Google Scholar]
- Wu, G.; Pond, W.G.; Flynn, S.P.; Ott, T.L.; Bazer, F.W. Maternal dietary protein deficiency decreases nitric oxide synthase and ornithine decarboxylase activities in placenta and endometrium of pigs during early gestation. J. Nutr 1998, 128, 2395–2402. [Google Scholar]
- Kwon, H.; Ford, S.P.; Bazer, F.W.; Spencer, T.E.; Nthanielsz, P.W.; Nijland, M.J.; Hess, B.W.; Wu, G. Maternal undernutrition reduces concentrations of amino acids and polyamines in ovine fetal plasma and fluids. Biol. Reprod 2004, 71, 901–908. [Google Scholar]
- Ozaki, T.; Hawkins, P.; Nishina, H.; Steyn, C.; Poston, L.; Hanson, M.A. Effects of undernutrition in early pregnancy on systemic small artery function in late-gestation fetal sheep. Am. J. Obstet. Gynecol 2000, 183, 1301–1307. [Google Scholar]
- Redmer, D.A.; Wallace, J.M.; Reynolds, L.P. Effect of nutrient intake during pregnancy on fetal and placental growth and vascular development. Domest. Anim. Endocrinol 2004, 27, 199–217. [Google Scholar]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am. J. Physiol 1998, 274, H1054–H1058. [Google Scholar]
- Babaei, S.; Teichert-Kuliszewska, K.; Monge, J.C.; Mohamed, F.; Bendeck, M.P.; Stewart, D.J. Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circ. Res 1998, 18, 1007–1015. [Google Scholar]
- Rutland, C.S.; Latunde-Dada, A.O.; Thorpe, A.; Plant, R.; Langley-Evans, S.; Leach, L. Effect of gestational nutrition on vascular integrity in the murine placenta. Placenta 2007, 28, 734–742. [Google Scholar]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal l-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar]
- Polyakov, A.; Cohen, S.; Baum, M.; Trickey, D.; Jolley, D.; Wallace, E.M. Patterns of antenatal corticosteroid prescribing 1998–2004. Aust. N. Z. J. Obstet. Gynaecol 2007, 47, 42–45. [Google Scholar]
- Huang, L.T. The link between perinatal glucocorticoids exposure and psychiatric disorders. Pediatr. Res 2011, 69, 19R–25R. [Google Scholar]
- Lui, C.C.; Wang, J.Y.; Tain, Y.L.; Chen, Y.C.; Chang, K.A.; Lai, M.C.; Huang, L.T. Prenatal stress in rat causes long-term spatial memory deficit and hippocampus MRI abnormality: Differential effects of postweaning enriched environment. Neurochem. Int 2011, 58, 434–441. [Google Scholar]
- Hewitt, D.P.; Mark, P.J.; Waddell, B.J. Glucocorticoids prevent the normal increase in placental vascular endothelial growth factor expression and placental vascularity during late pregnancy in the rat. Endocrinology 2006, 147, 5566–5574. [Google Scholar]
- Parenti, A.; Morbidelli, L.; Cui, X.L.; Douglas, J.G.; Hood, J.D.; Granger, H.J.; Ledda, F.; Ziche, M. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. J. Biol. Chem 1998, 273, 4220–4226. [Google Scholar]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar]
- Bussolati, B.; Dunk, C.; Grohman, M.; Kontos, C.D.; Mason, J.; Ahmed, A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am. J. Pathol 2001, 159, 993–1008. [Google Scholar]
- Kroll, J.; Waltenberger, J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR). Biochem. Biophys. Res. Commun 1998, 252, 743–746. [Google Scholar]
- Ribatti, D.; Nico, B.; Crivellato, E. Morphological and molecular aspects of physiological vascular morphogenesis. Angiogenesis 2009, 12, 101–111. [Google Scholar]
- Rössig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Förstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res 2002, 91, 837–844. [Google Scholar]
- Zeng, L.; Xiao, Q.; Margariti, A.; Zhang, Z.; Zampetaki, A.; Patel, S.; Capogrossi, M.C.; Hu, Y.; Xu, Q. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J. Cell Biol 2006, 174, 1059–1069. [Google Scholar]
- Kuhn, P.; Xu, W. Protein arginine methyltransferases: Nuclear receptor coregulators and beyond. Prog. Mol. Biol. Transl. Sci 2009, 87, 299–342. [Google Scholar]
- Chen, W.; Cao, M.; Yang, Y.; Nagahama, Y.; Zhao, H. Expression pattern of prmt5 in adult fish and embryos of medaka, Oryzias latipes. Fish Physiol. Biochem 2009, 35, 325–332. [Google Scholar]
- Tain, Y.L.; Huang, L.T.; Lin, I.C.; Lau, Y.T.; Lin, C.Y. Melatonin prevents hypertension and increased asymmetric dimethylarginine in young spontaneous hypertensive rats. J. Pineal Res 2010, 49, 390–398. [Google Scholar]
- Piecha, G.; Koleganova, N.; Ritz, E.; Müller, A.; Fedorova, O.V.; Bagrov, A.Y.; Lutz, D.; Schirmacher, P.; Gross-Weissmann, M.L. High salt intake causes adverse fetal programming-vascular effects beyond blood pressure. Nephrol. Dial. Transplant 2012, 27, 3464–3476. [Google Scholar]
- Tain, Y.L.; Freshour, G.; Dikalova, A.; Griendling, K.; Baylis, C. Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am. J. Physiol. Renal Physiol 2007, 292, F1404–F1410. [Google Scholar]
- Tain, Y.L.; Kao, Y.H.; Hsieh, C.S.; Chen, C.C.; Sheen, J.M.; Lin, I.C.; Huang, L.T. Melatonin blocks oxidative stress-induced increased asymmetric dimethylarginine. Free Radic. Biol. Med 2010, 49, 1088–1098. [Google Scholar]
- Germain, A.M.; Valdés, G.; Romanik, M.C.; Reyes, M.S. Evidence supporting a beneficial role for long-term l-arginine supplementation in high-risk pregnancies. Hypertension 2004, 44, e1. [Google Scholar]
- Cynober, L.; Moinard, C.; De Bandt, J.P. The 2009 ESPEN Sir David Cuthbertson. Citrulline: A new major signaling molecule or just another player in the pharmaconutrition game? Clin. Nutr 2010, 29, 545–551. [Google Scholar]
- Prieto, C.P.; Krause, B.J.; Quezada, C.; San Martin, R.; Sobrevia, L.; Casanello, P. Hypoxia-reduced nitric oxide synthase activity is partially explained by higher arginase-2 activity and cellular redistribution in human umbilical vein endothelium. Placenta 2011, 32, 932–940. [Google Scholar]
- Miller, S.L.; Wallace, E.M.; Walker, D.W. Antioxidant Therapies: A potential role in perinatal medicine. Neuroendocrinology 2012, 96, 13–23. [Google Scholar]
- Dennery, P.A. Oxidative stress in development: nature or nurture? Free Radic. Biol. Med 2010, 49, 1147–1151. [Google Scholar]
- Rumbold, A.; Duley, L.; Crowther, C.A.; Haslam, R.R. Antioxidants for preventing pre-eclampsia. Cochrane Database Syst. Rev. 2008, 1. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).