Recent Advances in Synthetic Bioelastomers

Abstract
:1. Introduction
2. Requirements of Degradable Bioelastomers
3. Synthetic Bioelastomers
3.1. Biodegradable Polyurethanes
3.1.1. Biodegradable Soft Segments
3.1.2. Diisocyanates
3.1.3. Biodegradable Chain Extenders
3.1.4. Biodegradation Mechanism and Degradation Rate
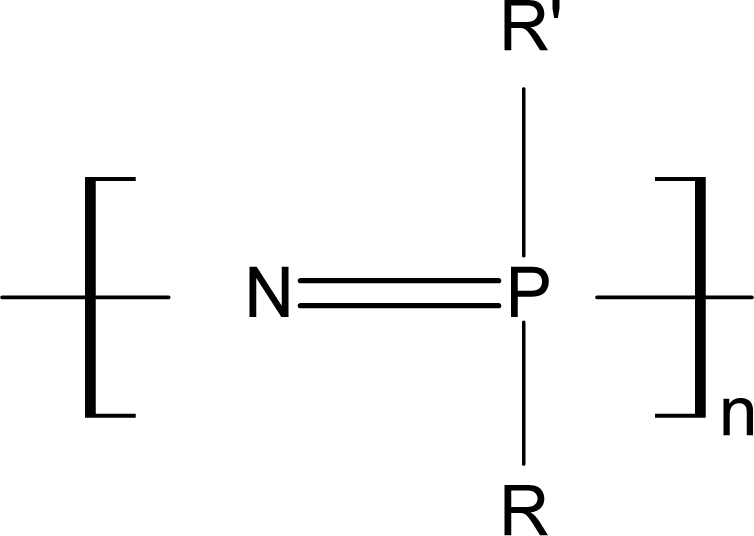
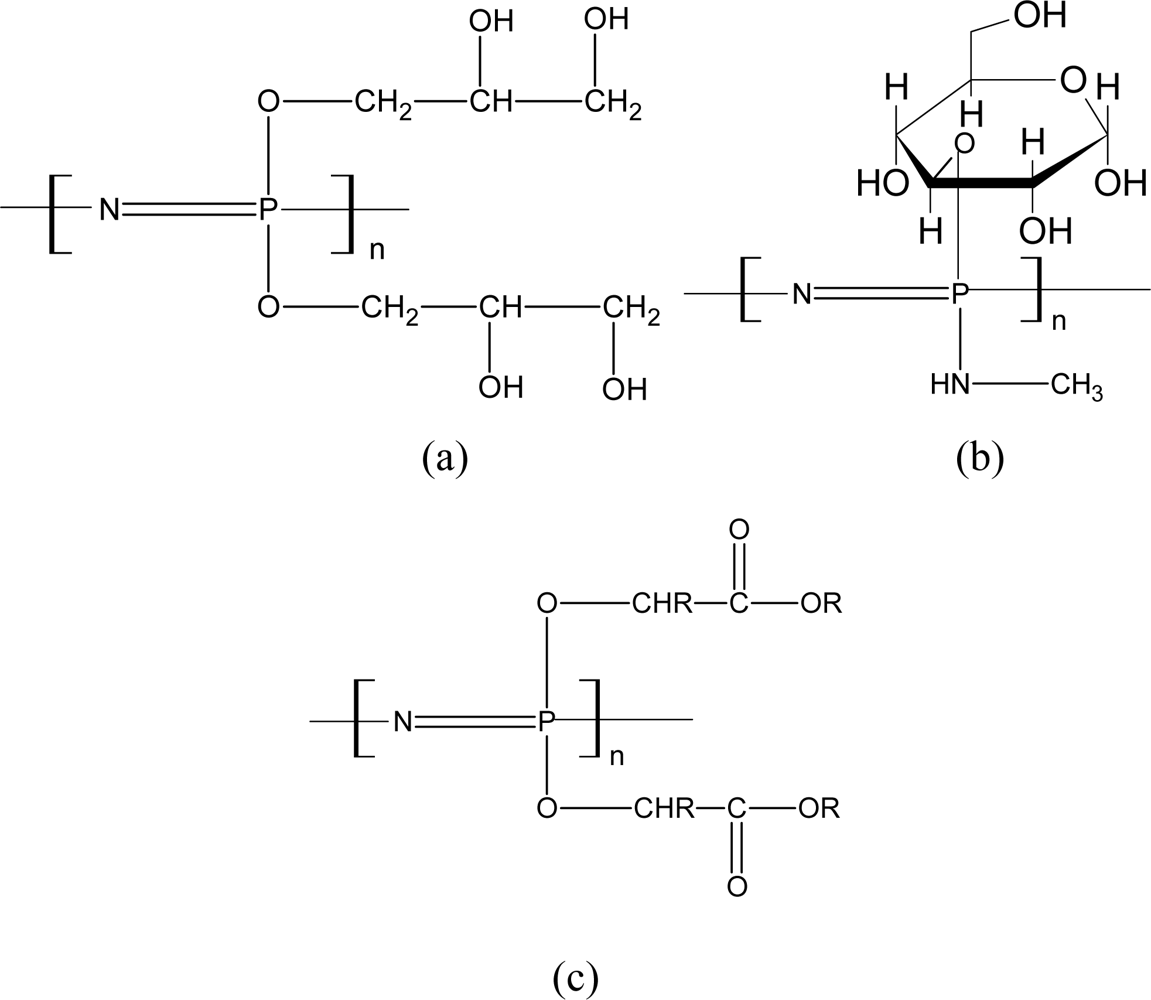
3.2. Biodegradable Polyphosphazenes
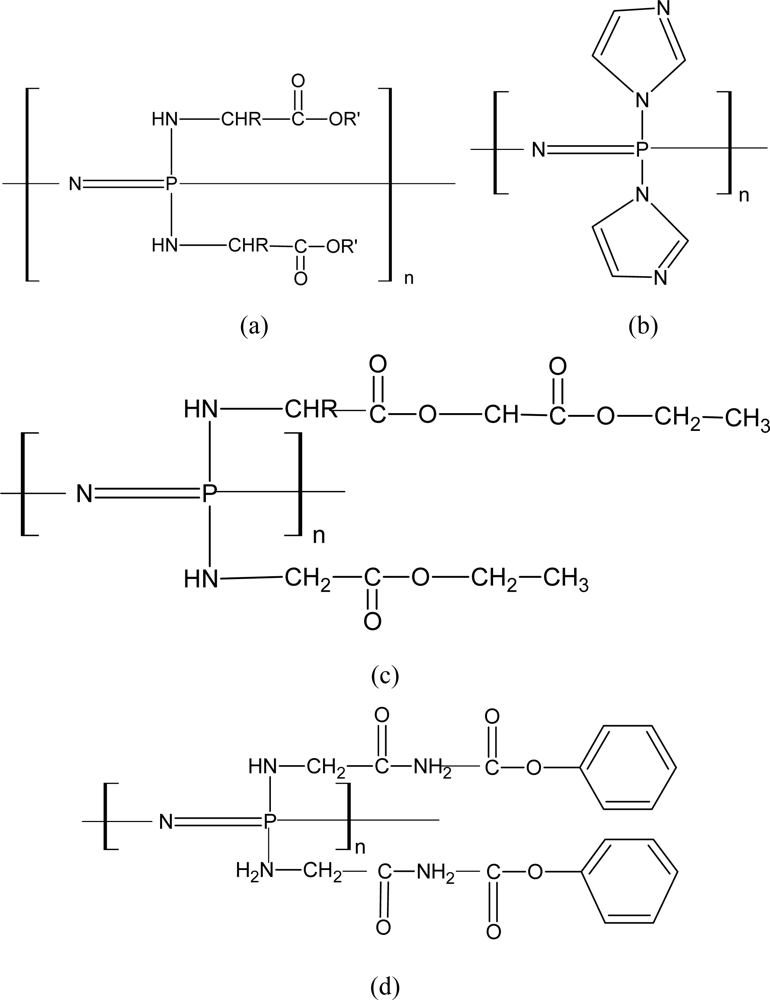
3.2.1. Aminated PNs
3.2.2. Alkoxy-Substituted PNs
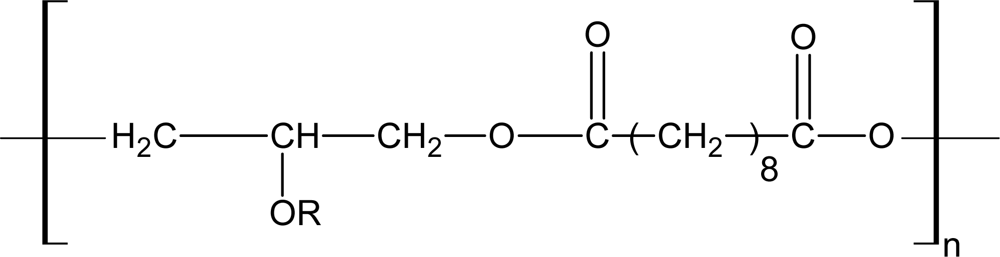
3.3. Network Polyesters Synthesized with Polyatomic Acids and Polyatomic Alcohols
3.3.1. Poly(polyol sebacate)
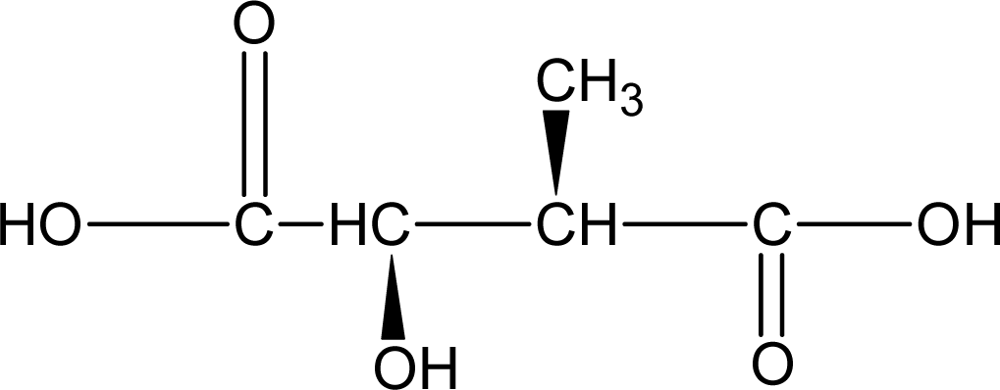
3.3.2. Poly(diol-citrates)
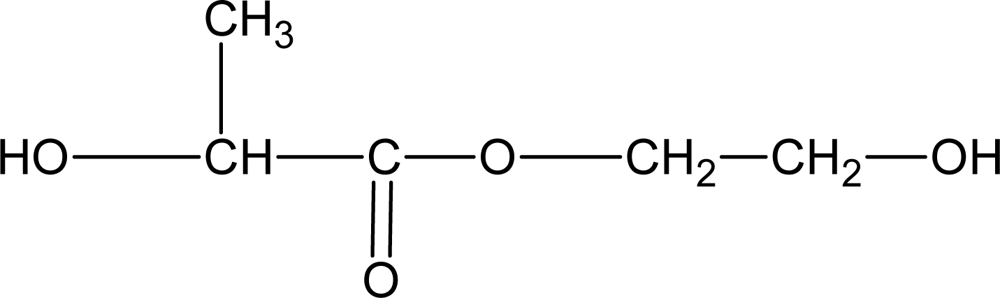
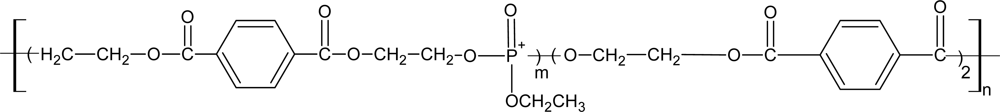
3.4. Biodegradable Poly(ether ester)
3.5. Poly(ε-caprolactone) Copolymers with Glycolide or Lactide
3.6. Poly(1,3-trimethylene carbonate) and Copolymers
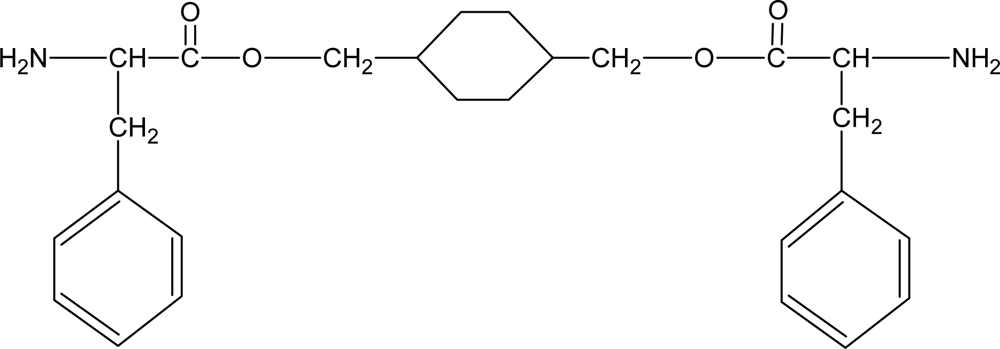
3.7. Poly(ester amide)s
4. Conclusions
Acknowledgments
References
- Wang, Y; Ameer, GA; Sheppard, BJ; Langer, R. A tough biodegradable elastomer. Nat. Biotechnol 2002, 20, 602–606. [Google Scholar]
- Tamura, T; Yamaoka, T; Kunugi, S; Panitch, A; Tirrell, Da. Effects of temperature and pressure on the aggregation of properties of an engineered elastin model polypeptide in aqueous solution. Biomacromolecules 2000, 1, 552–555. [Google Scholar]
- Karthik, N; William, T; Brinkman, BS; Thomas, JOP; Mohan, S; Elizabeth, W; Vince, PC; Elliot, LC. Viscoelastic and mechanical behavior of recombinant protein elastomers. Biomaterials 2005, 26, 4695–4706. [Google Scholar]
- O'Brien, JP; Yang, JJ. Peptide-based diblock and triblock dispersants and diblock polymers. US Pat Appl No: 20050054752 2005. [Google Scholar]
- Rachana, B; Dishma, S; Patel, KC; Trivedi, U. PHA–rubber blends: Synthesis, characterization and biodegradation. Bioresour. Technol 2008, 99, 4615–4620. [Google Scholar]
- Malafaya, PB; Silva, GA; Reis, RL. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv. Drug Deliv. Rev 2007, 59, 207–233. [Google Scholar]
