Multi-Step Usage of in Vivo Models During Rational Drug Design and Discovery

Abstract
:1. Introduction
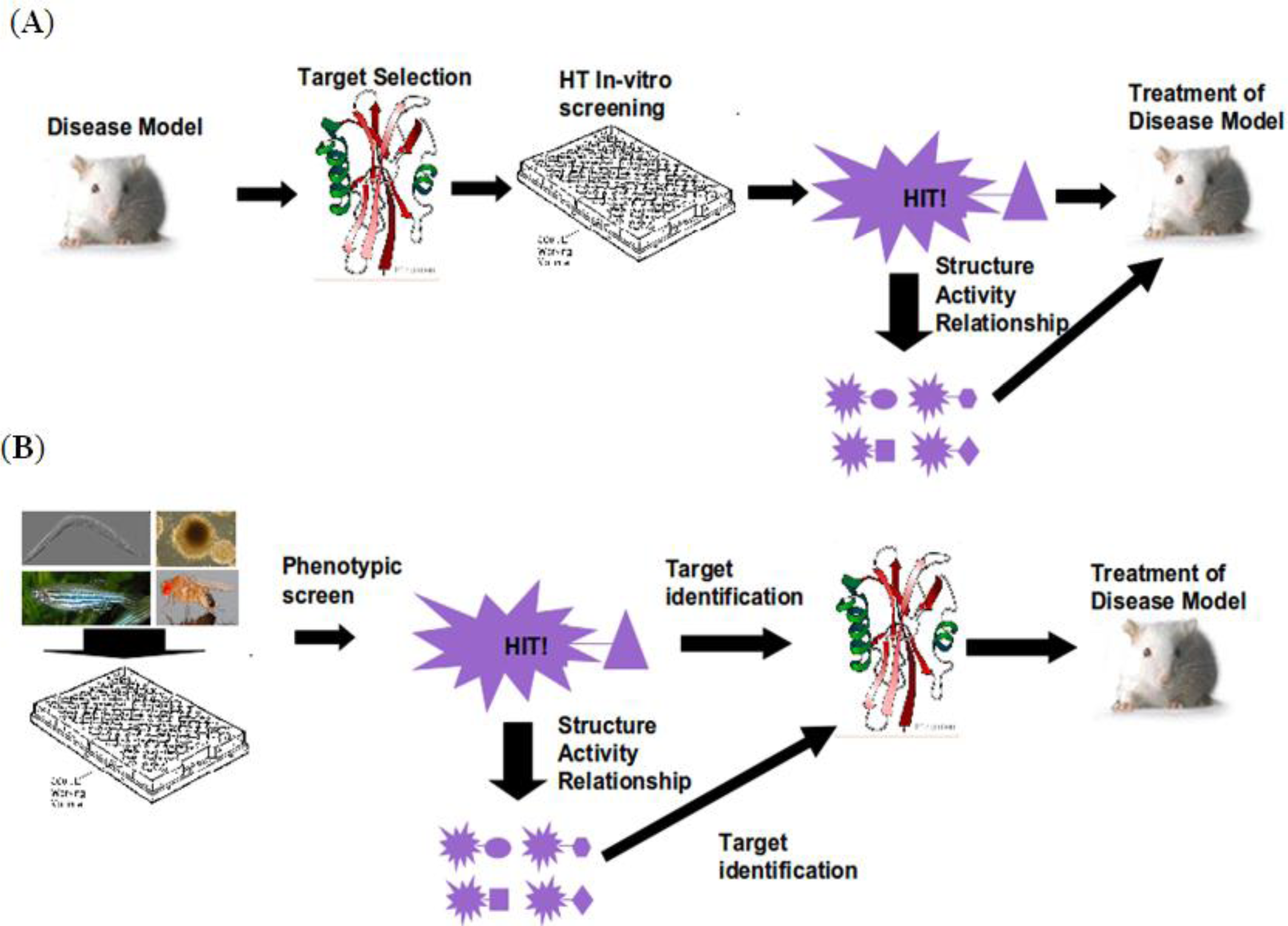
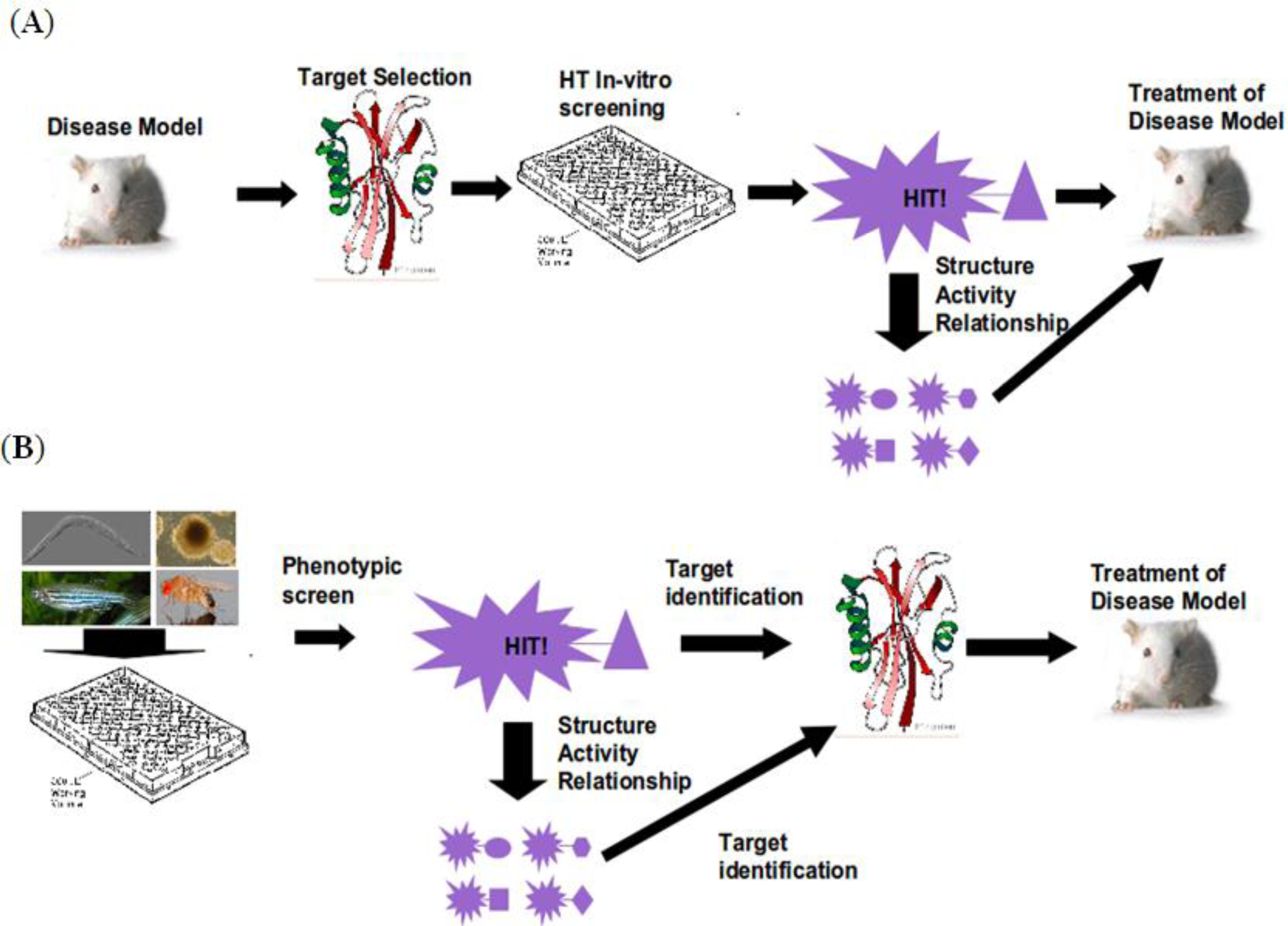
2. Workflow
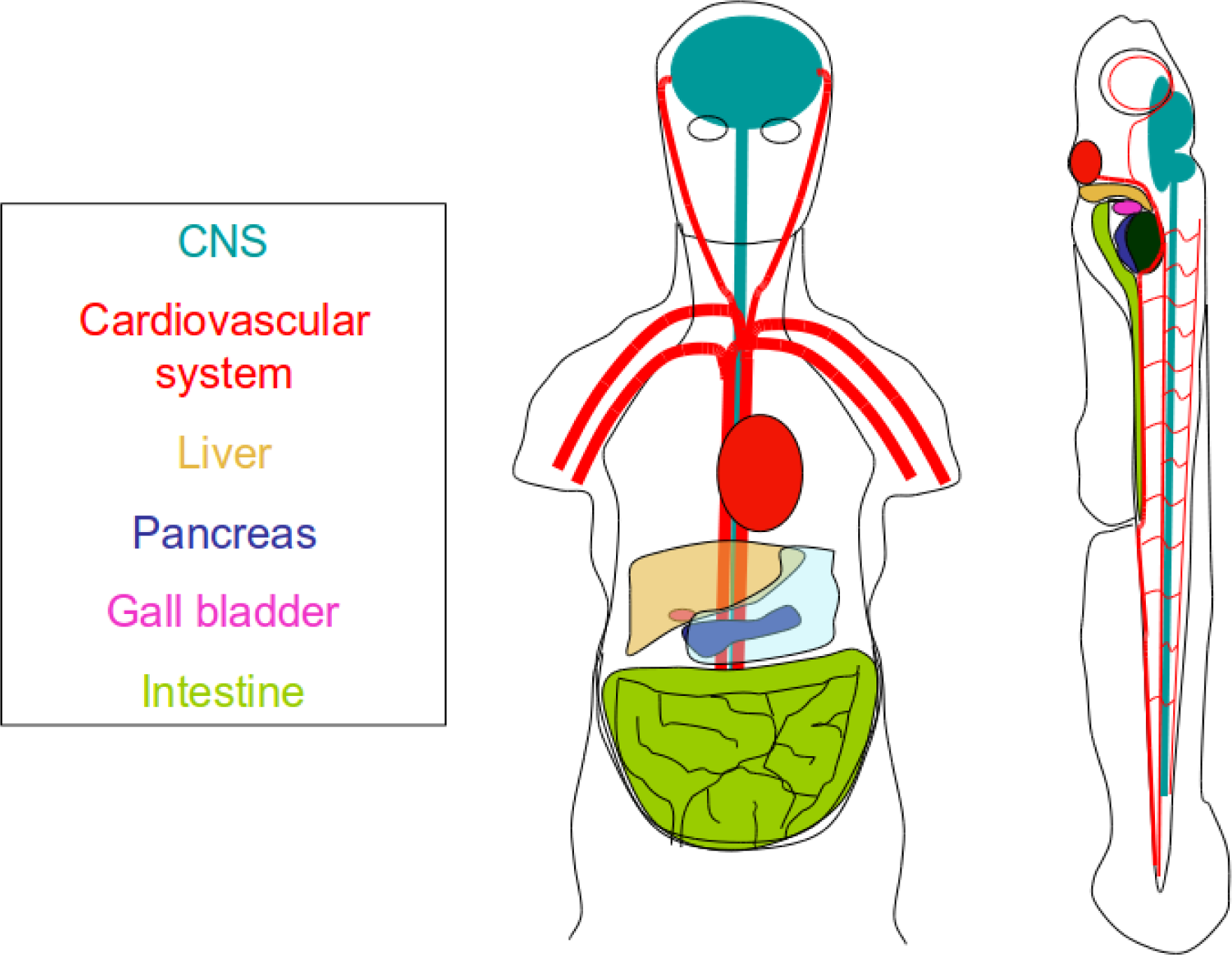
