Studies of New Fused Benzazepine as Selective Dopamine D3 Receptor Antagonists Using 3D-QSAR, Molecular Docking and Molecular Dynamics

Abstract
:1. Introduction
2. Results and Discussion
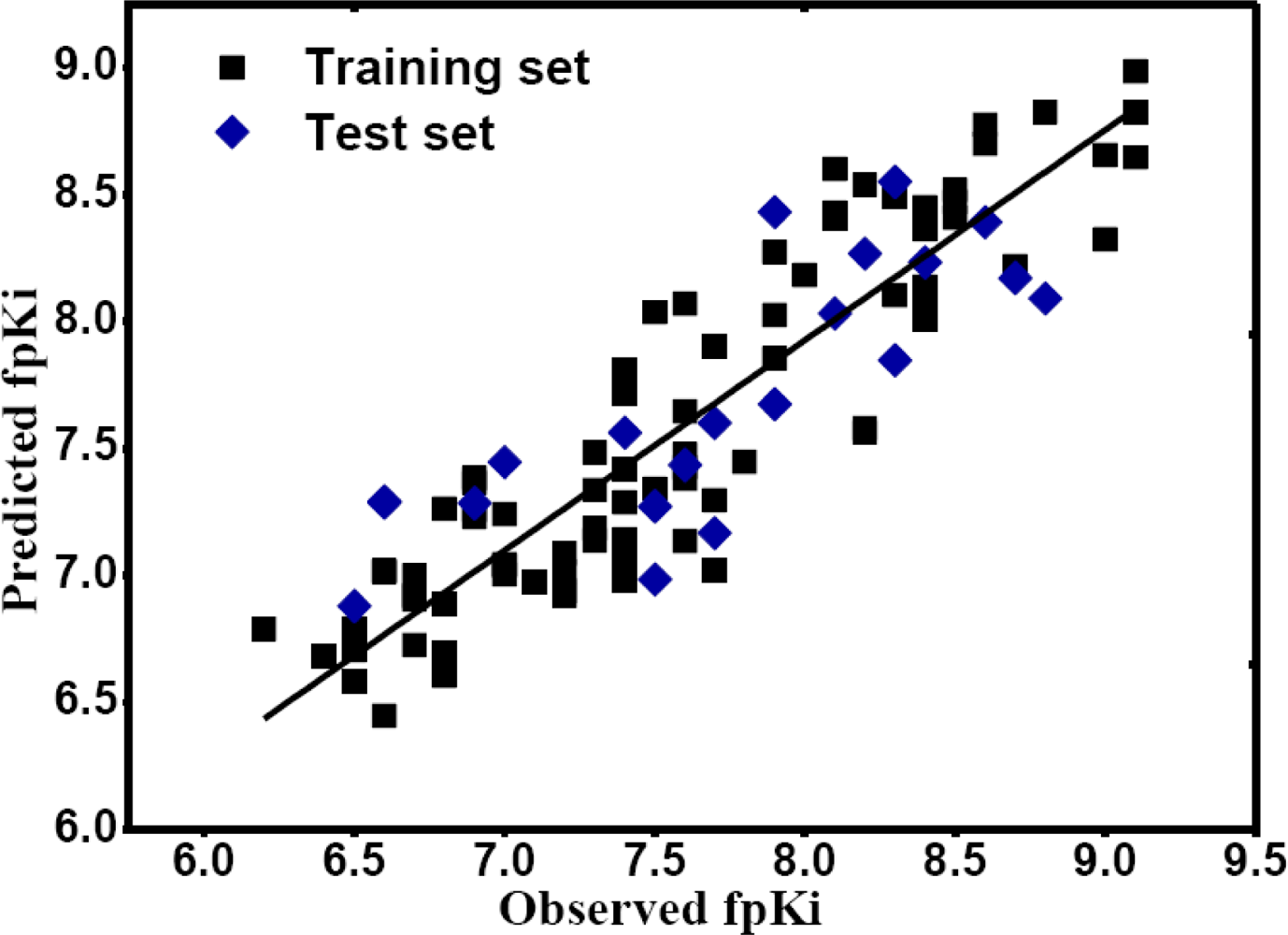
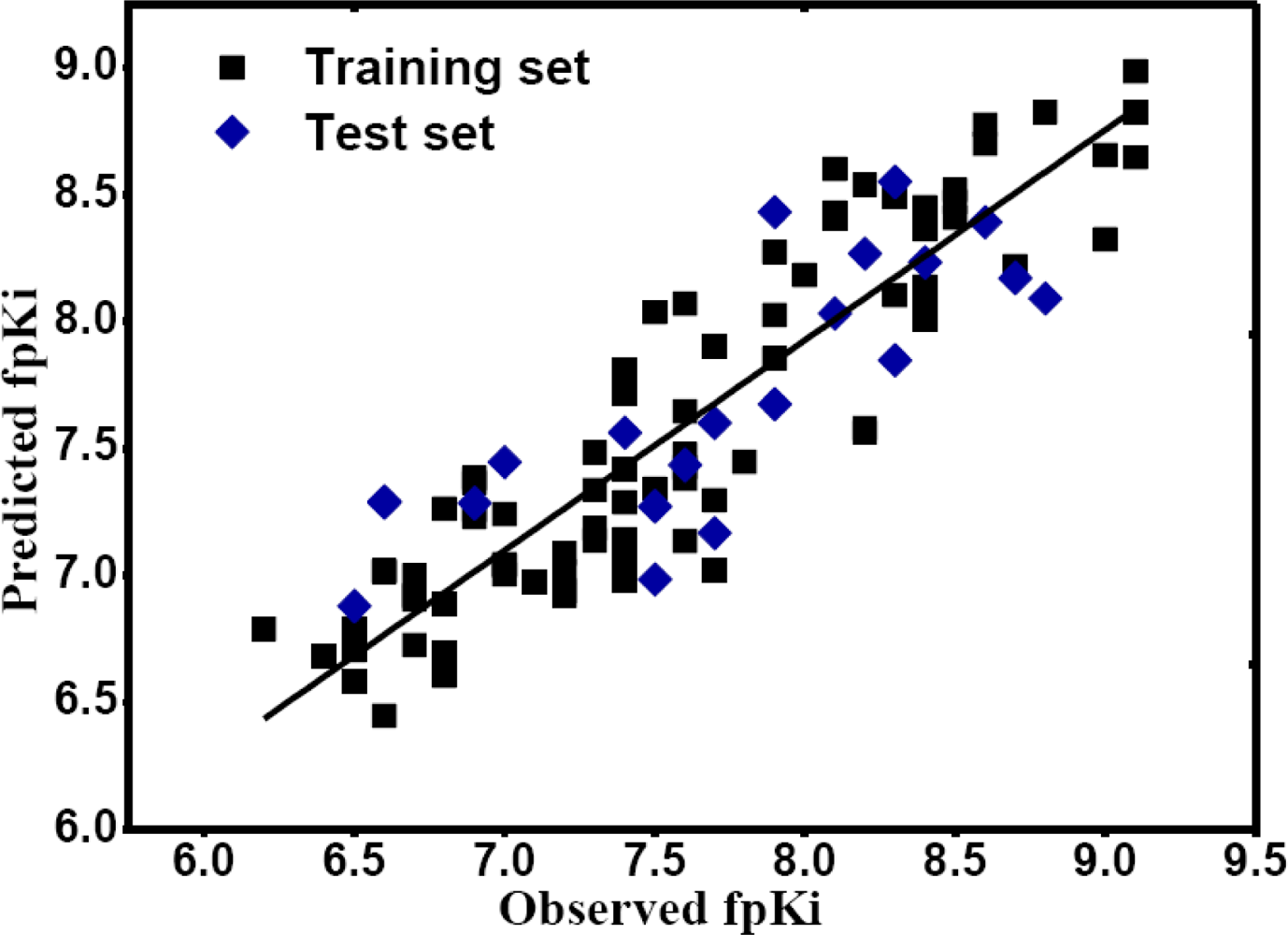
2.1. CoMFA and CoMSIA Statistical Results
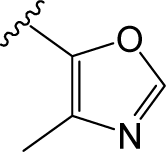 ) in skeleton type H group of compounds 76–100. This unique molecular structure may cause the larger standard deviations from the mean of the residuals for the two compounds.