- Sokolsky-Papkov, M; Agashi, K; Olaye, A; Shakesheff, K; Domb, AJ. Polymer carriers for drug delivery in tissue engineering. Adv. Drug Deliv. Rev 2007, 59, 187–206. [Google Scholar]
- Yang, J; Webb, A; Ameer, G. Biodegradable elastomeric polymers for tissue engineering. In Handbook of Biodegradable Polymeric Materials and Their Applications; Mallapragada, SK, Narasimhan, B, Eds.; American Scientific Publishers: Valencia, CA, USA, 2005; pp. 21–38. [Google Scholar]
- Hazer, B; Steinbüchel, A. Increased diversification of polyhydroxyalkanoates by modification reactions for industrial and medical applications. Appl. Microbiol. Biot 2007, 74, 1–12. [Google Scholar]
- Lenz, RW; Marchessault, RH. Bacterial polyesters: Biosynthesis, biodegradable plastics and biotechnology. Biomacromolecules 2005, 6, 1–8. [Google Scholar]
- Hazer, DB; Hazer, B; Kaymaz, F. Synthesis of microbial elastomers based on soybean oily acids. Biocompatibility studies. Biomed. Mater 2009, 4, 035011. [Google Scholar]
- Yoda, R. Elastomers for biomedical applications. J. Biomater. Sci., Polym. Ed 1998, 6, 561–526. [Google Scholar]
- Gopferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar]
- Langer, R; Peppas, NJ. Chemical and physical structure of polymers as carriers for controlled release of bioactive agents: A review. Macromol. Chem. Phys 1983, C23, 61. [Google Scholar]
- Gorna, K; Gogolewski, S. Novel biodegradable polyurethanes for biomedical applications. In Synthetic Bioabsorbable Polymers for Implants; Agrawal, CM, Parr, JE, Lin, ST, Eds.; ASTM International: West Conshohocken, PA, USA, 2000; Volume 1, p. 39. [Google Scholar]
- Tatai, L; Moore, TG; Adhikari, R; Malherbe, F; Jayasekara, R; Griffiths, I; Gunatillake, PA. Thermoplastic biodegradable polyurethanes: The effect of chain extender structure on properties and in-vitro degradation. Biomaterials 2007, 28, 5407–5417. [Google Scholar]
- Schacht, E; Vandorpe, J; Dejardin, S; Lemmouchi, Y; Seymour, L. Biomedical applications of degradable polyphosphazenes. Biotechnol. Bioeng 1996, 52, 102–108. [Google Scholar]
- Honarkar, H; Rahimi, A. Applications of inorganic polymer materials, III: Polyphosphazenes. Monatsh. Chem. Chem. Month 2007, 138, 923–933. [Google Scholar]