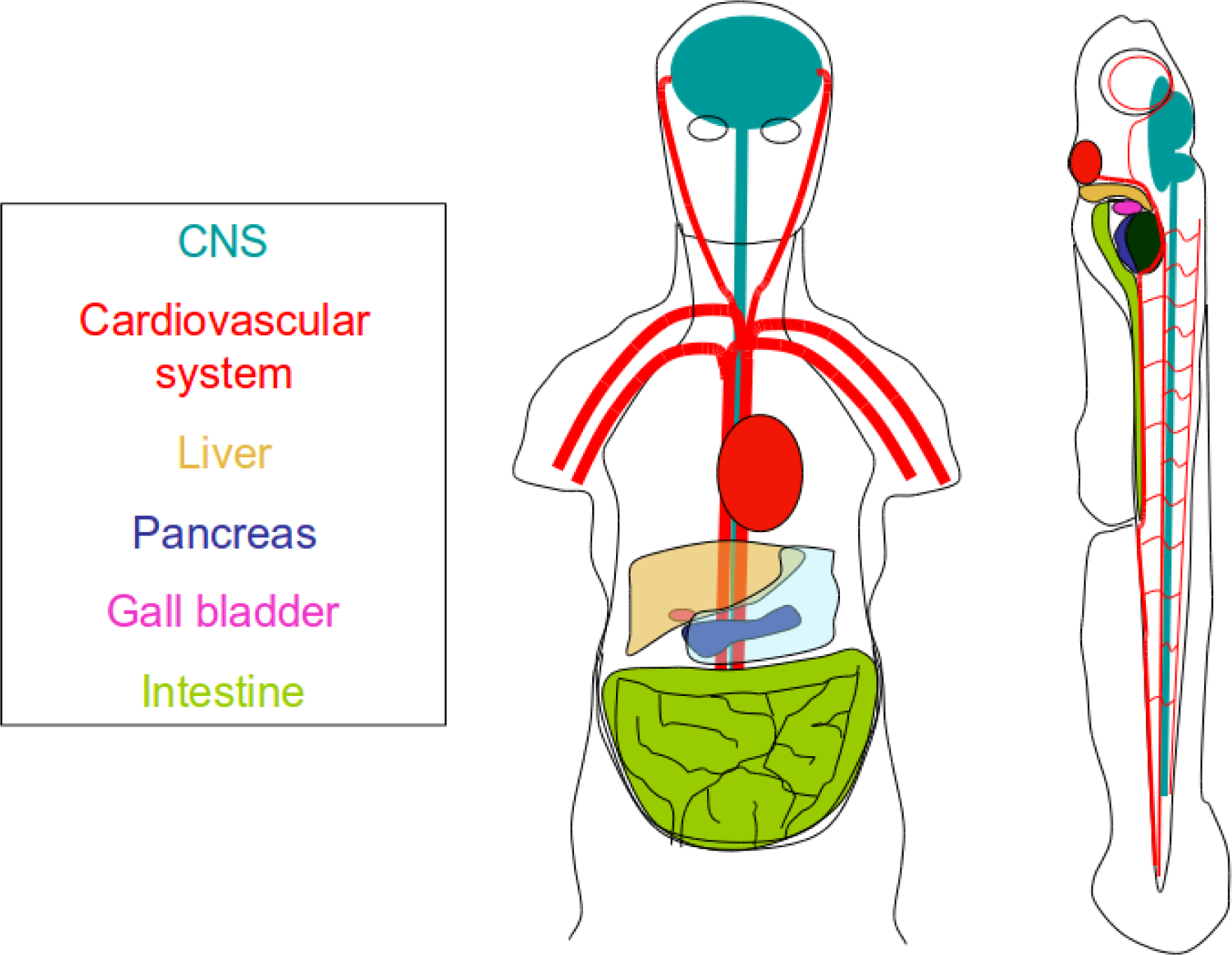
3. Organismal Models
4. Target Identification
5. SAR and Compound Optimization
6. Vertebrate Toxicity
7. Afterword
References
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945.
