p66Shc Aging Protein in Control of Fibroblasts Cell Fate


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
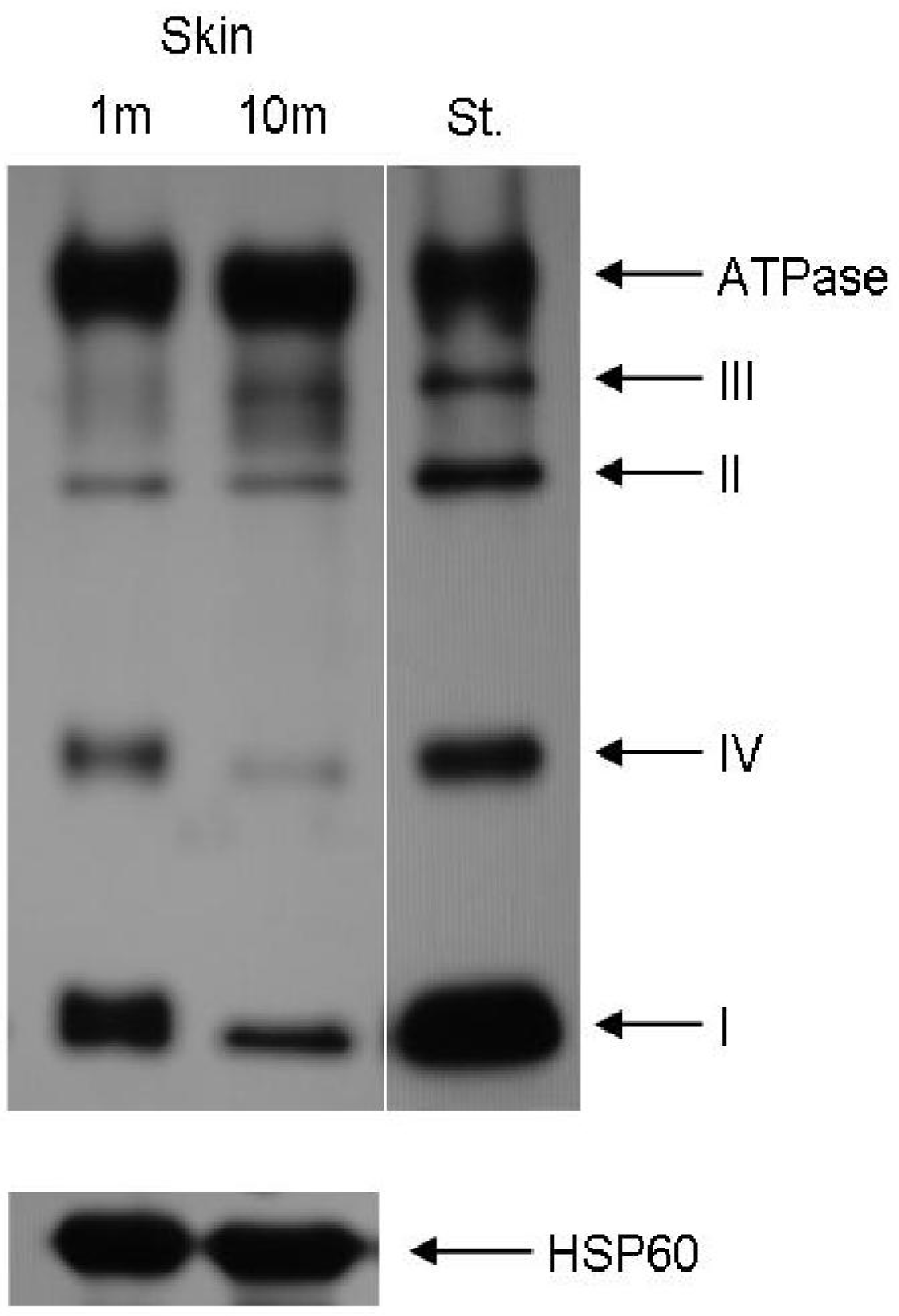
2. Age-Related Dysfunctions of the Mitochondrial Respiratory Chain and Antioxidant Defense
2.1. Antioxidant Defense and Aging
2.2. Antioxidant Supplementation
3. p66Shc—The Oxidative Stress and Longevity
