1. Introduction
In recent days, density functional theory (DFT) has been enjoying tremendous success in electronic structure calculations for molecules and solids alike [
1–
8]. The DFT methods are able to describe the electronic structure of these systems with accuracies comparable to traditional correlated molecular orbital methods at a decreased computational cost. Furthermore these techniques are observed to assign more bonding character to the Lewis system in which the nucleophilic reaction occurs [
9]. The DFT-based global and local properties (namely, DFT descriptors), such as Fukui functions, global and local hardness or softness [
10,
11], have already been used for reliable predictions in various types of electrophilic and nucleophilic reactions on a diversity of material structures [
1–
9,
12–
16]. In some sense, the DFT-descriptors provide us with more rigorous alternatives than the classical frontier orbital analysis. Chatterjee’s group have already used the DFT-descriptors for predictions in electrophilic and nucleophilic reactions in the case of zeolites and clay materials with or without solvent environment [
1–
7].
On the other hand, the bromine, chlorine, and fluorine oxides are known to be important in lower stratospheric ozone depletion, and have been the subjects of intense studies in recent years [
17–
26, and references cited therein]. Relevant bromine oxide fluorides, represent intriguing ternary molecules involving covalent bond between highly electronegative atoms, possessing a large number of unpaired electrons, resulting in strong lone pair-lone pair repulsions. Therefore, the hypervalent structures of these species could be characterized. As early as 1972, Johnson
et al. [
27] reported the thermodynamic properties of Br(VII) FBrO
3 species. In 1976, Appelman
et al. [
28] characterized the molecular structure of gaseous perbromyl fluoride (FBrO
3), and Gillespie and Spekkens [
29] prepared and characterized potassium difluorodioxobromate (BrO
2F
2−) and tetrafluoro-oxobromate (BrOF
4−). In 1978, Christe
et al. [
30] reported the vibrational frequencies and assignment of BrOF
3. In 2005, Lehmann
et al. [
31] reported synthesis and characterization of salts containing the bromine (VII) BrO
3F
2− anion; last year, Lehmann
et al. [
32] also reported the characterization of BrO
3F and ClO
3F to [XO
2][SbF
6] (X = Cl, Br) by single crystal X-ray diffraction, raman spectroscopy, and computational methods. The results showed that of a few computational methods, the DFT functional, B3LYP in combination with the aug-cc-pVTZ basis set, and the QCISD and CCSD(T) calculations provided the most reliable correlation with the experimental geometry and vibrational frequencies of BrO
2+ [
33] and likely provide reliable estimates of the geometric parameters and vibrational frequencies of BrO
3+, as well as benchmarks for calculations involving bromine fluoride and oxide fluoride species [
33]. Correspondingly, the density functional theory (DFT) in conjunction with DZP++ basis set has also localized these Br-hypervalent ternary structures to be minimum on the potential energy surfaces (PES) [
34,
35]. The planar/lineaer FBrO/FBrO-, pseudo-trigonal bipyramid F(F
2)Br=O (C
s symmetric) [
34] and [F-(:BrO
2)-F]
− (C
2v) anionic [
29], and quasi-octahedral (OBr-F
4)
− (C
4v) [
34,
29] Br(V) structures have been found to be the lowest-lying isomers. However, the hypervalent FBrO
2, FBrO
3 [
35], and their corresponding anionic isomers are local minima on the PES. These DFT methods, especially the hybrid DFT methods (BHLYP and B3LYP) are reliable to predict the bond lengths and bond angles [
32]. Besides the rich fluoride chemistry of the III and V oxidation states of Br oxides, the fluoride ion transfer reactions containing Br(VII) are scarce and have only been established by the syntheses of the ternary bromine oxide fluorides, BrO
3F
2− [
31]. In this work, we report the systemic theoretical investigation of the similar BrO
4F/BrO
4F
− species, which may be of importance in atmospheric chemistry.
DFT/DZP++ scheme has been shown to be successful in prediction of electron affinities (EAs) of many species, such as BrOF
n/BrOF
n−, FBrO
2/FBrO
3, Br
2O
n/Br
2O
n−, BrClF
n/BrClF
n and SF
5O
n/SF
5O
n− (n = 1–3) species [
34–
38]. These studies and others have demonstrated that the DFT/DZP++ methods can predict electron affinities (EAs) in a good accuracy [
39]. In addition, these methods are reliable for the geometry optimization of the neutral radicals and their anion.