) in skeleton type H group of compounds 76–100. This unique molecular structure may cause the larger standard deviations from the mean of the residuals for the two compounds.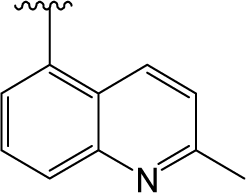 ) are always more active than those with b type of R3 group. However, 29 is an exception, exhibiting lower biological activity than corresponding molecule 30. Whether this disagreement in variance of the structure-activity is due to different binding conformation or specific interaction mechanism still needs further experiments to determine.
) are always more active than those with b type of R3 group. However, 29 is an exception, exhibiting lower biological activity than corresponding molecule 30. Whether this disagreement in variance of the structure-activity is due to different binding conformation or specific interaction mechanism still needs further experiments to determine.2.2. MlogP Contribution
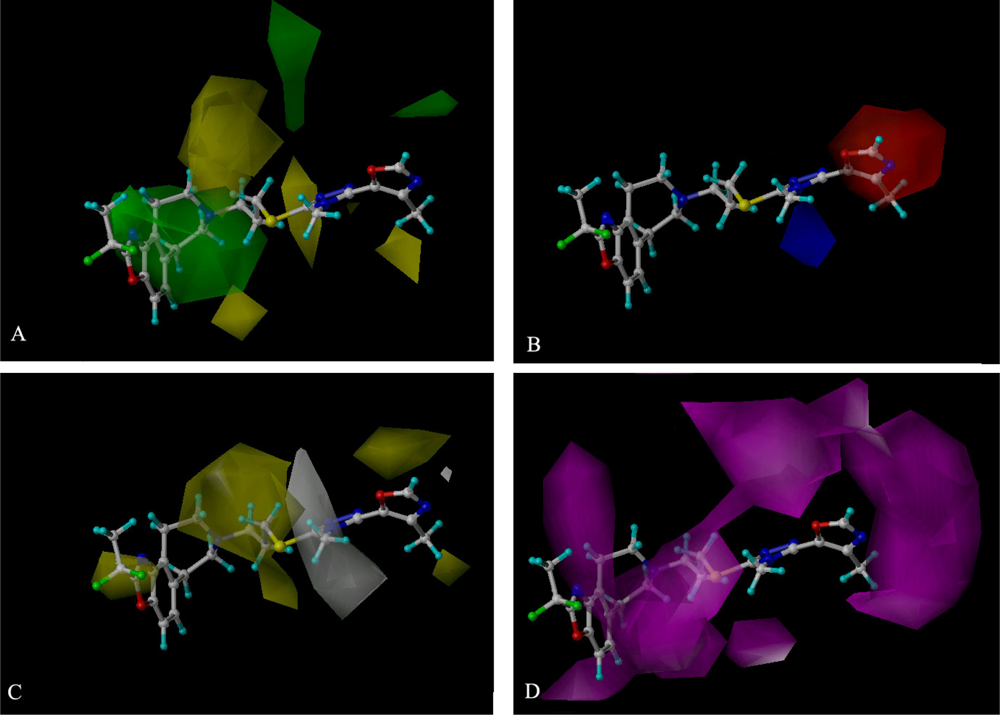
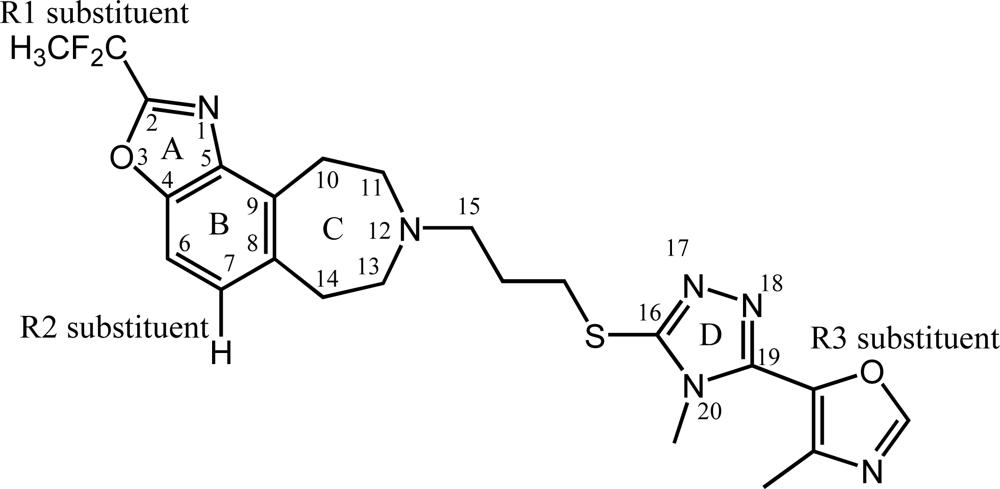
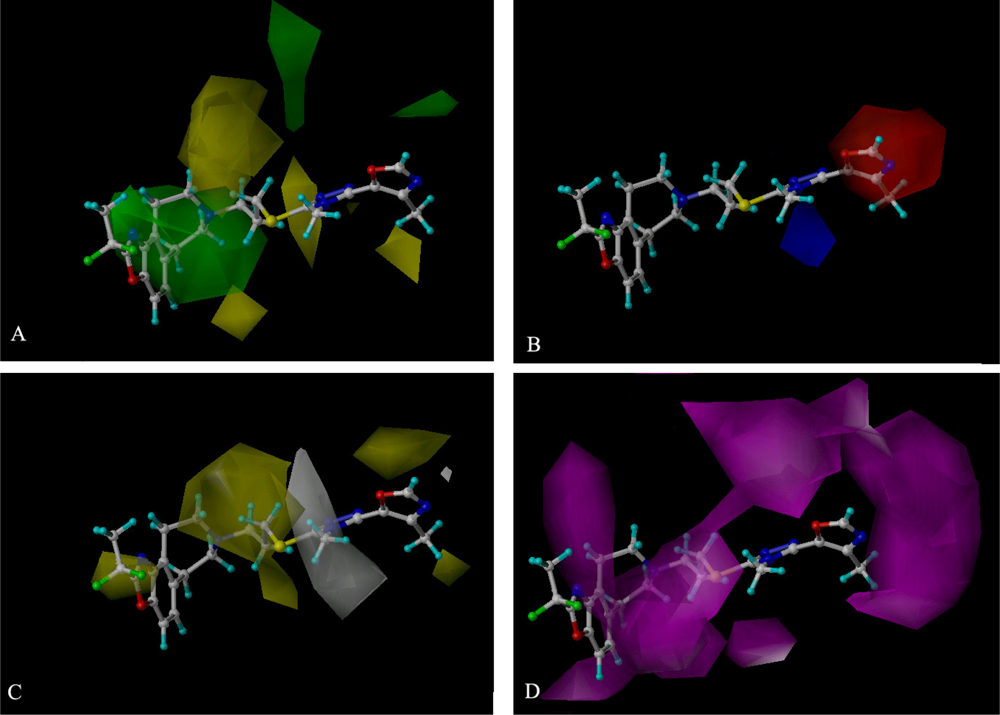
2.3. 3D-QSAR Contour Maps
2.4. CoMSIA Contour Maps Analysis
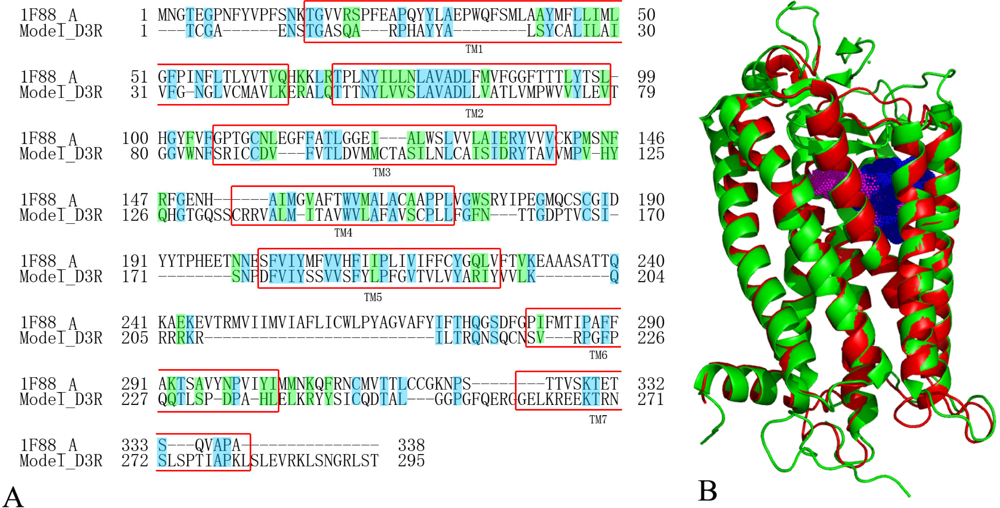
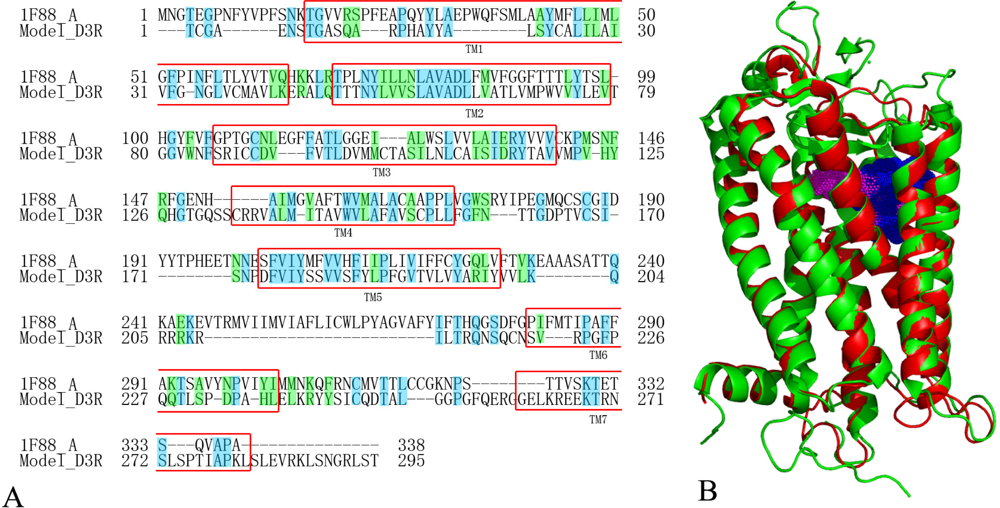
2.5. Homology Modeling Results
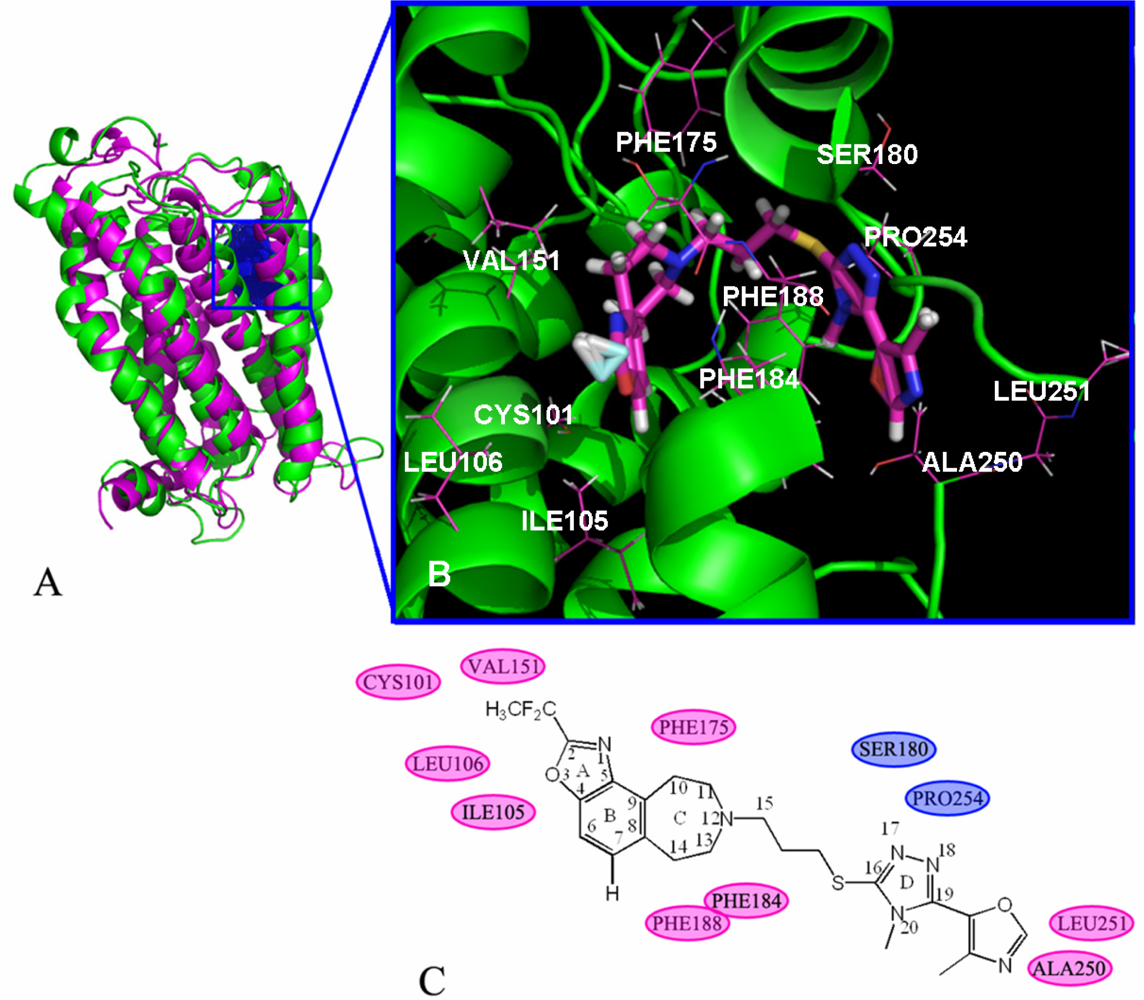
2.6. Docking Results
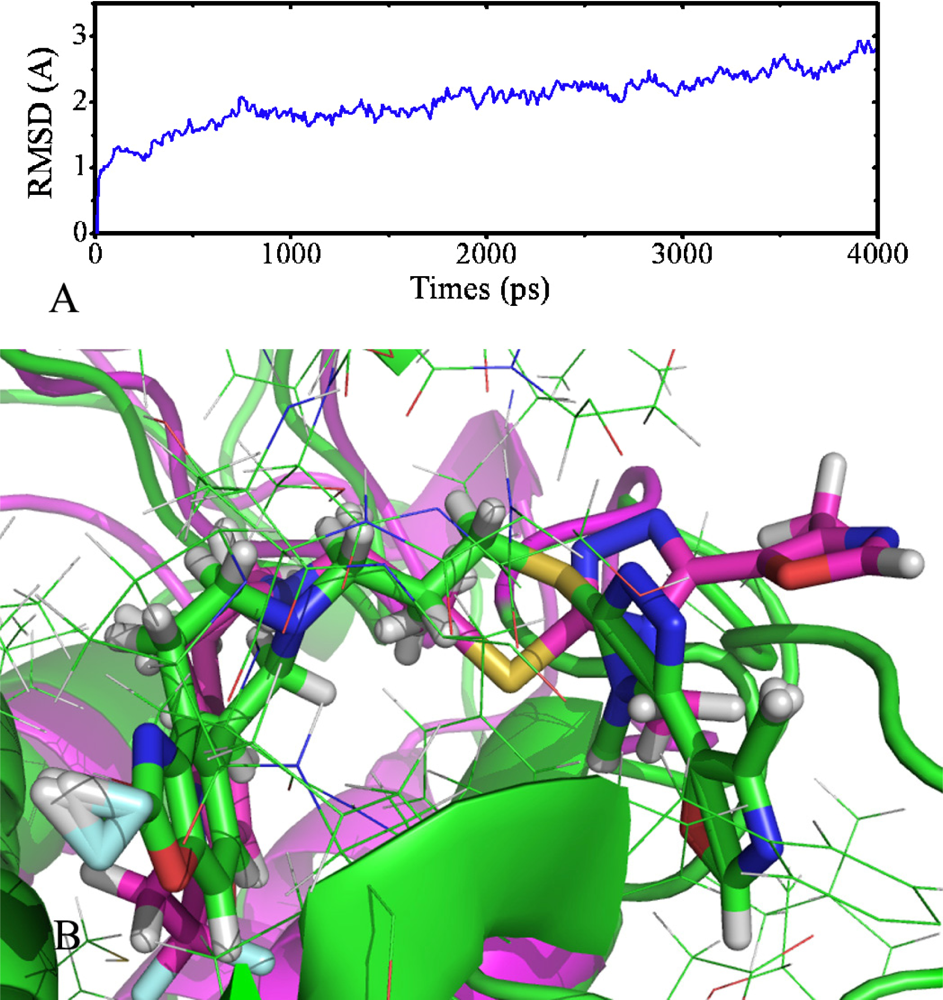
2.7. Molecular Dynamics Simulations
3. Materials and Methods
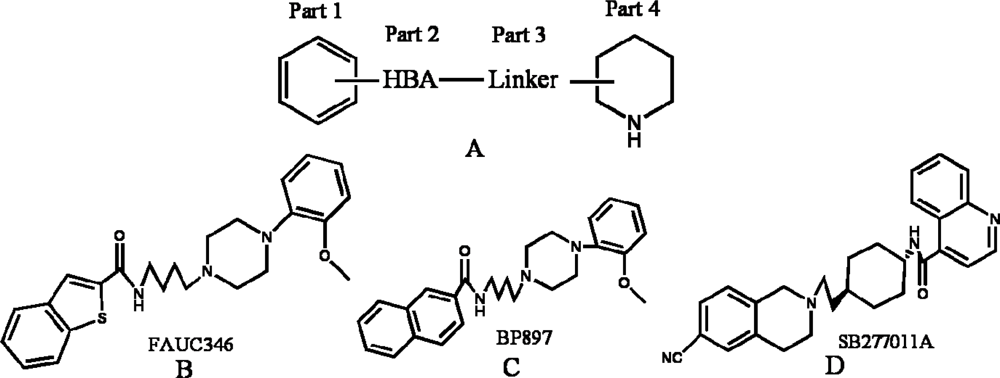
3.1. Compounds and Activity
3.2. Homology Modeling
3.3. Molecular Docking
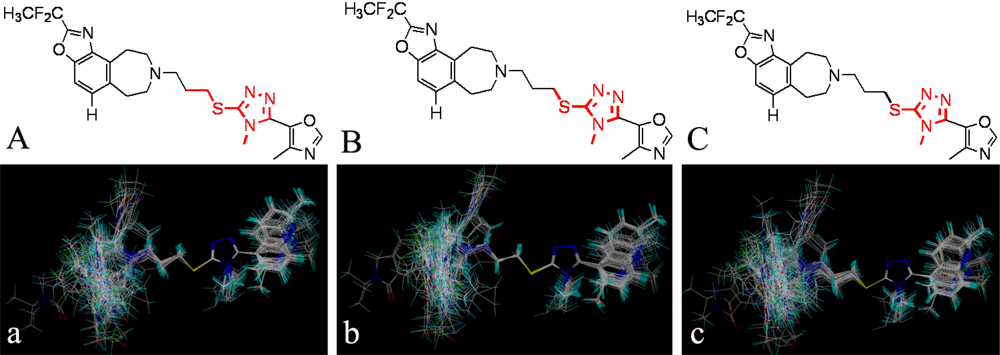
3.4. Conformational Sampling and Alignment
3.5. Descriptor Calculation
3.6. Calculation and Validation of 3D-QSAR Models
3.7. Molecular Dynamics Simulations
4. Conclusions
Acknowledgments
References
- Boeckler, F; Gmeiner, P. The structural evolution of dopamine d3 receptor ligands: Structure-activity relationships and selected neuropharmacological aspects. Pharmacol. Ther 2006, 112, 281–333. [Google Scholar]
- Sokoloff, P; Giros, B; Martres, MP; Bouthenet, ML; Schwartz, JC. Molecular cloning and characterization of a novel dopamine receptor (d3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar]
- Levesque, D. Aminotetralin drugs and d3 receptor functions. What may partially selective d3 receptor ligands tell us about dopamine d3 receptor functions? Biochem. Pharmacol 1996, 52, 511–518. [Google Scholar]