4. p66Shc—One Protein Two Functions
5. A New Job for p66Shc: Its Role in Adipocyte Metabolism
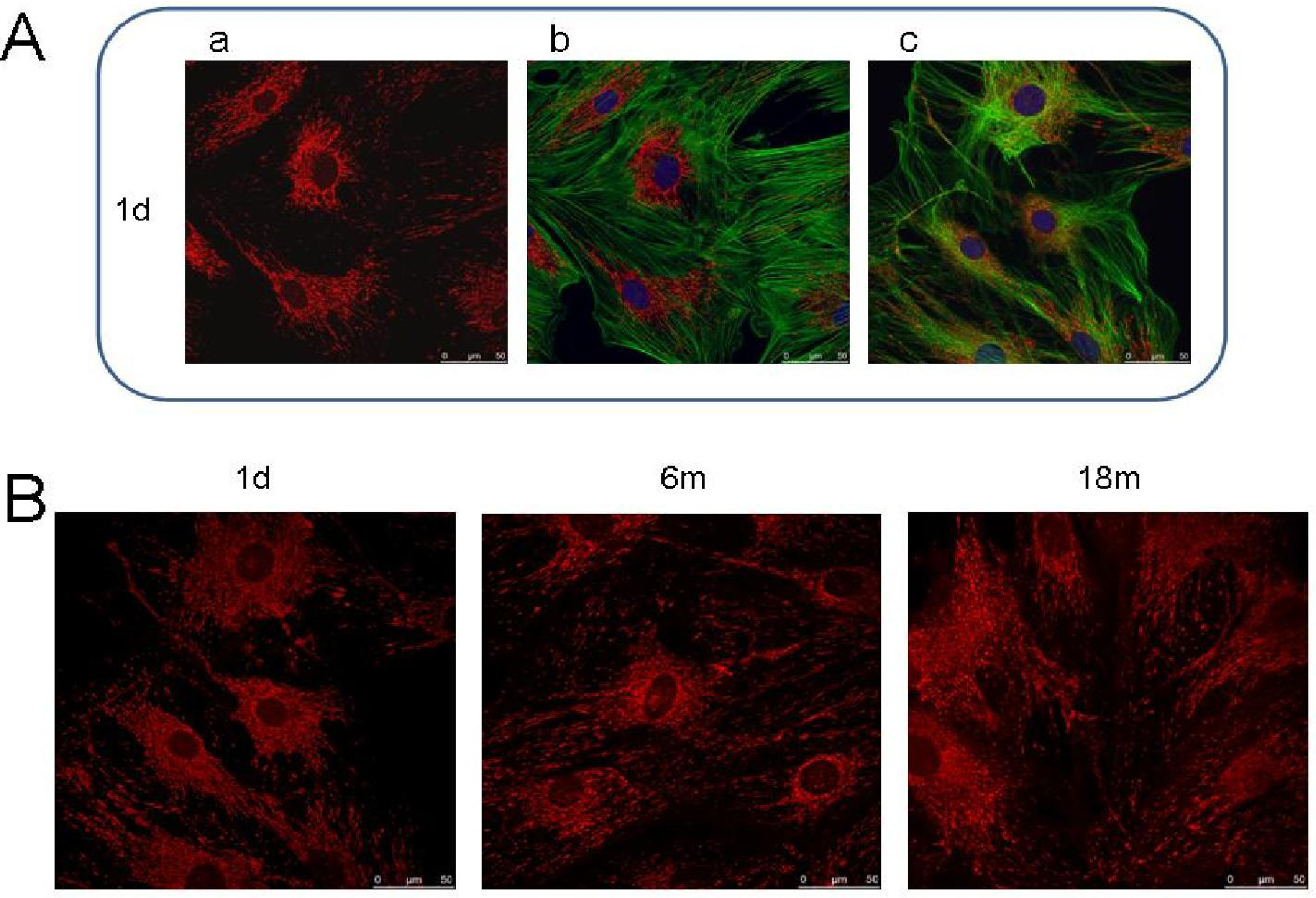
6. Studies on p66Shc in Fibroblast Models
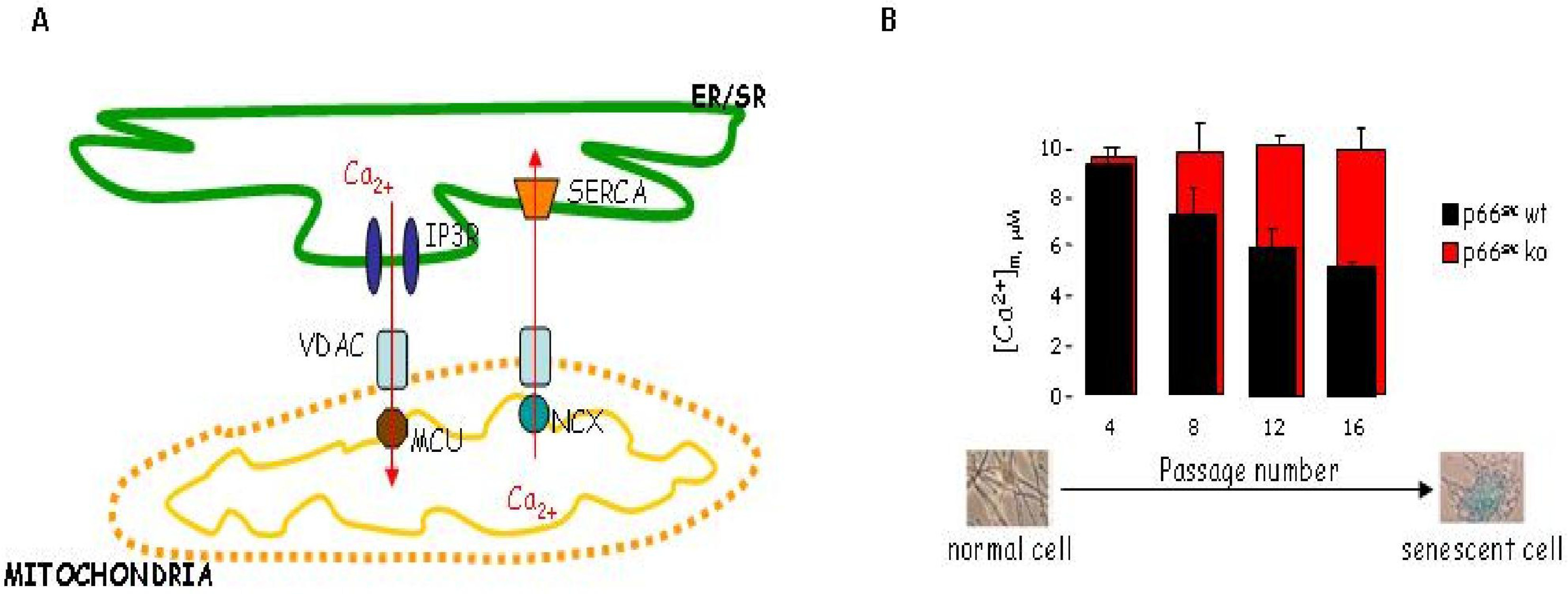
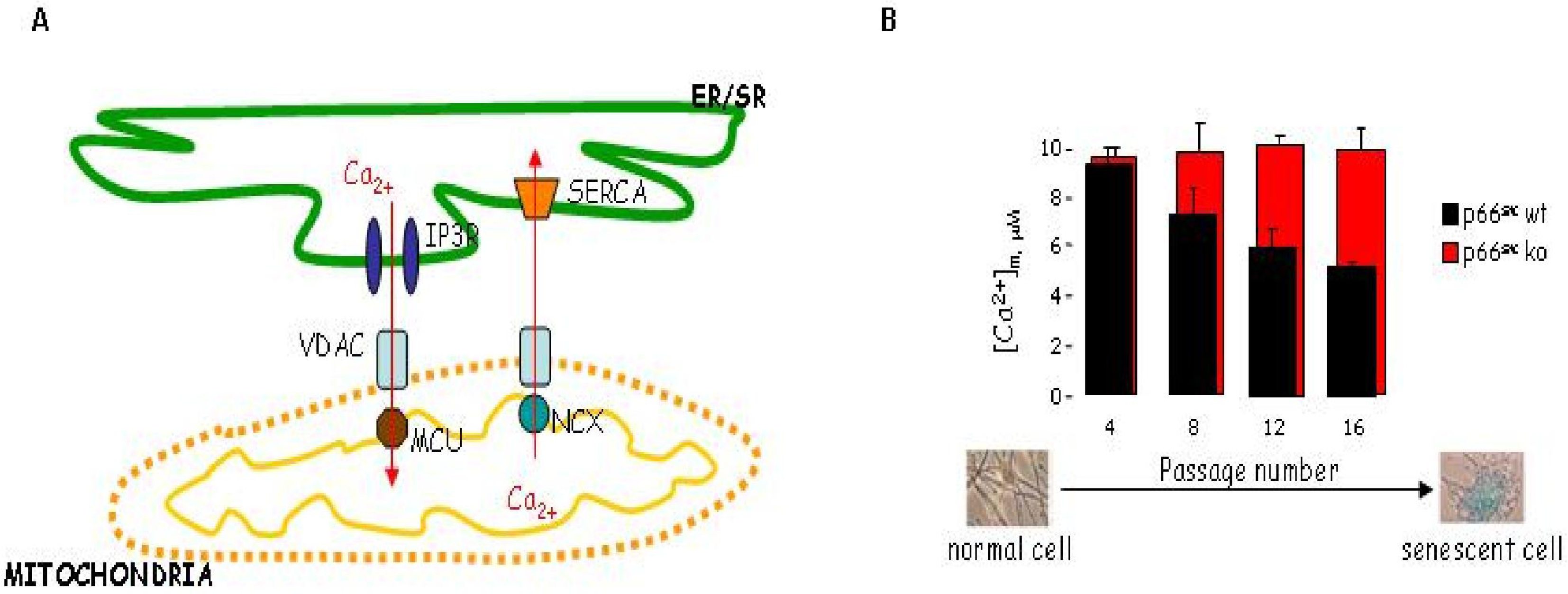
7. Mitochondrial Ca2+ Homeostasis is Compromised in Senescent Cells through a p66Shc Dependent Pathway