The aim of the present work is to apply five DFT methods to predict the electron affinities of ternary bromine oxide fluoride, BrO
4F, as well as the equilibrium geometries, harmonic vibrational frequencies, and bond dissociation energies. Four forms of the electron affinities are calculated, evaluated as the neutral–anion energy separations in the following manners. The adiabatic electron affinities (EA
ad) are determined by, EA
ad = E
(optimized neutral) – E
(optimized anion), zero-point corrected adiabatic electron affinities (EA
zero) are determined by, EA
zero = E
(zero-point corrected neutral) – E
(zero-point corrected anion), the vertical electron affinities (EA
vert) by, EA
vert =
E(optimized neutral) –
E(anion at optimized neutral geometry), and the vertical detachment energies (VDE) of the anion by, VDE =
E(neutral at optimized anion geometry) –
E(optimized anion). The DFT descriptors, such as Fukui functions, global and local hardness or softness [
10,
11], also have been used for the reliable predictions in the stability of BrO
4F isomers.
4. Results and Discussion
With the present five DFT methods, the optimized O-F bond length for single OF molecule ranges from 1.331 Å (BHLYP) to 1.385 Å (BLYP) (not shown). The trend of bond lengths predicted for O-F is BHLYP < B3P86 < B3LYP < BP86 < BLYP. The DZP++ B3LYP method gives the result closest to the experimental O-F bond length (r
e) of 1.3541 Å, obtained from Raman spectroscopy [
18 and references cited therein]. The B3LYP method also obtain the best prediction result for dissociation energy (D
e) of OF [
23] and BrO [
21]. For a discussion of the reliability of B3LYP thermochemistry, see the recent work of Boese, Martin, and Handy [
54]. Therefore, in the following discussion, unless otherwise stated, we use the B3LYP result for molecular structures and energetics.
For neutral BrO4F species, the molecular chain FBr…OO…OO structure with a terminal F-Br moiety connected by OO…OO chain lies the lowest energetically. This structure in its 5A′ state (all of the five DFT methods) or 3A′ state (both BP86 and BLYP pure DFT methods) corresponds a very loose van der Waals complex between BrF…OO and O2, possessing a binding energy of about zero and the very long Br…O (2.719–3.004 Å in 5A′ state and 2.618, 2.719 Å in 3A′ state) and (O)O…O(O) (5.220–7.095 Å in 5A′ state, and 5.746, 6.014 Å in 3A′ state) distances (not shown). It is favorable to dissociate into BrF + 2O2 (3Σg−) or BrF + O2 (1Δg) + O2 (3Σg−).
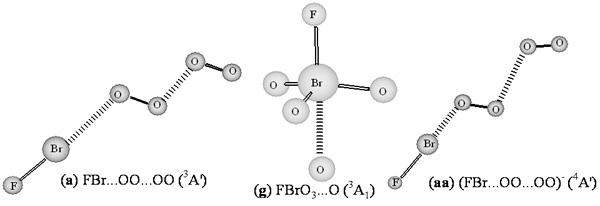
The FBr…OO…OO structures in
3A′ state (
a: 3A′) optimized by three hybrid DFT methods (BHLYP, B3P86 and B3LYP) and in
1A′ (
b: 1A′) (BHLYP) or
1A (
b: 1A) state (with the rest four DFT methods) are reported in
Figure 1. The optimized geometries for both Br- and F-terminal structures, including cis- and trans- BrOO…OOF (
c: 1A and
d: 1A), and BrOO(O)…OF (
e: 1A), and those of Br-hypervalent structures, O
2Br…OOF (
f: 1A) and FBrO
3...O (
g: C
3v,
3A
1) are also displayed in
Figure 1. The optimized geometries for anionic BrO
4F
− species, including (FBr…OO)
−…OO (
aa:
4A′) chain, and [FBr(O
2)...OO]
− (
ab:
2A′), (FO…BrO
3)
− (
ac:
2A′) Br-hypervalent structures are shown in
Figure 2. They may represent an important intermediate in atmospheric reactions.
The calculated energies (
Table 1) show that the FBr…OO…OO structure in its
5A′ state or its dissociation products (FBr...OO (
3A″) + O
2 (
3Σ
g−)) lies lower than the corresponding
3A′ (
a) and
1A′ or
1A (
b) states by about 33 and 60 kcal/mol respectively with the B3LYP method. This state also lies much lower than the cis-, trans- BrOO…OOF (
c: 1A and
d: 1A) and BrOO
2…OF (
e: 1A) isomers by ca.64, 64, and 95 kcal/mol (
Table 1) respectively (B3LYP). The O
2Br…OOF (
f: 1A) and FBrO
3...O (
g: C
3v,
3A
1) Br-hypervalent structures lie much higher than the
5A′ state by ca. 78 and 130 kcal/mol (
Table 1) respectively. With a few exceptions, the two pure DFT methods (BP86 and BLYP) predict much smaller relative energies and the bond dissociation energies than three hybrid DFT methods. All these discrepancies indicate that BrO
4F is a challenging target for DFT methods.