- Salama, I; Schlotter, K; Utz, W; Hübner, H; Gmeiner, P; Boeckler, F. Comfa and comsia investigations of dopamine d3 receptor ligands leading to the prediction, synthesis, and evaluation of rigidized fauc 365 analogues. Bioorg. Med. Chem 2006, 14, 5898–5912. [Google Scholar]
- Heidbreder, CA; Gardner, EL; Xi, ZX; Thanos, PK; Mugnaini, M; Hagan, JJ; Ashby, CR, Jr. The role of central dopamine d3 receptors in drug addiction: A review of pharmacological evidence. Brain Res. Rev 2005, 49, 77–105. [Google Scholar]
- Wood, MD; Boyfield, I; Nash, DJ; Jewitt, FR; Avenell, KY; Riley, GJ. Evidence for antagonist activity of the dopamine d3 receptor partial agonist, bp 897, at human dopamine d3 receptor. Eur. J. Pharmacol 2000, 407, 47–51. [Google Scholar]
- Micheli, F; Bonanomi, G; Braggio, S; Capelli, AM; Celestini, P; Damiani, F; Di Fabio, R; Donati, D; Gagliardi, S; Gentile, G; Hamprecht, D; Petrone, M; Radaelli, S; Tedesco, G; Terreni, S; Worby, A; Heidbreder, C. New fused benzazepine as selective d3 receptor antagonists. Synthesis and biological evaluation. Part one: [h]-fused tricyclic systems. Bioorg. Med. Chem. Lett 2008, 18, 901–907. [Google Scholar]
- Micheli, F; Heidbreder, C. Selective dopamine d3 receptor antagonists: A review 2001–2005. Recent Pat. CNS Drug Discov 2006, 1, 271–288. [Google Scholar]
- Bettinetti, L; Schlotter, K; Hubner, H; Gmeiner, P. Interactive sar studies: Rational discovery of super-potent and highly selective dopamine d3 receptor antagonists and partial agonists. J. Med. Chem 2002, 45, 4594–4597. [Google Scholar]
- Southam, E; Lloyd, A; Jennings, CA; Cluderay, JE; Cilia, J; Gartlon, JE; Jones, DN. Effect of the selective dopamine d3 receptor antagonist sb-277011-a on regional c-fos-like expression in rat forebrain. Brain Res 2007, 1149, 50–57. [Google Scholar]
- Micheli, F; Bonanomi, G; Braggio, S; Capelli, AM; Damiani, F; Di Fabio, R; Donati, D; Gentile, G; Hamprecht, D; Perini, O; Petrone, M; Tedesco, G; Terreni, S; Worby, A; Heidbreder, C. New fused benzazepine as selective d3 receptor antagonists. Synthesis and biological evaluation. Part 2: [g]-fused and hetero-fused systems. Bioorg. Med. Chem. Lett 2008, 18, 908–912. [Google Scholar]
- Kapetanovic, IM. Computer-aided drug discovery and development (caddd): In silico-chemicobiological approach. Chem. Biol. Interact 2008, 171, 165–176. [Google Scholar]