- Wang, Y; Ameer, GA; Sheppard, BJ; Langer, R. A tough biodegradable elastomer. Nat. Biotechnol 2002, 20, 602–606. [Google Scholar]
- Wang, Y; Kim, YM; Langer, R. In vivo degradation characteristics of poly(glycerol sebacate). J. Biomed. Mater. Res. A 2003, 66, 192–197. [Google Scholar]
- Yang, J; Webb, AR; Ameer, GA. Novel citric acid-based biodegradable elastomers for tissue engineering. Adv Mater 2004, 16, 511–516. [Google Scholar]
- Yang, J; Webb, AR; Pickerill, SJ; Hageman, G; Ameer, GA. Synthesis and evaluation of poly(diol citrate) biodegradable elastomers. Biomaterials 2006, 27, 1889–1898. [Google Scholar]
- Deschamps, AA; Claase, MB; Sleijster, WJ; Bruijn, JD; Grijpma, DW; Feijen, J. Design of segmented poly(ether ester) materials and structures for the tissue engineering of bone. J. Control. Release 2002, 78, 175–186. [Google Scholar]
- Webb, AR; Yang, J; Ameer, GA. Biodegradability polyester elastomers in tissue engineering. Exp. Opin. Biol. Ther 2004, 4, 801–812. [Google Scholar]
- Lee, SH; Kim, BS; Kim, SH. Elastic biodegradable poly(glycolide-co-caprolactone) scaffold for tissue engineering. J. Biomed. Mater. Res 2003, 66A, 29–37. [Google Scholar]
- Pěgo, AP; Poot, AA; Grijpma, DW; Feijen, J. Biodegradable elastomeric scaffolds for soft tissue engineering. J. Control. Release 2003, 87, 69–79. [Google Scholar]
- Pêgo, AP; van Luyn, MJA; Brouwer, LA; van Wachem, PB; Poot, AA; Grijpma, DW; Feijen, J. In vivo behaviour of poly(1,3-trimethylene carbonate) and copolymers of 1,3-trimethylene carbonate with d,l-lactide or ε-caprolactone: Degradation and tissue response. J. Biomed. Mater. Res 2003, 67A, 1044–1054. [Google Scholar]
- Pego, AP; Poot, AA; Grijpma, DW; Feijen, J. Physical properties of high molecular weight 1,3-trimethylene carbonate and d,l-lactide copolymers. J. Mater. Sci.—Mat. Med 2003, 14, 767–773. [Google Scholar]
- Tsitlanadze, G; Kviria, T; Katsarava, R; Chu, CC. In vitro enzymatic biodegradation of amino acid based poly(ester amide)s biomaterials. J. Mater. Sci.—Mat. Med 2004, 15, 185–190. [Google Scholar]