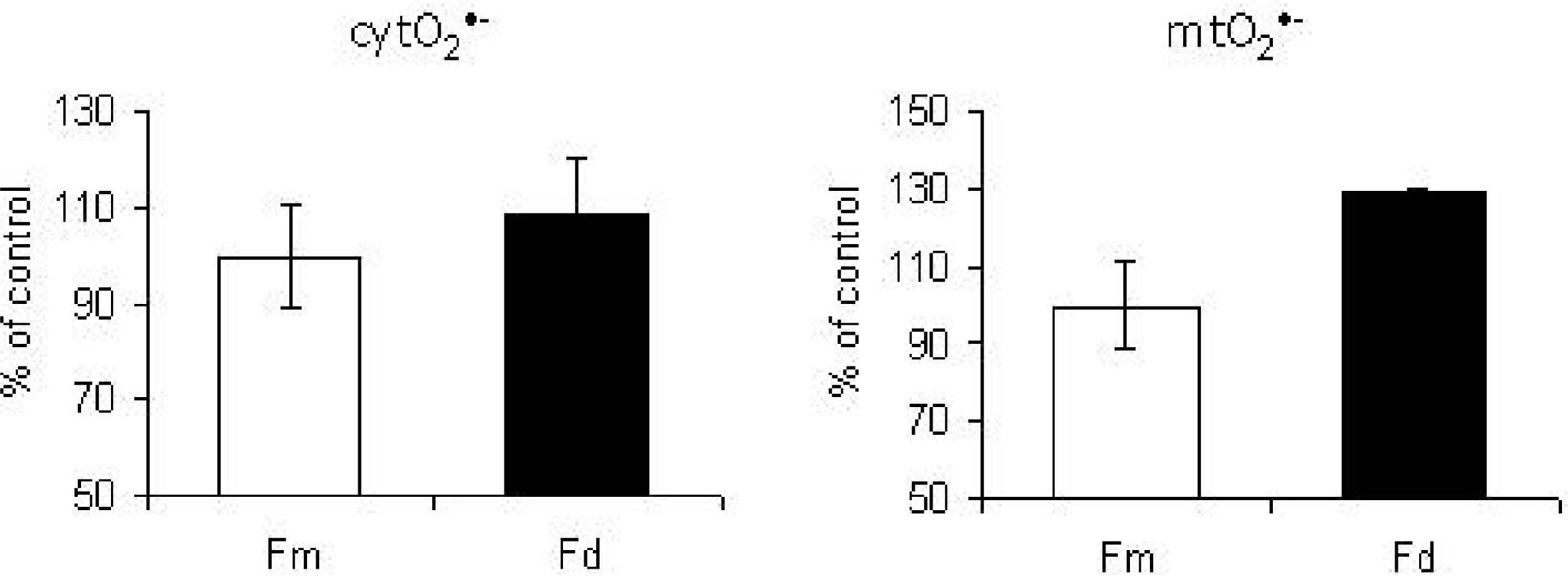
8. p66Shc Pathway Is Activated by Intracellular Oxidative Stress—Studies on Fibroblasts from Patients Harboring Mitochondrial Defects
9. Concluding Remarks
Ethics
Acknowledgments
- Conflict of InterestThe authors have declared no conflicts of interest.