- Papa, E; Battaini, F; Gramatica, P. Ranking of aquatic toxicity of esters modelled by qsar. Chemosphere 2005, 58, 559–570. [Google Scholar]
- Wang, Z; Li, Y; Ai, C; Wang, Y. In silico prediction of estrogen receptor subtype binding affinity and selectivity using statistical methods and molecular docking with 2-arylnaphthalenes and 2-arylquinolines. Int. J. Mol. Sci 2010, 11, 3434–3458. [Google Scholar]
- Wang, X; Yang, W; Xu, X; Zhang, H; Li, Y; Wang, Y. Studies of benzothiadiazine derivatives as hepatitis c virus ns5b polymerase inhibitors using 3d-qsar, molecular docking and molecular dynamics. Curr. Med. Chem 2010, 17, 2788–2803. [Google Scholar]
- Ai, C; Li, Y; Wang, Y; Chen, Y; Yang, L. Insight into the effects of chiral isomers quinidine and quinine on cyp2d6 inhibition. Bioorg. Med. Chem. Lett 2009, 19, 803–806. [Google Scholar]
- Ai, C; Wang, Y; Li, Y; Yang, L. A 3-d qsar study of catechol-O-methyltransferase inhibitors using comfa and comsia. QSAR Comb. Sci 2008, 27, 1183–1192. [Google Scholar]
- Wang, Y; Li, Y; Ma, Z; Yang, W; Ai, C. Mechanism of microrna-target interaction: Molecular dynamics simulations and thermodynamics analysis. PLoS Comput. Biol 1010, 6, e1000866. [Google Scholar]
- Hansch, C; Verma, RP; Kurup, A; Mekapati, SB. The role of qsar in dopamine interactions. Bioorg. Med. Chem. Lett 2005, 15, 2149–2157. [Google Scholar]
- Artico, M; Botta, M; Corelli, F; Mai, A; Massa, S; Ragno, R. Investigation on qsar and binding mode of a new class of human rhinovirus-14 inhibitors by comfa and docking experiments. Bioorg. Med. Chem 1996, 4, 1715–1724. [Google Scholar]
- Gasteiger, J; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar]
- Del Re, G; Pullman, B; Yonezawa, T. Electronic structure of the [alpha]-amino acids of proteins: I. Charge distributions and proton chemical shifts. Biochim. Biophys. Acta 1963, 75, 153–182. [Google Scholar]
- Becker, ED; Charney, E; Anno, T. Molecular vibrations of quinones. Vi. A vibrational assignment for p-benzoquinone and six isotopic derivatives. Thermodynamic functions of p-benzoquinone. J. Chem. Phys 1965, 42, 942–949. [Google Scholar]
- Dixit, A; Kashaw, SK; Gaur, S; Saxena, AK. Development of comfa, advance comfa and comsia models in pyrroloquinazolines as thrombin receptor antagonist. Bioorg. Med. Chem 2004, 12, 3591–3598. [Google Scholar]