- Santerre, JP; Woodhouse, K; Laroche, G; Labow, RS. Understanding the biodegradation of polyurethanes: From classical implants to tissue engineering materials. Biomaterials 2005, 26, 7457–7470. [Google Scholar]
- Saad, B; Ciardelli, G; Matter, S; Welti, M; Uhlschmid, GK; Neuenschwander, P; Suter, UW. Degradable and highly porous polyestherurethane foam as biomaterial: Effects and phagocytosis of degradation products in osteoblasts. J. Biomed. Mater. Res 1998, 39, 594–602. [Google Scholar]
- Skarja, GA; Woodhouse, KA. Structure-property relationships of degradable polyurethane elastomers containing an amino acid based chain extender. J. Appl. Polym. Sci 2000, 75, 1522–1534. [Google Scholar]
- Skarja, GA; Woodhouse, KA. In vitro degradation and erosion of degradable, segmented polyurethanes containing amino acid based chain extender. J. Biomater. Sci. Polym. Ed 2001, 12, 851–873. [Google Scholar]
- Guan, J; Fujimoto, KL; Sacks, MS; Wagner, WR. Preparation and characterization of highly porous, biodegradable polyurethane scaffolds for soft tissue applications. Biomaterials 2005, 26, 3961–3971. [Google Scholar]
- Saad, B; Hirt, TD; Welti, M; Uhlschmid, GK; Neuenschwander, P; Suter, UW. Development of degradable polyesterurethanes for medical applications. J. Biomed. Mater. Res 1997, 36, 65–74. [Google Scholar]
- Saad, B; Kuboki, M; Matter, S; Welti, M; Uhlschmid, GK; Neuenschwander, P; Suter, UW. DegraPol-foam: A degradable and highly porous polyesterurethane as a new substrate for bone formation. Artif. Organs 2000, 24, 939–945. [Google Scholar]
- Zhang, JY; Beckman, EJ; Hu, J; Piesco, NP; Agarwal, S. A new peptide-based urethane polymer: Synthesis degradation and potential to support cell growth in vitro. Biomaterials 2000, 21, 1247–1258. [Google Scholar]
- Woo, GLY; Mittelman, MW; Santerre, JP. Synthesis and characterization of a novel biodegradable antimicrobial polymer. Biomaterials 2000, 21, 1235–1246. [Google Scholar]
- Guan, JJ; Sacks, MS; Beckman, EJ; Wagner, WR. Synthesis, characterization, and cytocompatibility of elastomeric, biodegradable poly(ester-urethane) ureas based on poly(caprolactone) and putrescine. J. Biomed. Mater. Res 2002, 61, 493–503. [Google Scholar]