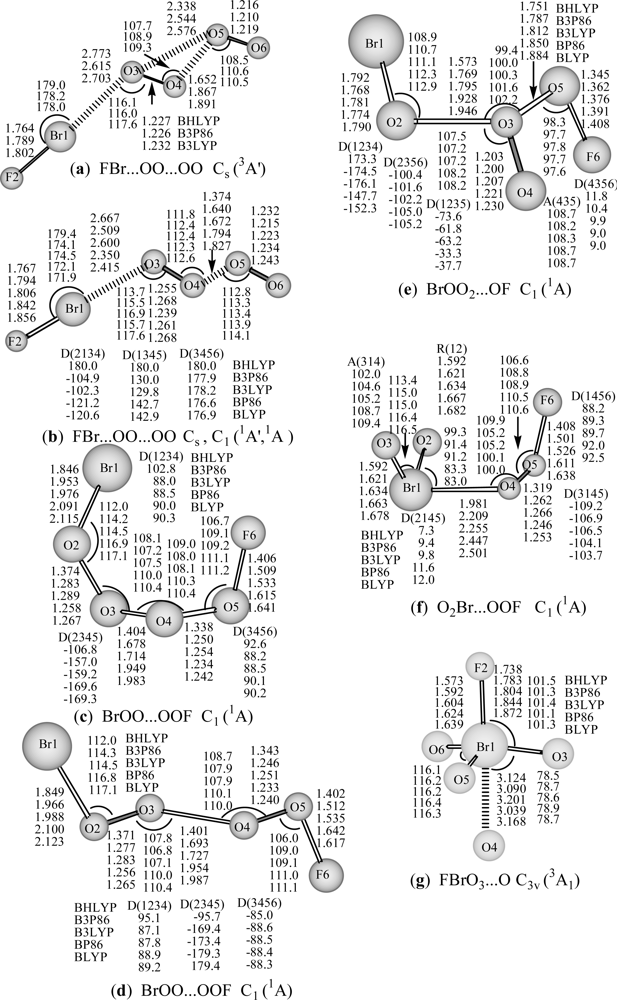
As can be seen from
Figure 1, for the FBr…OO…OO structure in its
3A′ state, the covalent bond lengths are predicted to be 1.764–1.802 Å for the Br-F bond, and 1.227–1.232 Å for interim O-O and 1.216–1.219 Å for the terminal O-O bond, and the complex bond distances are 2.615–2.773 Å for Br…O, and 1.652–1.891 Å for (O)O
…O(O) with three hybrid DFT methods. At the B3LYP level, Br-F bond length, the interim O-O and terminal O-O bond lengths in the
3A′ state (
a in
Figure 1) are 1.802, 1.232, and 1.219 Å respectively, and Br…O or (O)O
…O(O) complex distance is 2.703 or 1.891 Å respectively. These structure parameters are similar to those of the corresponding
1A′ state or
1A state of (
b in
Figure 1), in which, the Br-F bond distance, the interim O-O and terminal O-O bond lengths (
b in
Figure 1) are slightly elongated (1.806, 1.239, and 1.223 Å respectively at B3LYP level), and Br…O or (O)O
…O(O) complex distance is significantly shortened (2.600 or 1.672 Å respectively). The geometric and electronic structures show that the F-Br terminal moiety connected by OO…OO chain structure in
3A′ (hybrid DFT methods) or in singlet state may be viewed as a van der Waals complex between BrF moiety and OO-OO covalent-like chain respectively. NBO analyses (B3LYP) show that the
3A′ state possesses stronger single Br-F (WBI: 0.795 vs 0.781) and double O-O (WBI: 1.462 vs 1.436 for interim O-O; 1.524 vs 1.519 for terminal O-O) bonds than the singlet state, and that the covalent OO-OO (WBI: 0.418 vs 0.717) and complex Br…O (WBI: 0.063 vs 0.089) bonds in
3A′ state are weaker than those in singlet state. Compared with the
3A″ state of FBr...OO [
35], the Br-F and interim O-O bonds in
3A′ state of FBr
...OO-OO are slightly elongated by 0.01 Å, whereas Br
...O distance is significantly shorter by 0.26 Å (B3LYP), and the terminal O-O bond distance is very similar to that in free O
2 (
3Σ
g−) (1.194–1.245 Å) [
55].