- Golbraikh, A; Tropsha, A. Beware of q2! J. Mol. Graph. Model 2002, 20, 269–276. [Google Scholar]
- Ashek, A; Cho, SJ. A combined approach of docking and 3d qsar study of [beta]-ketoacyl-acyl carrier protein synthase iii (fabh) inhibitors. Bioorg. Med. Chem 2006, 14, 1474–1482. [Google Scholar]
- Gilbert, KM; Boos, TL; Dersch, CM; Greiner, E; Jacobson, AE; Lewis, D; Matecka, D; Prisinzano, TE; Zhang, Y; Rothman, RB; Rice, KC; Venanzi, CA. Dat/sert selectivity of flexible gbr 12909 analogs modeled using 3d-qsar methods. Bioorg. Med. Chem 2007, 15, 1146–1159. [Google Scholar]
- Wiese, M; Pajeva, IK. Structure-activity relationships of multidrug resistance reversers. Curr. Med. Chem 2001, 8, 685–713. [Google Scholar]
- Li, Y; Wang, Y; Ling, Y; Zhang, S; Liu, C. Impact of molecular hydrophobic field on passive diffusion, p–glycoprotein active efflux, and p–glycoprotein modulation of steroids. Int. Electron. J. Mol. Des 2006, 5, 60–78. [Google Scholar]
- 2006. (accesed on 26 January 2011). Talete srl. Dragon for Windows (Software for Molecular Descriptor Calculations), Version 5.4. Available on line: http://www.Talete.Mi.It/.
- Moriguchi, I; Hirono, S; Liu, Q; Nakagome; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull 1992, 40, 127–130. [Google Scholar]
- Viswanadhan, VN; Ghose, AK; Revankar, GR; Robins, RK. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comp. Sci 1989, 29, 163–172. [Google Scholar]
- Cross, S; Cruciani, G. Molecular fields in drug discovery: Getting old or reaching maturity? Drug Discov. Today 2010, 15, 23–32. [Google Scholar]
- Nayana, MR; Sekhar, YN; Nandyala, H; Muttineni, R; Bairy, SK; Singh, K; Mahmood, SK. Insight into the structural requirements of proton pump inhibitors based on comfa and comsia studies. J. Mol. Graph. Model 2008, 27, 233–243. [Google Scholar]
- Austin, NE; Avenell, KY; Boyfield, I; Branch, CL; Hadley, MS; Jeffrey, P; Johnson, CN; Macdonald, GJ; Nash, DJ; Riley, GJ; Smith, AB; Stemp, G; Thewlis, KM; Vong, AK; Wood, M. Novel 2,3,4,5-tetrahydro-1h-3-benzazepines with high affinity and selectivity for the dopamine d3 receptor. Bioorg. Med. Chem. Lett 2000, 10, 2553–2555. [Google Scholar]