References
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol 1956, 11, 298–300. [Google Scholar]
- Boffoli, D; Scacco, SC; Vergari, R; Solarino, G; Santacroce, G; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar]
- Feuers, RJ. The effects of dietary restriction on mitochondrial dysfunction in aging. Ann. N. Y. Acad. Sci 1998, 854, 192–201. [Google Scholar]
- Kwong, LK; Sohal, RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys 2000, 373, 16–22. [Google Scholar]
- Lenaz, G; Bovina, C; Castelluccio, C; Fato, R; Formiggini, G; Genova, ML; Marchetti, M; Pich, MM; Pallotti, F; Parenti Castelli, G; et al. Mitochondrial complex I defects in aging. Mol. Cell. Biochem 1997, 174, 329–333. [Google Scholar]
- Nakahara, H; Kanno, T; Inai, Y; Utsumi, K; Hiramatsu, M; Mori, A; Packer, L. Mitochondrial dysfunction in the senescence accelerated mouse (SAM). Free Radic. Biol. Med 1998, 24, 85–92. [Google Scholar]
- Navarro, A; Sanchez Del Pino, MJ; Gomez, C; Peralta, JL; Boveris, A. Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice. Am. J. Physiol. Regul. Integr. Comp. Physiol 2002, 282, R985–R992. [Google Scholar]
- Van Remmen, H; Richardson, A. Oxidative damage to mitochondria and aging. Exp. Gerontol 2001, 36, 957–968. [Google Scholar]
- Hsieh, RH; Hou, JH; Hsu, HS; Wei, YH. Age-dependent respiratory function decline and DNA deletions in human muscle mitochondria. Biochem. Mol. Biol. Int 1994, 32, 1009–1022. [Google Scholar]
- Yen, TC; Chen, YS; King, KL; Yeh, SH; Wei, YH. Liver mitochondrial respiratory functions decline with age. Biochem. Biophys. Res. Commun 1989, 165, 944–1003. [Google Scholar]
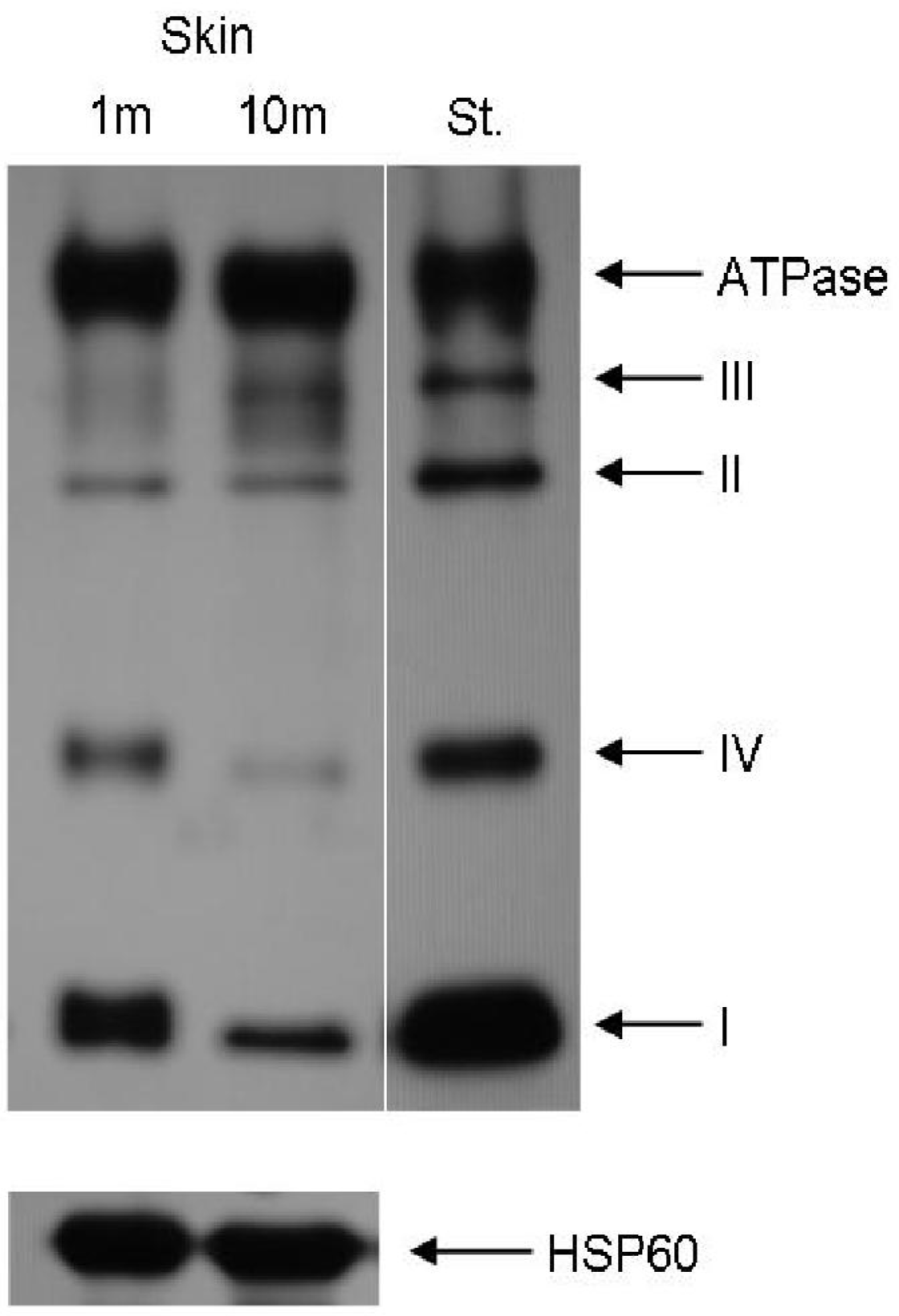
- Greco, M; Villani, G; Mazzucchelli, F; Bresolin, N; Papa, S; Attardi, G. Marked aging-related decline in efficiency of oxidative phosphorylation in human skin fibroblasts. FASEB J 2003, 17, 1706–1708. [Google Scholar]
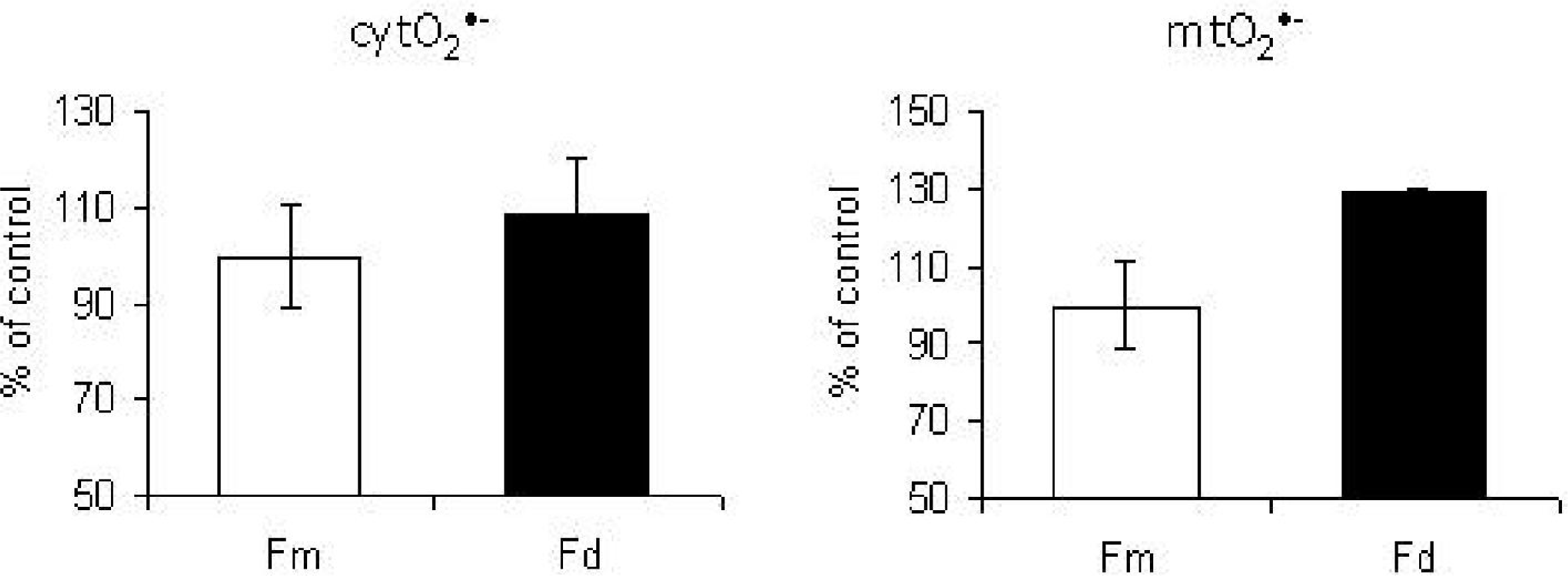
- Lebiedzinska, M; Karkucinska-Wieckowska, A; Giorgi, C; Karczmarewicz, E; Pronicka, E; Pinton, P; Duszynski, J; Pronicki, M; Wieckowski, MR. Oxidative stress-dependent p66Shc phosphorylation in skin fibroblasts of children with mitochondrial disorders. Biochim. Biophys. Acta 2010, 1797, 952–960. [Google Scholar]
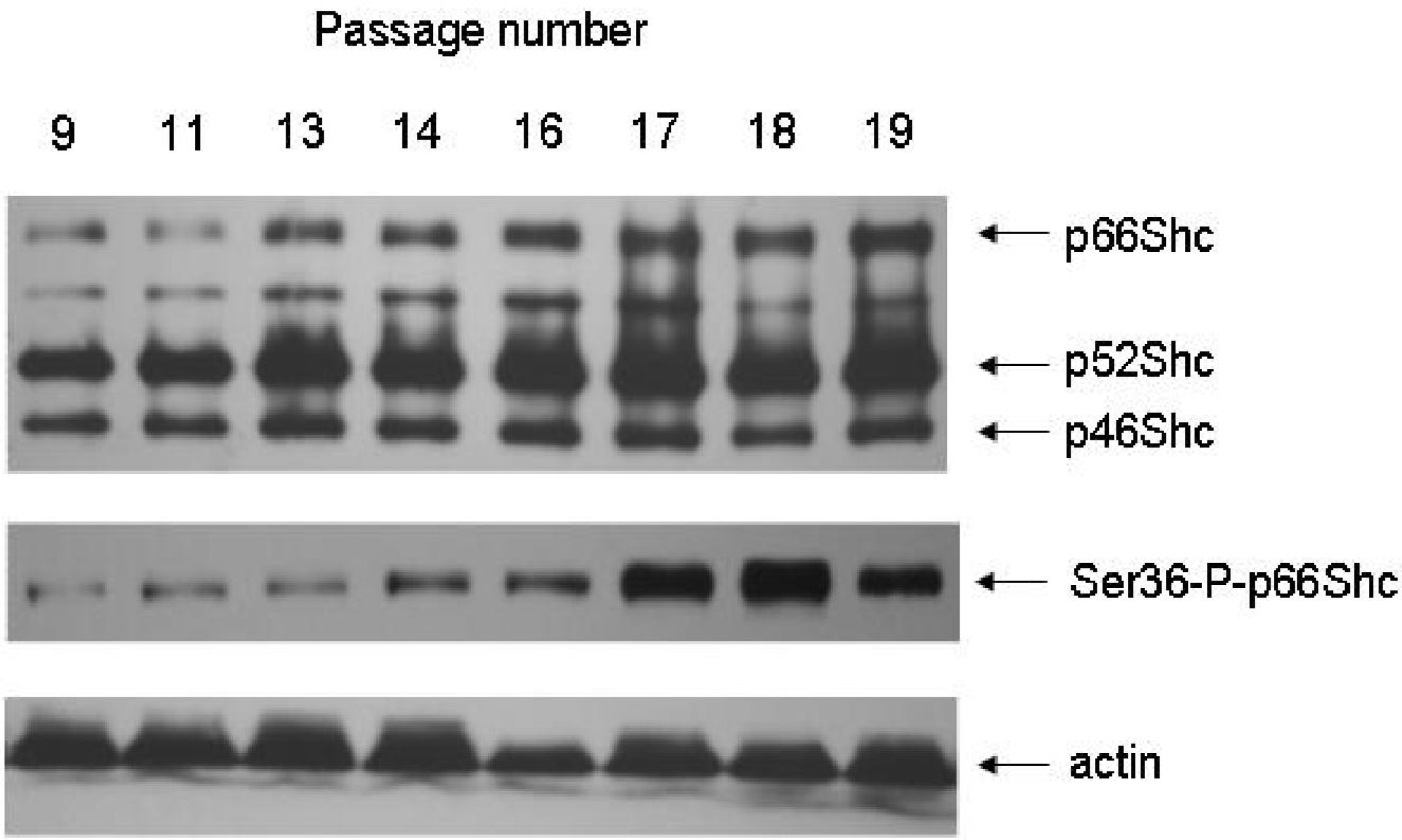
- Lebiedzinska, M; Duszynski, J; Rizzuto, R; Pinton, P; Wieckowski, MR. Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch. Biochem. Biophys 2009, 486, 73–80. [Google Scholar]
- Wilson, PD; Franks, LM. The effect of age on mitochondrial ultrastructure. Gerontology 1975, 21, 81–94. [Google Scholar]
- Szczepanowska, J; Zablocki, K; Duszynski, J. Influence of a mitochondrial genetic defect on capacitative calcium entry and mitochondrial organization in the osteosarcoma cells. FEBS Lett 2004, 578, 316–322. [Google Scholar]