- Hopkins, AL; Groom, CR. The druggable genome. Nat. Rev. Drug Discov 2002, 1, 727–730. [Google Scholar]
- Adams, CP; Brantner, VV. Spending on new drug development. Health Econ 2010, 19, 130–141. [Google Scholar]
- Talele, TT; Khedkar, SA; Rigby, AC. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top Med. Chem 2010, 10, 127–141. [Google Scholar]
- Zon, LI; Peterson, RT. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov 2005, 4, 35–44. [Google Scholar]
- Cohen, NC. Structure-based drug design and the discovery of aliskiren (Tekturna): Perseverance and creativity to overcome a R&D pipeline challenge. Chem. Biol. Drug Des 2007, 70, 557–565. [Google Scholar]
- Brenner, S. In the beginning was the worm. Genetics 2009, 182, 413–415. [Google Scholar]
- Poulin, G; Nandakumar, R; Ahringer, J. Genome-wide RNAi screens in Caenorhabditis elegans: Impact on cancer research. Oncogene 2004, 23, 8340–8345. [Google Scholar]
- Putcha, GV; Johnson, EM. Men are but worms: Neuronal cell death in C. elegans and vertebrates. Cell Death Differ 2004, 11, 38–48. [Google Scholar]
- Smith, KR; Kieserman, EK; Wang, PI; Basten, SG; Giles, RH; Marcotte, EM; Wallingford, JB. A role for central spindle proteins in cilia structure and function. Cytoskeleton 2010, 68, 112–124. [Google Scholar]
- Ewald, CY; Li, C. Understanding the molecular basis of Alzheimer’s disease using a Caenorhabditis elegans model system. Brain Struct. Funct 2010, 214, 263–283. [Google Scholar]
- Harrington, AJ; Hamamichi, S; Caldwell, GA; Caldwell, KA. C. elegans as a model organism to investigate molecular pathways involved with Parkinson’s disease. Dev. Dyn 2010, 239, 1282–1295. [Google Scholar]
- Puccio, H. Multicellular models of Friedreich ataxia. J Neurol 2009, 256(Suppl. 1), 18–24. [Google Scholar]
- Morcos, M; Hutter, H. The model Caenorhabditis elegans in diabetes mellitus and Alzheimer’s disease. J. Alzheimers Dis 2009, 16, 897–908. [Google Scholar]
- Burns, AR; Kwok, TCY; Howard, A; Houston, E; Johanson, K; Chan, A; Cutler, SR; McCourt, P; Roy, PJ. High-throughput screening of small molecules for bioactivity and target identification in Caenorhabditis elegans. Nat. Protoc 2006, 1, 1906–1914. [Google Scholar]
- Kwok, TCY; Ricker, N; Fraser, R; Chan, AW; Burns, A; Stanley, EF; McCourt, P; Cutler, SR; Roy, PJ. A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature 2006, 441, 91–95. [Google Scholar]
- Gosai, SJ; Kwak, JH; Luke, CJ; Long, OS; King, DE; Kovatch, KJ; Johnston, PA; Shun, TY; Lazo, JS; Perlmutter, DH; et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One 2010, 5, e15460. [Google Scholar]
- Harris, TW; Chen, N; Cunningham, F; Tello-Ruiz, M; Antoshechkin, I; Bastiani, C; Bieri, T; Blasiar, D; Bradnam, K; Chan, J; et al. WormBase: A multi-species resource for nematode biology and genomics. Nucleic Acids Res 2004, 32(Suppl. 1), D411–D417. [Google Scholar]
- Wells, WA. High-throughput worms. NemaPharm, Inc. Chem. Biol 1998, 5, R147–R148. [Google Scholar]
- Kokel, D; Li, Y; Qin, J; Xue, D. The nongenotoxic carcinogens naphthalene and para-dichlorobenzene suppress apoptosis in Caenorhabditis elegans. Nat. Chem. Biol 2006, 2, 338–345. [Google Scholar]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev Genet 2005, 6, 9–23. [Google Scholar]