- Storey, RF; Hickey, TP. Degradable polyurethane networks based on d,l-lactide, glycolide, e-caprolactone, and trimethylene carbonate homopolyester and copolyester triols. Polymer 1994, 35, 830–838. [Google Scholar]
- Grad, S; Kupcsik, L; Gorna, K; Gogolewski, S; Alini, M. The use of biodegradable polyurethane scaffolds for cartilage tissue engineering: Potential and limitations. Biomaterials 2003, 24, 5163–5171. [Google Scholar]
- Ganta, SR; Piesco, NP; Long, P; Gassner, R; Motta, LF; Papworth, GD; Stolz, DB; Watkins, SC; Agarwal, S. Vascularisation and tissue infiltration of a biodegradable polyurethane matrix. J. Biomed. Mater. Res 2003, 64A, 238–242. [Google Scholar]
- Zhang, JY; Beckman, EJ; Hu, J; Yang, GG; Agarwal, S; Hollinger, JO. Synthesis, biodegradability, and biocompatibility of lysine diisocyanate-glucose polymers. Tissue Eng 2002, 8, 771–785. [Google Scholar]
- Zhang, JY; Doll, BA; Beckman, EJ; Hollinger, JO. Three dimensional biocompatible ascorbic acid-containing scaffold for bone tissue engineering. Tissue Eng 2003, 9, 1143–1157. [Google Scholar]
- Guelcher, SA; Gallagher, KM; Didier, JE; Klinedinst, DB; Doctor, JS; Goldstein, AS; Wilkes, GL; Beckman, EJ; Hollinger, JO. Synthesis of biocompatible segmented polyurethanes from aliphatic diisocyanates and diurea diol chain extenders. Acta Biomater 2005, 1, 471–484. [Google Scholar]
- Guelcher, SA; Srinivasan, A; Hafeman, AE; Gallagher, KM; Doctor, JS; Khetan, S; McBride, SB; Hollinger, JO. Synthesis, in vitro biocompatibility and biodegradation, and mechanical properties of two-component polyurethane scaffolds: Effects of water and polyol composition. Tissue Eng 2007, 13, 2321–2333. [Google Scholar]
- Guelcher, SA; Patel, V; Gallagher, K; Connolly, S; Didier, JE; Doctor, J; Hollinger, JO. Synthesis and biocompatibility of polyurethane foam scaffolds from lysine diisocyanate and polyester polyols. Tissue Eng 2006, 12, 1247. [Google Scholar]
- Borkenhagen, M; Stoll, RC; Neuenschwander, P; Suter, UW; Aebischer, P. In vivo performance of a new biodegradable polyester urethane system used a nerve guidance channel. Biomaterials 1998, 19, 2155–2165. [Google Scholar]
- Guelcher, SA. Biodegradable polyurethanes: Synthesis and applications in regenerative medicine. Tissue Eng. Part B 2008, 14, 3–17. [Google Scholar]