It is worthy to note that the geometries predicted using the five functionals are all similar, with small variations in bond lengths and angles. The general trend for the covalent bond lengths is BLYP > BP86 > B3LYP > B3P86 > BHLYP. According to previous studies on geometries of BrOF
n/BrOF
n−, FBrO
2/FBrO
3, BrClF
n/BrClF
n and BrF
n species [
34,
35,
37,
56], the hybrid DFT methods (BHLYP, B3P86 or B3LYP method) are excellent methods for the prediction of covalent bond lengths. The B3LYP method taking the median position may be regarded as a compromise between the reliabilities of geometry and thermochemical parameter predictions. This order coincides with that predicted for the FO molecule [
25] where comparison with experiment indicates the B3LYP method to be the most accurate in prediction of geometry, and for BrO in predictions of bond dissociation and adiabatic electron affinity (EA
ad) [
21].
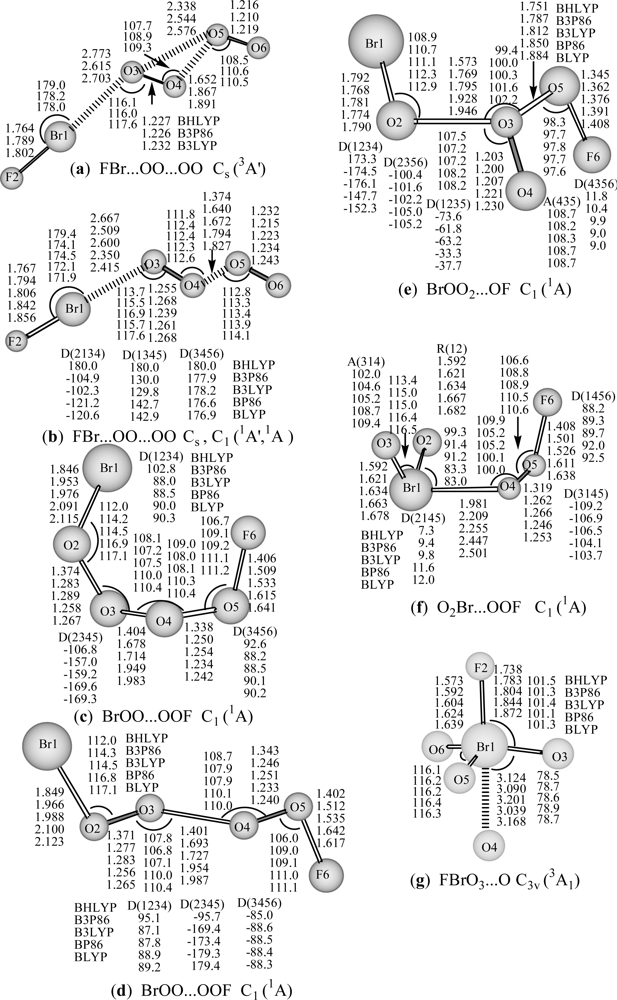
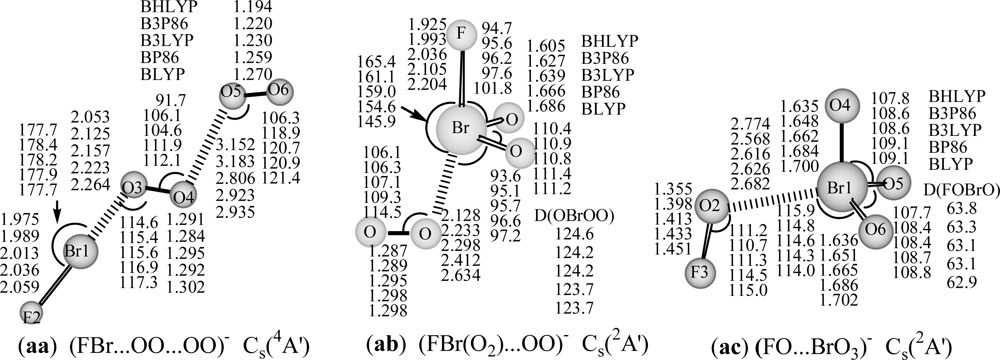
The attachment of an electron to FBr…OO…OO complex, results in the
4A′ ground state for anion (
aa:
4A′ in
Figure 2). As might be expected, this structure is more stable than other anionic BrO
4F
− Br-hypervalent structures (
ab:
2A′ and
ac:
2A′ in
Figure 2) by 32 and 78 kcal/mol at B3LYP/DZP++ level. The covalent bond lengths are predicted to be 1.975–2.059 Å for the Br-F bond, and 1.284–1.302 Å for interim O-O and 1.194–1.270 Å for the terminal O-O bond, and the complex bond distances are 2.053–2.264 Å for Br…O, and 2.806–3.183 Å for (O)O
…O(O) in the
4A′ state of BrO
4F
−. Comparison with the similar neutral isomer shows that there is a substantial change in geometry between neutral
3A′ state and anionic
4A′ state. The Br-F bond (2.013 Å at B3LYP level), the interim O-O bond (1.295 Å) and Br…O bond (2.157 Å) in anionic
4A′ state are analogous to those of (FBr-OO)
− (2.038 Å for Br-F, 1.302 Å for O-O, and 2.135 Å for Br…O) [
35]; the terminal O-O bond in the
4A′ state of BrO
4F
− (1.230 Å) is similar to that of free O
2 (1.219 Å at B3LYP level) [
55]; the (O)O
…O(O) distance of 2.806 Å in anionic BrO
4F
− is substantially longer than the corresponding (O)O
…O(O) distance (1.891 Å) in
3A′ state of neutral BrO
4F. Thus, this BrO
4F
− structure in
4A′ state could be regarded as a van der Waals complex between (FBr-OO)
− [
35] and O
2 (
3Σ
g−) due to suitable Br…O and (O)O…O(O) bonding distances, and the high negative charge of FBr-OO moiety (near to −1 from NBO analysis). Neither theoretical nor experimental values of BrO
4F/BrO
4F
− are available for comparison. For this structure in its doublet