- van der Plas, MC; Pilgram, GSK; de Jong, AW; Bansraj, MR; Fradkin, LG; Noordermeer, JN. Drosophila Dystrophin is required for integrity of the musculature. Mech. Dev 2007, 124, 617–630. [Google Scholar]
- Nichols, CD. Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol. Ther 2006, 112, 677–700. [Google Scholar]
- Chang, S; Bray, SM; Li, Z; Zarnescu, DC; He, C; Jin, P; Warren, ST. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol 2008, 4, 256–263. [Google Scholar]
- Nikonenko, BV; Samala, R; Einck, L; Nacy, CA. Rapid, simple in vivo screen for new drugs active against Mycobacterium tuberculosis. Antimicrob. Agents Chemother 2004, 48, 4550–4555. [Google Scholar]
- Williams, C; Kim, S; Ni, TT; Mitchell, L; Ro, H; Penn, JS; Baldwin, SH; Solnica-Krezel, L; Zhong, TP. Hedgehog signaling induces arterial endothelial cell formation by repressing venous cell fate. Dev. Biol 2010, 341, 196–204. [Google Scholar]
- Hong, CC; Kume, T; Peterson, RT. Role of crosstalk between phosphatidylinositol 3-kinase and extracellular signal-regulated kinase/mitogen-activated protein kinase pathways in artery-vein specification. Circ. Res 2008, 103, 573–579. [Google Scholar]
- Takada, N; Kucenas, S; Appel, B. Sox10 is necessary for oligodendrocyte survival following axon wrapping. Glia 2010, 58, 996–1006. [Google Scholar]
- Goessling, W; North, TE; Zon, LI. New waves of discovery: Modeling cancer in zebrafish. J. Clin. Oncol 2007, 25, 2473–2479. [Google Scholar]
- Richardson, J; Lundegaard, PR; Reynolds, NL; Dorin, JR; Porteous, DJ; Jackson, IJ; Patton, EE. Mc1r Pathway regulation of zebrafish melanosome dispersion. Zebrafish 2008, 5, 289–295. [Google Scholar]
- Hao, J; Williams, CH; Webb, ME; Hong, CC. Large scale zebrafish-based in vivo small molecule screen. J. Vis. Exp 2010, 46, 2243. [Google Scholar]
- Yu, PB; Hong, CC; Sachidanandan, C; Babitt, JL; Deng, DY; Hoyng, SA; Lin, HY; Bloch, KD; Peterson, RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol 2008, 4, 33–41. [Google Scholar]
- Tran, TC; Sneed, B; Haider, J; Blavo, D; White, A; Aiyejorun, T; Baranowski, TC; Rubinstein, AL; Doan, TN; Dingledine, R; et al. Automated, quantitative screening assay for antiangiogenic compounds using transgenic zebrafish. Cancer Res 2007, 67, 11386–11392. [Google Scholar]
- Molina, G; Vogt, A; Bakan, A; Dai, WX; de Oliveira, PQ; Znosko, W; Smithgall, TE; Bahar, IB; Lazo, JS; Day, BW; et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol 2009, 5, 680–687. [Google Scholar]
- Ao, A; Williams, CH; Hao, J; Hong, CC. Modified Mouse Embryonic Stem Cell based Assay for Quantifying Cardiogenic Induction Efficiency. J Vis Exp 2011, in press. [Google Scholar]
- Zhang, X. Modulation of embryonic stem cell fate and somatic cell reprogramming by small molecules. Reprod. Biomed. Online 2010, 21, 26–36. [Google Scholar]
- Wang, H; Hao, J; Hong, CC. Cardiac Induction of Embryonic Stem Cells by a Small Molecule Inhibitor of Wnt/β-Catenin Signaling. ACS Chem. Biol 2010, 6, 192–197. [Google Scholar]
- Zhu, S; Wurdak, H; Wang, J; Lyssiotis, CA; Peters, EC; Cho, CY; Wu, X; Schultz, PG. A small molecule primes embryonic stem cells for differentiation. Cell Stem Cell 2009, 4, 416–426. [Google Scholar]
- Saxe, JP; Wu, H; Kelly, TK; Phelps, ME; Sun, YE; Kornblum, HI; Huang, J. A phenotypic small-molecule screen identifies an orphan ligand-receptor pair that regulates neural stem cell differentiation. Chem. Biol 2007, 14, 1019–1030. [Google Scholar]