- Skarja, GA; Woodhouse, KA. In vitro degradation and erosion of degradable segmented polyurethanes containing an amino acid-based chain extender. J Biomater Sci, Polym Ed 2001, 12, 851–873. [Google Scholar]
- Dahiyat, BI; Posadas, EM; Hirosue, S; Hostin, E; Leong, W. Degradable biomaterials with elastomeric characteristics and drugcarrier function. React. Polym 1995, 25, 101–109. [Google Scholar]
- Gunatillake, P; Mayadunne, R; Adhikari, R. Recent developments in biodegradable polymers. Biotech. Annu. Rev 2006, 12, 1387–2656. [Google Scholar]
- Gorna, K; Gogolewski, S. Preparation, degradation, and calcification of biodegradable polyurethane foams for bone graft substitutes. J. Biomed. Mater. Res 2003, 67A, 813–827. [Google Scholar]
- Guan, J; Sacks, M; Beckman, E; Wagner, W. Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: Synthesis, characterization and cytocompatibility. Biomaterials 2003, 25, 85–96. [Google Scholar]
- Skarja, GA; Woodhouse, KA. Synthesis and characterization of degradable polyurethane elastomers containing an amino-acid based chain extender. J. Biomater. Sci. Polym. Ed 1998, 9, 271–295. [Google Scholar]
- Gorna, K; Gogolewski, S. Biodegradable polyurethanes for implants. II. In vitro degradation and calcification of materials from poly(ecaprolactone)-poly(ethylene oxide) diols and various chain extenders. J. Biomed. Mater. Res 2002, 60, 592–606. [Google Scholar]
- Hiltunen, K; Tuominen, J; Seppälä, JV. Hydrolysis of lactic acid-based poly(ester-urethane)s. Polym. Int 1998, 47, 186–192. [Google Scholar]
- Lendlein, A; Colussi, M; Neuenschwander, P; Suter, UW. Hydrolytic degradation of phase-segragated multiblock copoly(ester urethane)s containing weak links. Macromol. Chem. Phys 2001, 13, 2702–2711. [Google Scholar]
- Deschamps, AA; van Apeldoorn, AA; Hayen, H; de Bruijn, JD; Karst, U; Grijpma, DW; Feijen, J. In vivo and in vitro degradation of poly(ether ester) block copolymers based on poly(ethylene glycol) and poly(butylene terephthalate). Biomaterials 2004, 25, 247–258. [Google Scholar]