2A′ state, the results of all five DFT methods are suspect due to the large spin contamination, with <S
2> = 1.77 or 1.76.
For the cis- and trans- BrOO…OOF (
c: 1A and
d: 1A in
Figure 1) structures, the bond lengths are calculated to be 1.846–2.123 Å for the Br-O bond (that in cis-form tinily shorter than in trans-like), 1.402–1.642 Å for the F-O bond, 1.401–1.987 Å for the central single O–O bond, and 1.233–1.374 Å for outer O–O bonds connected by Br and F. In the cis- BrOO…OOF, the O…OO fragment nearly in a planar, both BrO and FO bonds are almost perpendicular to this planar, however, in the trans- BrOO…OOF, the OO…OO chain nearly in a planar, the BrO and FO bonds are also almost perpendicular to this planar. At B3LYP level, both BrOO…OOF isomers nearly possess the same stability. This BrOO…OOF conformation could be viewed as a complex comprising of unstable BrOO and FOO molecules, furthermore, the DFT methods predict it thermodynamic instability with respect to dissociation into BrOO + FOO (not shown).
For the BrOO
2…OF (
e: 1A) structures, the bond lengths are calculated to be 1.768–1.792 Å for the Br-O bond, 1.345–1.408 Å for the F-O bond, 1.200–1.230 Å for the central double O–O bond and 1.573–1.946 Å, 1.7514–1.884 Å for outer single O–O bonds connected by Br and F, respectively. At B3LYP level, the Br-O (1.781 Å) or F-O (1.376 Å) bond is slightly longer than that in BrO [
21] or FO [
25]. Thus, this BrOO
2…OF (
e: 1A) structures could be regarded as a complex comprising of simple BrO, O
2 and FO molecules. The hybrid DFT methods predict it thermodynamic instability with respect to dissociation into BrO + O
2 + OF (not shown), whereas the pure DFT methods predict the reaction energy of about 10 (BP86) and 6 kcal/mol (BLYP) for BrOO
2…OF (
e: 1A) → BrO + O
2 + OF (not shown).
For Br-hypervalent structures: O
2Br…OOF (
f: 1A), the bond lengths are predicted to be 1.592–1.682 Å for Br-O
term (with an oxygen atom at the terminal position), 1.981–2.501 Å for Br-O
mid (with O atom at the middle position), and 1.246–1.319 Å for O-O, and 1.408–1.638 Å for F-O. The predicted Br-O
term length is comparable to that of OBrO (1.649 Å) [
21], F-O or O-O bond distances are slightly shorter or longer than those in FOO (1.649 or 1.200 Å) [
18]. This structure could be thought as a complex between BrO
2 and FOO. Likewise, the hybrid DFT methods predict it thermodynamic instability with respect to dissociation into BrO
2 + FOO (not shown), and the pure DFT methods predict the dissociation energy of O
2Br…OOF (
f: 1A) → BrO
2 + FOO reaction is about 2.3 (BP86) and 0.5 kcal/mol (BLYP) respectively (not shown).