- Inarrea, P; Moini, H; Han, D; Rettori, D; Aguilo, I; Alava, MA; Iturralde, M; Cadenas, E. Mitochondrial respiratory chain and thioredoxin reductase regulate intermembrane Cu, Zn-superoxide dismutase activity: Implications for mitochondrial energy metabolism and apoptosis. Biochem. J 2007, 405, 173–179. [Google Scholar]
- Michiels, C; Raes, M; Toussaint, O; Remacle, J. Importance of Se-glutathione peroxidase, catalase, and Cu/Zn-SOD for cell survival against oxidative stress. Free Radic. Biol. Med 1994, 17, 235–248. [Google Scholar]
- Wei, YH; Lee, HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp. Biol. Med 2002, 227, 671–682. [Google Scholar]
- Van Remmen, H; Qi, W; Sabia, M; Freeman, G; Estlack, L; Yang, H; Guo, ZM; Huang, TT; Strong, R; Lee, S; et al. Multiple deficiencies in antioxidant enzymes in mice result in a compound increase in sensitivity to oxidative stress. Free Radic. Biol. Med 2004, 36, 1625–1634. [Google Scholar]
- Hsieh, CC; Papaconstantinou, J. Dermal fibroblasts from long-lived Ames dwarf mice maintain their in vivo resistance to mitochondrial generated reactive oxygen species (ROS). Aging (Albany NY) 2009, 1, 784–802. [Google Scholar]
- Hoppe, U; Bergemann, J; Diembeck, W; Ennen, J; Gohla, S; Harris, I; Jacob, J; Kielholz, J; Mei, W; Pollet, D; et al. Coenzyme Q10, a cutaneous antioxidant and energizer. Biofactors 1999, 9, 371–378. [Google Scholar]
- Sander, CS; Chang, H; Salzmann, S; Muller, CS; Ekanayake-Mudiyanselage, S; Elsner, P; Thiele, JJ. Photoaging is associated with protein oxidation in human skin in vivo. J. Invest. Dermatol 2002, 118, 618–625. [Google Scholar]
- Berneburg, M; Grether-Beck, S; Kurten, V; Ruzicka, T; Briviba, K; Sies, H; Krutmann, J. Singlet oxygen mediates the UVA-induced generation of the photoaging-associated mitochondrial common deletion. J. Biol. Chem 1999, 274, 15345–15349. [Google Scholar]
- Burke, KE. Interaction of vitamins C and E as better cosmeceuticals. Dermatol. Ther 2007, 20, 314–321. [Google Scholar]
- Packer, L; Smith, JR. Extension of the lifespan of cultured normal human diploid cells by vitamin E. Proc. Natl. Acad. Sci. USA 1974, 71, 4763–4767. [Google Scholar]
- Stockl, P; Hutter, E; Zwerschke, W; Jansen-Durr, P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol 2006, 41, 674–682. [Google Scholar]
- Ghneim, HK; Al-Sheikh, YA. The effect of aging and increasing ascorbate concentrations on respiratory chain activity in cultured human fibroblasts. Cell Biochem. Funct 2010, 28, 283–292. [Google Scholar]
- Nichols, JA; Katiyar, SK. Skin photoprotection by natural polyphenols: Anti-inflammatory, antioxidant and DNA repair mechanisms. Arch. Dermatol. Res 2010, 302, 71–83. [Google Scholar]
- Meng, Q; Velalar, CN; Ruan, R. Effects of epigallocatechin-3-gallate on mitochondrial integrity and antioxidative enzyme activity in the aging process of human fibroblast. Free Radic. Biol. Med 2008, 44, 1032–1041. [Google Scholar]