- Austin, NE; Avenell, KY; Boyfield, I; Branch, CL; Hadley, MS; Jeffrey, P; Johnson, CN; Macdonald, GJ; Nash, DJ; Riley, GJ; Smith, AB; Stemp, G; Thewlis, KM; Vong, AK; Wood, MD. Design and synthesis of novel 2,3-dihydro-1h-isoindoles with high affinity and selectivity for the dopamine d3 receptor. Bioorg. Med. Chem. Lett 2001, 11, 685–688. [Google Scholar]
- Maiti, R; van Domselaar, GH; Zhang, H; Wishart, DS. Superpose: A simple server for sophisticated structural superposition. Nucleic Acids Res 2004, 32, W590–W594. [Google Scholar]
- Hillisch, A; Pineda, LF; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar]
- Boeckler, F; Lanig, H; Gmeiner, P. Modeling the similarity and divergence of dopamine d2-like receptors and identification of validated ligand-receptor complexes. J. Med. Chem 2005, 48, 694–709. [Google Scholar]
- Ortore, G; Tuccinardi, T; Bertini, S; Martinelli, A. A theoretical study to investigate d2dar/d4dar selectivity: Receptor modeling and molecular docking of dopaminergic ligands. J. Med. Chem 2006, 49, 1397–1407. [Google Scholar]
- Varady, J; Wu, X; Fang, X; Min, J; Hu, Z; Levant, B; Wang, S. Molecular modeling of the three-dimensional structure of dopamine 3 (d3) subtype receptor: Discovery of novel and potent d3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J. Med. Chem 2003, 46, 4377–4392. [Google Scholar]
- Sudandiradoss, C; George Priya Doss, C; Rajasekaran, R; Purohit, R; Ramanathan, K; Sethumadhavan, R. Analysis of binding residues between scorpion neurotoxins and d2 dopamine receptor: A computational docking study. Comput. Biol. Med 2008, 38, 1056–1067. [Google Scholar]
- Ogata, K; Umeyama, H. An automatic homology modeling method consisting of database searches and simulated annealing. J Mol Graph Model 2000, 18, 258–272. [Google Scholar]
- Palczewski, K; Kumasaka, T; Hori, T; Behnke, CA; Motoshima, H; Fox, BA; Le Trong, I; Teller, DC; Okada, T; Stenkamp, RE; Yamamoto, M; Miyano, M. Crystal structure of rhodopsin: A g protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar]
- Lambert, C; Leonard, N; de Bolle, X; Depiereux, E. Esypred3d: Prediction of proteins 3d structures. Bioinformatics 2002, 18, 1250–1256. [Google Scholar]
- Laskowski, RA; MacArthur, MW; Moss, DS; Thornton, JM. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr 1993, 26, 283–291. [Google Scholar]