- Arrell, DK; Terzic, A. Network systems biology for drug discovery. Clin. Pharmacol. Ther 2010, 88, 120–125. [Google Scholar]
- Waring, JF; Ciurlionis, R; Jolly, RA; Heindel, M; Ulrich, RG. Microarray analysis of hepatotoxins in vitro reveals a correlation between gene expression profiles and mechanisms of toxicity. Toxicol. Lett 2001, 120, 359–368. [Google Scholar]
- Lamb, J; Crawford, ED; Peck, D; Modell, JW; Blat, IC; Wrobel, MJ; Lerner, J; Brunet, JP; Subramanian, A; Ross, KN; et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar]
- Rho, SB; Kim, B; Kang, S. A gene signature-based approach identifies thioridazine as an inhibitor of phosphatidylinositol-3′-kinase (PI3K)/AKT pathway in ovarian cancer cells. Gynecol. Oncol 2011, 120, 121–127. [Google Scholar]
- Lomenick, B; Hao, R; Jonai, N; Chin, RM; Aghajan, M; Warburton, S; Wang, J; Wu, RP; Gomez, F; Loo, JA; et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar]
- Rix, U; Superti-Furga, G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol 2009, 5, 616–624. [Google Scholar]
- Kley, N. Chemical dimerizers and three-hybrid systems: Scanning the proteome for targets of organic small molecules. Chem. Biol 2004, 11, 599–608. [Google Scholar]
- Senderowitz, H; Marantz, YG. Protein-coupled receptors: Target-based in silico screening. Curr. Pharm. Des 2009, 15, 49–68. [Google Scholar]
- Hao, J; Ho, JN; Lewis, JA; Karim, KA; Daniels, RN; Gentry, PR; Hopkins, CR; Lindsley, CW; Hong, CC. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem. Biol 2010, 5, 245–253. [Google Scholar]
- FDA ICH Guidance for industry: S7A Safety Pharmacology Studies for Human Pharmaceuticals: US Department of Health and Human Services. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm074959.pdf (accessed on 28 March 2011).
- Milan, DJ; Jones, IL; Ellinor, PT; MacRae, CA. In vivo recording of adult zebrafish electrocardiogram and assessment of drug-induced QT prolongation. Am. J. Physiol. Heart Circ. Physiol 2006, 291, H269–H273. [Google Scholar]
- Burns, CG; Milan, DJ; Grande, EJ; Rottbauer, W; MacRae, CA; Fishman, MC. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat. Chem. Biol 2005, 1, 263–264. [Google Scholar]
- Schwerte, T; Pelster, B. Digital motion analysis as a tool for analysing the shape and performance of the circulatory system in transparent animals. J. Exp. Biol 2000, 203, 1659–1669. [Google Scholar]
- Airhart, MJ; Lee, DH; Wilson, TD; Miller, BE; Miller, MN; Skalko, RG. Movement disorders and neurochemical changes in zebrafish larvae after bath exposure to fluoxetine (PROZAC). Neurotoxicol. Teratol 2007, 29, 652–664. [Google Scholar]
- Richards, FM; Kimber, G; Butler, P; Waldron, G; Knight, T; Goldsmith, P; Fleming, AL. Prediction of ophthalmic effects in man by screening drugs for the optokinetic and optomotor responses in zebrafish. Invest. Ophthalmol. Vis. Sci 2006, 2622, 123. [Google Scholar]
- Berghmans, S; Hunt, J; Roach, A; Goldsmith, P. Zebrafish offer the potential for a primary screen to identify a wide variety of potential anticonvulsants. Epilepsy Res 2007, 75, 18–28. [Google Scholar]
- Best, JD; Berghmans, S; Hunt, JJ; Clarke, SC; Fleming, A; Goldsmith, P; Roach, AG. Non-associative learning in larval zebrafish. Neuropsychopharmacology 2008, 33, 1206–1215. [Google Scholar]