For the rare Br(VII) FBrO
3...O (
g: C
3v,
3A
1) complex between FBrO
3 and O atom, the bond lengths are predicted to be 1.573–1.639 Å for Br-O, 1.738–1.832 Å for Br-F, and 3.039–3.168 Å for Br…O. At B3LYP level, Br-O bond long is 1.604 Å, analogous to that in BrO
4− (1.603 Å) [
21] or BrO
3F
2− (1.601 Å) [
31], and longer than that in FBrO
3 (1.582 Å) [
35], however, significantly shorter than that in BrO
3− (1.648 Å) [
21]. The Br-F bond length is 1.804 Å, being significantly shorter than that in BrO
3F
2− (1.872 or 1.849 Å) [
31], while substantially longer than that in FBrO
3 (1.708 Å) [
35]. ∠FBrO and ∠OBrO angles are 101.4 and 116.2° respectively, slightly narrower than those in FBrO
3 theoretically (101.9 and 115.9) or experimentally (103.3 and 114.9°) [
27]. Generally, the predicted lengths are comparable to those of FBrO
3 (C
3v) and BrO
3F
2− anion [
31]. The DFT methods predict the dissociation energy of FBrO
3...O (
g: C
3v,
3A
1) → FBrO
3 (C
3v) + O reaction is about 1 kcal/mol (
Table 1), demonstrating that this Br(VII) FBrO
3...O (
g: C
3v,
3A
1) hypervalent structure is bound for dissociation to FBrO
3 and O.
The corresponding anion eventually to dissociation into FBr(O
2)
−...OO (
ab:
2A′) complex structure, Br-F and Br-O bonds are elongated to be 2.036 and 1.639 Å (B3LYP), the Br…O complex distance and O-O bond length are about 2.3 and 1.30 Å. The DFT methods predict the dissociation energy of FBr(O
2)
−...OO (
ab:
2A′) → BrF
− + O
2 (
3Σ
g−) +O
2 (
1Δ
g) reaction being in the range of 7–48 kcal/mol, the BHLYP result is too small (7 kcal/mol). This is not unexpected, given the large fraction of exact exchange in the BHLYP method [
57]. For the global minimum FBr…OO…OO anion (
aa:
4A′), the predictions of five different DFT methods for the dissociation energies for
aa to dissociate to its components [FBr...OO
−(
2A″) + O
2, FBr...OO (
3A″) + O
2−, or BrF
−+ 2O
2(
3Σ
g−)] show the same trend, i.e. the pure DFT (BP86 and BLYP) methods predict higher dissociation energies than the hybrid DFT methods, and the BHLYP result is the smallest.
For the higher-lying hypervalent anionic (FO…BrO
3)
− complex structure (
ac:
2A′), the bond lengths are predicted to be 1.635–1.702 Å for Br-O bonds, 1.355–1.451Å for F-O bond, 1.355–1.451Å for F-O bond, and 2.568–2.774 Å for Br…O complex bond. The theoretical dissociation energies for (FO…BrO
3)
− → BrO
3− (C
3v) + FO is in the range of 2.8–17.9 kcal/mol (
Table 2). Likewise, the pure DFT methods predict higher dissociation energies, and the BHLYP result is the lowest.
Generally, the theoretical dissociation energies (D
e) for BrO
4F/BrO
4F
− species can be evaluated from the data in
Tables 1 and
2. For the anionic BrO
4F
− species, all of five DFT methods predict almost consistent relative energies and bond dissociation energies, with the exception of the lowest BHLYP results (
Table 2) (vide supra). In contrast, for the neutral BrO
4F species (
Table 1), the relative energies and bond dissociation energies predicted by BHLYP method are nearly the biggest. It is noted that BHLYP method perform poorly for bond-breaking process [
57] due to the large (50%) contribution from Hartree-Fock or exact exchange. Based on the previous studies of the BrO
n species [
21] and the anionic BrO
4F
− species (vide supra), the B3LYP methods should predict reasonable dissociation energies and relative energies, however, caution is urged because of the complex of BrO
4F ternary system.