- Ramachandran, S; Rajendra Prasad, N; Karthikeyan, S. Sesamol inhibits UVB-induced ROS generation and subsequent oxidative damage in cultured human skin dermal fibroblasts. Arch. Dermatol. Res 2010, 302, 733–744. [Google Scholar]
- Skulachev, VP; Anisimov, VN; Antonenko, YN; Bakeeva, LE; Chernyak, BV; Erichev, VP; Filenko, OF; Kalinina, NI; Kapelko, VI; Kolosova, NG; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar]
- Pinton, P; Rimessi, A; Marchi, S; Orsini, F; Migliaccio, E; Giorgio, M; Contursi, C; Minucci, S; Mantovani, F; Wieckowski, MR; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar]
- Migliaccio, E; Giorgio, M; Mele, S; Pelicci, G; Reboldi, P; Pandolfi, PP; Lanfrancone, L; Pelicci, PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar]
- Tomilov, AA; Bicocca, V; Schoenfeld, RA; Giorgio, M; Migliaccio, E; Ramsey, JJ; Hagopian, K; Pelicci, PG; Cortopassi, GA. Decreased superoxide production in macrophages of long-lived p66Shc knock-out mice. J. Biol. Chem 2010, 285, 1153–1165. [Google Scholar]
- Finetti, F; Pellegrini, M; Ulivieri, C; Savino, MT; Paccagnini, E; Ginanneschi, C; Lanfrancone, L; Pelicci, PG; Baldari, CT. The proapoptotic and antimitogenic protein p66SHC acts as a negative regulator of lymphocyte activation and autoimmunity. Blood 2008, 111, 5017–5027. [Google Scholar]
- Luzi, L; Confalonieri, S; Di Fiore, PP; Pelicci, PG. Evolution of Shc functions from nematode to human. Curr. Opin. Genet. Dev 2000, 10, 668–674. [Google Scholar]
- Trinei, M; Giorgio, M; Cicalese, A; Barozzi, S; Ventura, A; Migliaccio, E; Milia, E; Padura, IM; Raker, VA; Maccarana, M; et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar]
- Giorgio, M; Migliaccio, E; Orsini, F; Paolucci, D; Moroni, M; Contursi, C; Pelliccia, G; Luzi, L; Minucci, S; Marcaccio, M; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar]
- Okada, S; Kao, AW; Ceresa, BP; Blaikie, P; Margolis, B; Pessin, JE. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem 1997, 272, 28042–28049. [Google Scholar]
- Wieckowski, MR; Giorgi, C; Lebiedzinska, M; Duszynski, J; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc 2009, 4, 1582–1590. [Google Scholar]
- Lebiedzinska, M; Szabadkai, G; Jones, AW; Duszynski, J; Wieckowski, MR. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol 2009, 41, 1805–1816. [Google Scholar]
- Nemoto, S; Combs, CA; French, S; Ahn, BH; Fergusson, MM; Balaban, RS; Finkel, T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J. Biol. Chem 2006, 281, 10555–10560. [Google Scholar]
- Giorgio, M; Trinei, M; Migliaccio, E; Pelicci, PG. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol 2007, 8, 722–728. [Google Scholar]
- Napoli, C; Martin-Padura, I; de Nigris, F; Giorgio, M; Mansueto, G; Somma, P; Condorelli, M; Sica, G; de Rosa, G; Pelicci, P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc. Natl. Acad. Sci. USA 2003, 100, 2112–2116. [Google Scholar]