- Pack, M; Solnica-Krezel, L; Malicki, J; Neuhauss, SC; Schier, AF; Stemple, DL; Driever, W; Fishman, MC. Mutations affecting development of zebrafish digestive organs. Development 1996, 123, 321–328. [Google Scholar]
- Berghmans, S; Butler, P; Goldsmith, P; Waldron, G; Gardner, I; Golder, Z; Richards, FM; Kimber, G; Roach, A; Alderton, W; et al. Zebrafish based assays for the assessment of cardiac, visual and gut function-potential safety screens for early drug discovery. J. Pharmacol. Toxicol. Methods 2008, 58, 59–68. [Google Scholar]
- Barrett, R; Chappell, C; Quick, M; Fleming, A. A rapid, high content, in vivo model of glucocorticoid-induced osteoporosis. Biotechnol. J 2006, 1, 651–655. [Google Scholar]
- Rihel, J; Prober, DA; Arvanites, A; Lam, K; Zimmerman, S; Jang, S; Haggarty, SJ; Kokel, D; Rubin, LL; Peterson, RT; et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 2010, 327, 348–351. [Google Scholar]
- Snaar-Jagalska, BE. ZF-CANCER: Developing high-throughput bioassays for human cancers in zebrafish. Zebrafish 2009, 6, 441–443. [Google Scholar]

{kind=link}
{kind=link}
| C. elegans | D. melanogaster | D. rerio | M. musculus | |
|---|---|---|---|---|
| Generation Time | 3–5 days | 10–14 days | 3–4 months | 6–8 weeks |
| Media | Solid or liquid | Solid | Liquid | N/A |
| Ease of Obtaining Embryos | +++++ | +++++ | ++++ | N/A |
| Number of Genes | ∼19,000 | ∼13,000 | ∼25,000 | ∼25,000 |
| Homology to Human Genome | >50% | >60% | >70% | >90% |
| Annual Cost | + | + | ++ | ++++ |
| Embryo | Type of Screen | Readout |
|---|---|---|
 Wild type |  Chemical genetic |  Morphological defect |
 Disease model (eg. Blood pooling) |  Therapeutic |  Restored to wild type |
 Trangenic Embryo Tg(Flk:dsRed) |  Transgene assisted |  Altered anatomy visualized through transgenic marker |
 Transgenic Reporter Line Tg(Top:dGFP) |  Pathway Reporter inhibitor /enhancer |  Down regulated Reporter gene |
| Strategy | Advantages | Disadvantages |
|---|---|---|
| Affinity | Traditionally used and readily accepted | Requires sophisticated equipment |
| Chromatography | Low throughput | |
| Requires chemical modification | ||
| Expression Profiling | No chemical modifications | Data can be noisy |
| High throughput | Requires sophisticated bioinformatics | |
| Imprecise | ||
| Yeast 3 Hybrid | High throughput | Non native environment |
| Not suitable for membrane bound proteins | ||
| Requires chemical modifications | ||
| DARTS | No chemical modifications | Low throughput |
| Does not require high affinity |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Williams, C.H.; Hong, C.C. Multi-Step Usage of in Vivo Models During Rational Drug Design and Discovery. Int. J. Mol. Sci. 2011, 12, 2262-2274. https://doi.org/10.3390/ijms12042262
Williams CH, Hong CC. Multi-Step Usage of in Vivo Models During Rational Drug Design and Discovery. International Journal of Molecular Sciences. 2011; 12(4):2262-2274. https://doi.org/10.3390/ijms12042262
Chicago/Turabian StyleWilliams, Charles H., and Charles C. Hong. 2011. "Multi-Step Usage of in Vivo Models During Rational Drug Design and Discovery" International Journal of Molecular Sciences 12, no. 4: 2262-2274. https://doi.org/10.3390/ijms12042262
APA StyleWilliams, C. H., & Hong, C. C. (2011). Multi-Step Usage of in Vivo Models During Rational Drug Design and Discovery. International Journal of Molecular Sciences, 12(4), 2262-2274. https://doi.org/10.3390/ijms12042262