- Elliott, SL; Fromstein, JD; Santerre, JP; Woodhouse, KA. Identification of biodegradation products formed by lphenylalanine based segmented polyurethaneureas. J. Biomater. Sci. Polym. Ed 2002, 13, 691–711. [Google Scholar]
- Dey, J; Xu, H; Shen, JH; Thevenot, P; Gondi, SR; Nguyen, KT; Sumerlin, BS; Tang, LP; Yang, J. Development of biodegradable crosslinked urethane-doped polyester elastomers. Biomaterials 2008, 29, 4637–4649. [Google Scholar]
- Nair, LS; Laurencin, CT. Biodegradable polymers as biomaterials. Prog. Polym. Sci 2007, 32, 762–798. [Google Scholar]
- Allen, RW; Allcock, HR. Conformational analysis of poly(alkoxy- and aryloxyphosphazene). Macromolecules 1976, 9, 956–961. [Google Scholar]
- Allcock, HR; Mang, MN; Dembek, AA. Poly[(aryloxy)phosphazenes] with phenylphenoxy and related bulky side groups, synthesis, thermal transition behavior, and optical properties. Macromolecules 1989, 22, 4179–4190. [Google Scholar]
- Potin, P; Jaeger, RD. Polyphosphazenes: Synthesis, structures, properties, applications. Eur. Polym. J 1991, 415, 341–348. [Google Scholar]
- Allcock, HR. Small–molecule phosphazene reings as models for high polymeric chains. Acc. Chem. Res 1979, 12, 351–358. [Google Scholar]
- Allcock, HR. Controlled synthesis of organic-inorganic polymers that possess a backbone of phosphorus and nitrogen atoms. Makromol. Chem 1981, 4, 3–19. [Google Scholar]
- Allcock, HR. Organometallic and bioactive phosphazenes. J. Polym. Sci. Polym. Symp 1983, 70, 71–77. [Google Scholar]
- Allcock, HR. Poly(organophosphazenes): Synthesis, unique properties and applications. Makromol. Chem. Makromol. Symp 1986, 6, 101–108. [Google Scholar]
- Allcock, HR; Kwon, S; Riding, GH; Fitzpatrick, RJ; Bennett, JL. Hydrophilic polyphosphazenes as hydrogels: Radiation cross-linking and hydrogel characteristics of poly[bis(methoxyethoxyethoxy)phosphazene]. Biomaterials 1988, 9, 509–513. [Google Scholar]
- Allcock, HR; Austin, PE; Neenan, TX; Langex, R; Shriver, DF. Covalent linkage of proteins to surface-modified poly(organophosphazenes): Immobilization of glucose-6-phosphate dehydrogenase and trypsin. Macromolecules 1986, 19, 1502–1508. [Google Scholar]