At B3LYP level, for the lowest energies species, the theoretical bond dissociation energies for neutral BrO
4F refer to the reactions: BrO
4F→ BrO
4-mF + O
m (m = 1–4). For BrO
4F → BrO
2F (
3A″) [
35] + O
2, the theoretical reaction energies (ca. zero) are much smaller than those of BrO
4F→ BrO
3F (
1A’) + O (range from 84 to 109 kcal/mol, about 100 kcal/mol at B3LYP level) and BrO
4F→ BrOF (
1A’) + O
3 (range from 48 to 101 kcal/mol, ca. 71 kcal/mol at B3LYP level), indicating the dissociation reaction is favored, which is consistent with the FBr…O
2…O
2 complex structure.
The most reliable B3LYP method predicts the dissociation energy (D
e) for F-Br…O
2…O
2 (
5A′) → BrF + 2O
2 and (F-Br…O
2…O
2)
− (
4A′) → BrF
− + 2O
2 are only 0.0 and 9.1 kcal/mol, respectively (
Tables 1 and
2), suggesting a weak van der Waals interaction between the BrF or BrF
− and O
2 moieties.
For the anionic BrO
4F
− species, the D
e of BrO
4F
− → BrO
4-mF
− + O
m and BrO
4F
− → BrO
4-mF + O
m−predicted (
Table 2). The bond dissociation energies for BrO
4F
− → BrO
2F
− + O
2 are smaller positive values, from 1.0 to 1.5 kcal/mol for three hybrid DFT methods and 4.4 or 4.9 kcal/mol for BP86 or BLYP (two pure DFT) methods. The D
e values predicted by BHLYP method are too low to be reliable. The D
e value of 1.4 kcal/mol predicted by B3LYP is much smaller than those of BrO
4F
− → BrO
3F
− + O (71 kcal/mol) and BrO
4F
− → OBrF
− + O
3 (60 kcal/mol).
For BrO
4F
− → BrO
4-mF + O
m− reactions, the higher bond dissociation energies are predicted, the D
e value (58 kcal/mol) of BrO
4F
− → BrO
2F + O
2− is also smaller than those of BrO
4F
− → BrO
3F + O
− (136 kcal/mol) and BrO
4F
− → OBrF + O
3− (81 kcal/mol), and demonstrating that complex BrO
nF [
34,
35] species have higher electron affinities than the free O
m species [
55]. For the challenging BrO
mF/BrO
mF
− (m = 1–4) species, minima on PES were found with all of DFT methods employed. However, the thermodynamic stabilities decrease with n (vide supra).
The EA
ad for FBr-O
2-O
2 (a:
3A′
← aa:
4A′) are predicted to be 4.95 eV(BHLYP), 4.97 eV(B3P86), and 4.52 eV(B3LYP), zero-point corrected EA
ad (EA
zero) is only increased about 0.05 eV. At B3LYP level, EA
zero is 4.57 eV, larger than those of FBr-OOO [
35] and FBrO [
34] by about 0.1 and 1.9 eV respectively, and much smaller than those of FBr-OO by 1.3 eV (B3LYP). Those with odd n (n = 1 and 3, closed shell) have smaller EAs than those of species for the even number of n (n = 2 and 4), which are open-shell triplet state. The EA
vert values range from 2.13 to 3.55 eV. The range of VDE is from 4.49 to 4.98eV. No experimental data are available.
The harmonic vibrational frequencies and IR active intensities of BrO
4F/BrO
4F
− species predicted by B3LYP method are available in
Tables 3 and
4. For triplet state FBr...O
2...O
2 (
a) (
Cs,
3A′), the calculated infrared spectrum is characterized by three strong bands around 1561 (terminal O-O symmetri stretch(s.s.)), 1440 (middle O-O s.s.), 628 cm
−1(F-Br s.s.), all other modes give rise to weak intensities. For singlet state FBr...OOOO (b) (C
1,
1A), the bands of ca. 1508 (terminal O-O s.s.), 1391 (middle O-O s.s.), and 620 cm
−1(F-Br s.s.) possess the stronger intensities. For BrOO...OOF chain structures (
c) and (
d), the predicted infrared spectrum are characterized by three stronger bands around 1223, 1376 (F-connected O-O s.s.), 1107, 1276 (Br-connected O-O s.s.), and 720, 667 cm
−1(F-O-O bend), respectively, the rest modes yield weak intensities. For BrOO2...OF structure (e), four bands around 1209, 934, 862, and 718 cm
−1 exist the stronger intensities, the corresponding modes are O-O (O2) s.s., F-O s.s., Br-O s.s., and O...O stretch.