- Berniakovich, I; Trinei, M; Stendardo, M; Migliaccio, E; Minucci, S; Bernardi, P; Pelicci, PG; Giorgio, M. p66Shc-generated oxidative signal promotes fat accumulation. J. Biol. Chem 2008, 283, 34283–34293. [Google Scholar]
- Ranieri, SC; Fusco, S; Panieri, E; Labate, V; Mele, M; Tesori, V; Ferraraa, A; Mauluccib, G; de Spiritob, M; Martorana, GE; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar]
- Tomilov, AA; Ramsey, JJ; Hagopian, K; Giorgio, M; Kim, KM; Lam, A; Migliaccio, E; Lloyd, KC; Berniakovich, I; Prolla, TA; et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011, 10, 55–65. [Google Scholar]
- Nemoto, S; Finkel, T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002, 295, 2450–2452. [Google Scholar]
- Pani, G; Koch, OR; Galeotti, T. The p53-p66shc-Manganese Superoxide Dismutase (MnSOD) network: A mitochondrial intrigue to generate reactive oxygen species. Int. J. Biochem. Cell Biol 2009, 41, 1002–1005. [Google Scholar]
- Sohal, RS; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar]
- Sohal, RS; Agarwal, S; Sohal, BH. Oxidative stress and aging in the Mongolian gerbil (Meriones unguiculatus). Mech. Ageing Dev 1995, 81, 15–25. [Google Scholar]
- Pandolfi, S; Bonafe, M; Di Tella, L; Tiberi, L; Salvioli, S; Monti, D; Sorbi, S; Franceschi, C. p66(shc) is highly expressed in fibroblasts from centenarians. Mech. Ageing Dev 2005, 126, 839–844. [Google Scholar]
- Lebiedzinska, M; Karkucinska-Wieckowska, A; Suski, JM; Szabadkai, G; Wilczyński, G; Wlodarczyk, J; Pronicki, M; Duszynski, J; Pinton, P; Wieckowski, MR. p66Shc-related response to the oxidative stress in fibroblasts of NARP patients. Mitochondrion 2011. to be submitted for publication. [Google Scholar]
- Tian, L; Cai, Q; Wei, H. Alterations of antioxidant enzymes and oxidative damage to macromolecules in different organs of rats during aging. Free Radic. Biol. Med 1998, 24, 1477–1484. [Google Scholar]
- Palomero, J; Galan, AI; Munoz, ME; Tunon, MJ; Gonzalez-Gallego, J; Jimenez, R. Effects of aging on the susceptibility to the toxic effects of cyclosporin A in rats. Changes in liver glutathione and antioxidant enzymes. Free Radic. Biol. Med 2001, 30, 836–845. [Google Scholar]
- Tatone, C; Carbone, MC; Falone, S; Aimola, P; Giardinelli, A; Caserta, D; Marci, R; Pandolfi, A; Ragnelli, AM. Amicarelli F Age-dependent changes in the expression of superoxide dismutases and catalase are associated with ultrastructural modifications in human granulosa cells. Mol. Hum. Reprod 2006, 12, 655–660. [Google Scholar]
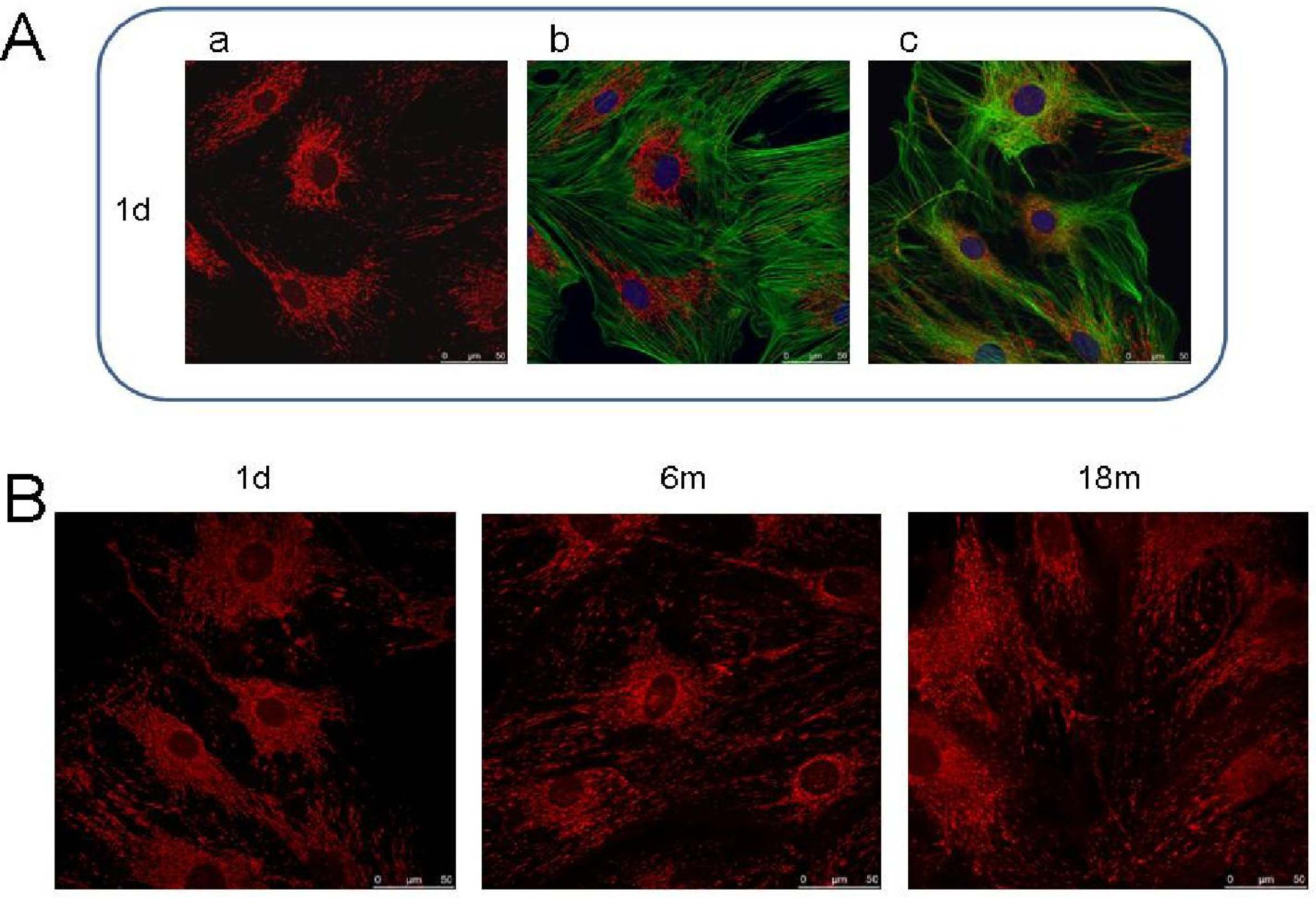
- Lee, HC; Yin, PH; Chi, CW; Wei, YH. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci 2002, 9, 517–526. [Google Scholar]
- Wei, YH; Lee, CF; Lee, HC; Ma, YS; Wang, CW; Lu, CY; Pang, CY. Increases of mitochondrial mass and mitochondrial genome in association with enhanced oxidative stress in human cells harboring 4, 977 BP-deleted mitochondrial DNA. Ann. N. Y. Acad. Sci 2001, 928, 97–112. [Google Scholar]