- Fratev, F; Benfenati, E. A combination of 3d-qsar, docking, local-binding energy (lbe) and grid study of the species differences in the carcinogenicity of benzene derivatives chemicals. J. Mol. Graph. Model 2008, 27, 147–160. [Google Scholar]
- Lv, Y; Yin, C; Liu, H; Yi, Z; Wang, Y. 3d-qsar study on atmospheric half-lives of pops using comfa and comsia. J. Environ. Sci 2008, 20, 1433–1438. [Google Scholar]
- Li, Y; Wang, Y-H; Yang, L; Zhang, S-W; Liu, C-H; Yang, S-L. Comparison of steroid substrates and inhibitors of p-glycoprotein by 3d-qsar analysis. J. Mol. Struct 2005, 733, 111–118. [Google Scholar]
- Xu, M; Zhang, A; Han, S; Wang, L. Studies of 3d-quantitative structure-activity relationships on a set of nitroaromatic compounds: Comfa, advanced comfa and comsia. Chemosphere 2002, 48, 707–715. [Google Scholar]
- Kovalishyn, VV; Kholodovych, V; Tetko, IV; Welsh, WJ. Volume learning algorithm significantly improved pls model for predicting the estrogenic activity of xenoestrogens. J. Mol. Graph. Model 2007, 26, 591–594. [Google Scholar]
- Thaimattam, R; Daga, P; Rajjak, SA; Banerjee, R; Iqbal, J. 3d-qsar comfa, comsia studies on substituted ureas as raf-1 kinase inhibitors and its confirmation with structure-based studies. Bioorg. Med. Chem 2004, 12, 6415–6425. [Google Scholar]
- Case, DA; Cheatham, TE, III; Darden, T; Gohlke, H; Luo, R; Merz, KM, Jr; Onufriev, A; Simmerling, C; Wang, B; Woods, RJ. The amber biomolecular simulation programs. J. Comput. Chem 2005, 26, 1668–1688. [Google Scholar]
- Wang, J; Wolf, RM; Caldwell, JW; Kollman, PA; Case, DA. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Jakalian, A; Jack, DB; Bayly, CI. Fast, efficient generation of high-quality atomic charges. Am1-bcc model: Ii. Parameterization and validation. J. Comput. Chem 2002, 23, 1623–1641. [Google Scholar]
- Duan, Y; Wu, C; Chowdhury, S; Lee, MC; Xiong, G; Zhang, W; Yang, R; Cieplak, P; Luo, R; Lee, T; Caldwell, J; Wang, J; Kollman, P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003, 24, 1999–2012. [Google Scholar]
- Ryckaert, J-P; Ciccotti, G; Berendsen, HJC. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Essmann, U; Perera, L; Berkowitz, ML; Darden, T; Lee, H; Pedersen, LG. A smooth particle mesh ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
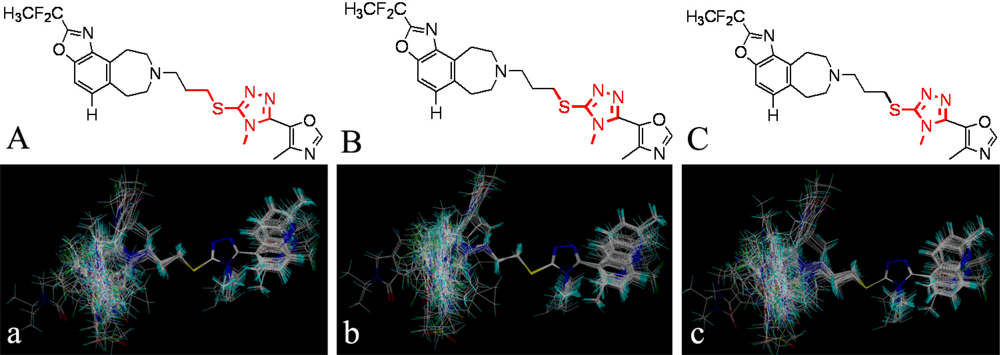
 , respectively.
, respectively.
, respectively.