For O
2Br...OOF structure (
f), four bands around 1529, 1055, 732, and 618 cm
−1 possess the stronger intensities, the corresponding modes are O-O stretch, OBrO asymmetric bend, OBrO symmetric bend, and FOO bend. For the highest symmetric Br(VII) FBrO
3...O (
g), theoretical infrared spectrum are characterized by the stronger bands around θ: 955 cm
−1 (BrO
3 asymmetric stretch (a.s.)); η: 864 cm
−1 (BrO
3 symm.bend); ζ: 567 cm
−1(F-Br s.s.); ɛ: 364 cm
−1(OBrO in the planar bend); δ: 345 cm
−1 (OBrO out of planar bend), the harmonic vibrational frequencies of BrO
3 radical are larger than the corresponding BrO
3+ [
32] (966, 850, 329, and 231 cm
−1). For anionic quartet state FBr...OO...OO (
aa) (C
s,
4A′) species, four bands around 1532, 1226, 383, and 227 cm
−1 possess the stronger intensities. For anionic hypervalency structure [FO...Br(O)O
2]
− (
ac) (C
s,
2A′), four bands around 957, 812, 805, 789 cm
−1 possess the stronger intensities.
Isodesmic reactions, which have been typically used to obtain the heats of formation for many molecules, are those in which the reactants and products contain the same types of bonds, i.e., the number of bonds broken and formed is conserved [
58]. An isodesmic reaction scheme requires that the heats of formation of all the molecules involved in the reaction to be known with the exception of the heat of formation of the particular isomer. Because of this property, errors in the energy that might occur due to defects in the basis set and electron correlation cancel, to a large extent. The isodesmic scheme used here is BrOOOOF + 4HOH → 3HOOH + HOBr + HOF. During the calculation of the heat of formation of BrOOOOF using the isodesmic scheme, literature values for the heats of formation of HOH (−57.10 kcal mol
−1) [
59], HOOH (−31.02 kcal mol
−1) [
59], and HOBr (−10.93 kcal mol
−1) [
60], HOF (−22.47 kcal mol
−1) [
61], were used. Using these results we were able to calculate the heats of reaction. For cis BrOOOOF (
c), the heat of formation is predicted to be 50 kcalmol
−1 at B3LYP level of theory (
Table 5). Using the relative energies (
Table 1) along with the heat of formation of BrOOOOF (
c), we obtained a value of 19 kcal mol
−1 for FBrOOOO(
a), 83 kcal mol
−1 for BrOO2…OF (
e), 64 kcal mol
−1 for O
2Br…OOF (
f), and 116 kcal mol
−1 for FBrO
3…O (
g) (shown in
Table 6). To further assess these results, we have listed all five DFT methods heats of formation of the isomers in
Table 6. At present, there are no experimental measurements to which be mainly due to the incompleteness of the basis sets and only partial allowance for electron correlation.
For these complexes of Lewis acid (BrF) and base (lone pair O
m chains), we treated as a local version of the hard and soft acid base (HSAB) principle [
40]. The DFT-based local reactivity descriptors such as the global or local softness or hardness, condensed Fukui functions can be used to explain the stability of isomers. The predicted global hardness (
η) and softness (
GS) for the minimum-energy BrO
4F structures (
a,
b,
c,
d,
e,
f, and
g isomers) with five DFT methods are shown in
Tables 7 and
8 respectively. The local softness (S
x+ and S
x−), and ratios of them (S
x−/S
x+) for the minimum BrO
4F structures (
a,
b,
c,
d,
e,
f, and
g isomers) at the B3LYP/DZP++ level are tabulated in
Table 9. According the Pearson’s PMH suggestion [
41], the Br(VII) structure (
g) FBrO
3…O in this work has the largest global hardness (
Table 7), and smallest global softness (
Table 8), thus triplet state FBrO
3…O structure is the most stable isomer. For BrO
4F isomers, the maximum value (from 5.1 to 8.2, at B3LYP/DZZ++ level, as 8.2) of global hardness (
Table 7) set in the highest symmetric Br(VII) FBrO
3...O structure (
g), whereas the minimum value (from 2.9 to 3.2) of hardness assign to singlet BrOO...OOF isomer (
b), inversely, the isomers (
g) or (
b) possesses the smallest or largest global softness (
Table 8), respectively, namely, from 0.06 to 1.0, or from 0.16 to 0.17. For Br in the different isomers presents almost either the largest or smallest S
x−/S
x+ values (
Table 9), corresponding to different bonds stabilities. An important finding from this investigation is that Br may reveal the flexibility in which the bromine atom shares valence electrons and orbitals to form a variety of hypervalent species, even the extend hypervalent system.


{kind=link}
{kind=link}
{kind=link}
{kind=link}