- Lee, HC; Yin, PH; Lu, CY; Chi, CW; Wei, YH. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J 2000, 348, 425–432. [Google Scholar]
- Rizzuto, R; Bernardi, P; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol 2000, 529, 37–47. [Google Scholar]
- Rimessi, A; Giorgi, C; Pinton, P; Rizzuto, R. The versatility of mitochondrial calcium signals: From stimulation of cell metabolism to induction of cell death. Biochim. Biophys. Acta 2008, 1777, 808–816. [Google Scholar]
- Giorgi, C; Romagnoli, A; Pinton, P; Rizzuto, R. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med 2008, 8, 119–130. [Google Scholar]
- Giorgi, C; de Stefani, D; Bononi, A; Rizzuto, R; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol 2009, 41, 1817–1827. [Google Scholar]
- Guillery, O; Malka, F; Frachon, P; Milea, D; Rojo, M; Lombes, A. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul. Disord 2008, 18, 319–330. [Google Scholar]
- Cameron, JM; Levandovskiy, V; MacKay, N; Robinson, BH. Respiratory chain analysis of skin fibroblasts in mitochondrial disease. Mitochondrion 2004, 4, 387–394. [Google Scholar]
- Robinson, BH. Cell culture studies on patients with mitochondrial diseases: Molecular defects in pyruvate dehydrogenase. J. Bioenerg. Biomembr 1988, 20, 313–323. [Google Scholar]
- Robinson, BH; Ward, J; Goodyer, P; Baudet, A. Respiratory chain defects in the mitochondria of cultured skin fibroblasts from three patients with lacticacidemia. J. Clin. Invest 1986, 77, 1422–1427. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suski, J.M.; Karkucinska-Wieckowska, A.; Lebiedzinska, M.; Giorgi, C.; Szczepanowska, J.; Szabadkai, G.; Duszynski, J.; Pronicki, M.; Pinton, P.; Wieckowski, M.R. p66Shc Aging Protein in Control of Fibroblasts Cell Fate. Int. J. Mol. Sci. 2011, 12, 5373-5389. https://doi.org/10.3390/ijms12085373
Suski JM, Karkucinska-Wieckowska A, Lebiedzinska M, Giorgi C, Szczepanowska J, Szabadkai G, Duszynski J, Pronicki M, Pinton P, Wieckowski MR. p66Shc Aging Protein in Control of Fibroblasts Cell Fate. International Journal of Molecular Sciences. 2011; 12(8):5373-5389. https://doi.org/10.3390/ijms12085373
Chicago/Turabian StyleSuski, Jan M., Agnieszka Karkucinska-Wieckowska, Magdalena Lebiedzinska, Carlotta Giorgi, Joanna Szczepanowska, Gyorgy Szabadkai, Jerzy Duszynski, Maciej Pronicki, Paolo Pinton, and Mariusz R. Wieckowski. 2011. "p66Shc Aging Protein in Control of Fibroblasts Cell Fate" International Journal of Molecular Sciences 12, no. 8: 5373-5389. https://doi.org/10.3390/ijms12085373
APA StyleSuski, J. M., Karkucinska-Wieckowska, A., Lebiedzinska, M., Giorgi, C., Szczepanowska, J., Szabadkai, G., Duszynski, J., Pronicki, M., Pinton, P., & Wieckowski, M. R. (2011). p66Shc Aging Protein in Control of Fibroblasts Cell Fate. International Journal of Molecular Sciences, 12(8), 5373-5389. https://doi.org/10.3390/ijms12085373