, respectively.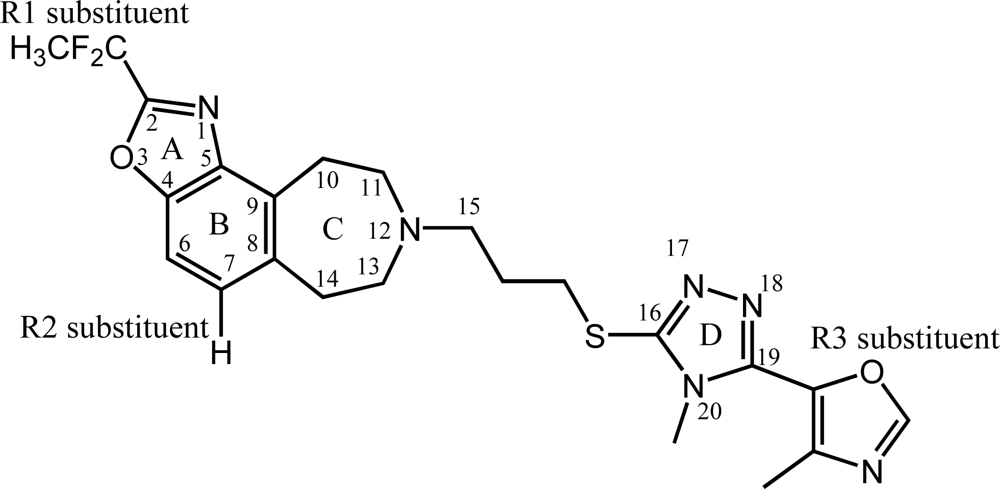




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistics | A | B | C | |||
|---|---|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Q2 | 0.371 | 0.404 | 0.364 | 0.373 | 0.506 | 0.511 |
| R2ncv | 0.805 | 0.780 | 0.815 | 0.780 | 0.838 | 0.819 |
| SEE | 0.346 | 0.366 | 0.338 | 0.366 | 0.316 | 0.331 |
| F | 43.739 | 44.388 | 46.437 | 44.427 | 54.837 | 56.746 |
| R2pre | 0.779 | 0.790 | 0.725 | 0.690 | 0.794 | 0.715 |
| SEP | 0.305 | 0.318 | 0.336 | 0.361 | 0.296 | 0.367 |
| OPN | 7 | 6 | 7 | 6 | 7 | 6 |
| Contribution: | ||||||
| Steric | 0.435 | 0.115 | 0.434 | 0.110 | 0.456 | 0.110 |
| Electrostatic | 0.501 | 0.337 | 0.502 | 0.335 | 0.481 | 0.341 |
| Hydrophobic | 0.240 | 0.238 | 0.233 | |||
| HB acceptor | 0.236 | 0.244 | 0.242 | |||
| MlogP | 0.064 | 0.072 | 0.064 | 0.072 | 0.064 | 0.072 |
| PLS Statistics | A | B | C | D | ||||
|---|---|---|---|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Q2 | 0.506 | 0.511 | 0.485 | 0.414 | 0.437 | 0.395 | 0.417 | 0.475 |
| R2ncv | 0.838 | 0.819 | 0.772 | 0.695 | 0.783 | 0.833 | 0.827 | 0.930 |
| SEE | 0.316 | 0.331 | 0.373 | 0.425 | 0.363 | 0.321 | 0.327 | 0.212 |
| F | 54.837 | 56.746 | 42.301 | 43.834 | 45.149 | 52.739 | 50.525 | 94.645 |
| R2pre | 0.794 | 0.715 | 0.712 | 0.554 | 0.735 | 0.539 | 0.791 | 0.465 |
| SEP | 0.296 | 0.367 | 0.339 | 0.451 | 0.330 | 0.459 | 0.295 | 0.527 |
| OPN | 7 | 6 | 6 | 4 | 6 | 7 | 7 | 10 |
| Contribution: | ||||||||
| Steric | 0.456 | 0.110 | 0.450 | 0.106 | 0.445 | 0.100 | 0.449 | 0.112 |
| Electrostatic | 0.481 | 0.341 | 0.485 | 0.351 | 0.493 | 0.312 | 0.493 | 0.335 |
| Hydrophobic | 0.233 | 0.228 | 0.247 | 0.256 | ||||
| HB acceptor | 0.242 | 0.226 | 0.287 | 0.249 | ||||
| MlogP | 0.064 | 0.072 | 0.065 | 0.089 | 0.063 | 0.054 | 0.058 | 0.047 |
| PLS Statistics | Ligand-based Model | Receptor-based Model | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Q2 | 0.506 | 0.511 | 0.418 | 0.603 |
| Q2cv(10) | 0.493 | 0.343 | 0.388 | 0.599 |
| R2ncv | 0.838 | 0.819 | 0.856 | 0.829 |
| R2boot | 0.892 | 0.872 | 0.899 | 0.882 |
| SEE | 0.316 | 0.331 | 0.292 | 0.316 |
| F | 54.837 | 56.746 | 114.875 | 125.886 |
| R2pre | 0.794 | 0.715 | 0.481 | 0.690 |
| SEP | 0.296 | 0.367 | 0.548 | 0.406 |
| OPN | 7 | 6 | 4 | 3 |
| Q2scrambling | −0.526 | −0.498 | 0.471 | 0.521 |
| CSDEP | 0.972 | 0.957 | 0.559 | 0.528 |
| dq2/dr2yy | −0.145 | 0.046 | 1.048 | 0.678 |
| Contribution: | ||||
| Steric | 0.456 | 0.110 | 0.433 | 0.121 |
| Electrostatic | 0.481 | 0.341 | 0.546 | 0.383 |
| Hydrophobic | 0.233 | 0.181 | ||
| HB acceptor | 0.242 | 0.294 | ||
| MlogP | 0.064 | 0.072 | 0.022 | 0.022 |
| PLS Statistics | Ligand-based Model | Receptor-based Model | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Q2 | 0.351 | 0.443 | 0.366 | 0.576 |
| R2ncv | 0.792 | 0.838 | 0.923 | 0.892 |
| SEE | 0.356 | 0.314 | 0.213 | 0.251 |
| F | 47.659 | 64.772 | 231.880 | 215.375 |
| R2pre | 0.668 | 0.471 | 0.416 | 0.604 |
| SEP | 0.384 | 0.537 | 0.606 | 0.475 |
| OPN | 6 | 6 | 4 | 3 |
| Contribution: | ||||
| Steric | 0.510 | 0.122 | 0.486 | 0.140 |
| Electrostatic | 0.490 | 0.318 | 0.514 | 0.367 |
| Hydrophobic | 0.288 | 0.205 | ||
| HB acceptor | 0.272 | 0.287 | ||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Li, Y.; Zhang, S.; Xiao, Z.; Ai, C. Studies of New Fused Benzazepine as Selective Dopamine D3 Receptor Antagonists Using 3D-QSAR, Molecular Docking and Molecular Dynamics. Int. J. Mol. Sci. 2011, 12, 1196-1221. https://doi.org/10.3390/ijms12021196
Liu J, Li Y, Zhang S, Xiao Z, Ai C. Studies of New Fused Benzazepine as Selective Dopamine D3 Receptor Antagonists Using 3D-QSAR, Molecular Docking and Molecular Dynamics. International Journal of Molecular Sciences. 2011; 12(2):1196-1221. https://doi.org/10.3390/ijms12021196
Chicago/Turabian StyleLiu, Jing, Yan Li, Shuwei Zhang, Zhengtao Xiao, and Chunzhi Ai. 2011. "Studies of New Fused Benzazepine as Selective Dopamine D3 Receptor Antagonists Using 3D-QSAR, Molecular Docking and Molecular Dynamics" International Journal of Molecular Sciences 12, no. 2: 1196-1221. https://doi.org/10.3390/ijms12021196
APA StyleLiu, J., Li, Y., Zhang, S., Xiao, Z., & Ai, C. (2011). Studies of New Fused Benzazepine as Selective Dopamine D3 Receptor Antagonists Using 3D-QSAR, Molecular Docking and Molecular Dynamics. International Journal of Molecular Sciences, 12(2), 1196-1221. https://doi.org/10.3390/ijms12021196