- Laurencin, CT; Koh, HJ; Neenan, TX; Allcock, HR; Langer, R. Controlled release using a new bioerodible polyphosphazenes system. J. Biomed. Mat. Res 1987, 21, 1231–1246. [Google Scholar]
- Ibim, SEM; Ambrosio, AMA; Kwon, MS; El-Amin, SF; Allcock, HR; Laurencin, CT. Novel polyphosphazene/poly(lactide-co-glycolide) blends: Miscibility and degradation studies. Biomaterials 1997, 18, 1565–1569. [Google Scholar]
- Lakshmi, S; Katti, DS; Laurencin, CT. Biodegradable polyphosphazenes for drug delivery applications. Adv. Drug Deliver. Rev 2003, 55, 467–482. [Google Scholar]
- Allcock, HR; Kugel, RL. Phosphonitrilic compounds. VII. High molecular weight poly(diaminophosphazenes). Inorg. Chem 1966, 5, 1716–1718. [Google Scholar]
- Allcock, HR; Fuller, TJ; Mack, DP; Matsumura, K; Smeltz, KM. Synthesis of poly[(amino acid alkyl ester) phosphazenes]. Macromolecules 1977, 10, 824–830. [Google Scholar]
- Allcock, HR; Pucher, SR; Scopelianos, AG. Poly[(amino acid ester)phosphazenes]: Synthesis, crystallinity and hydrolytic sensitivity in solution and the solid state. Macromolecules 1994, 27, 1071–1075. [Google Scholar]
- Crommen, JHL; Schacht, EH; Mense, EHG. Biodegradable polymers. II. Degradation characteristics of hydrolysis sensitive poly[(organo)phosphazenes]. Biomaterials 1992, 13, 601–611. [Google Scholar]
- Qui, LY; Zhu, KJ. Novel biodegradable polyphosphazenes containing glycine ethylester and benzyl ester of amino acethydroxamic acid as co substituents: Synthesis, characterization and degradation properties. J. Appl. Polym. Sci 2000, 77, 2987–2995. [Google Scholar]
- Allcock, HR; Fuller, TJ; Matsumura, K. Hydrolysis pathways for aminophosphazenes. Inorg. Chem 1982, 21, 515–521. [Google Scholar]
- Ibim, SM; Ambrosio, AA; Larrier, D; Allcock, HR; Laurencin, CT. Controlled macromolecule release from poly(phosphazene) matrices. J. Control. Release 1996, 40, 31–39. [Google Scholar]
- Allcock, HR; Kwon, S. Glyceryl polyphosphazenes: Synthesis, properties and hydrolysis. Macromolecules 1988, 21, 1980–1985. [Google Scholar]
- Allcock, HR; Pucher, SR. Polyphosphazenes with glycosyl and methyl amino, trifluoroethoxy, phenoxy or (methox-yethoxy)ethoxy side groups. Macromolecules 1991, 24, 23–34. [Google Scholar]
- Allcock, HR; Pucher, SR; Scopelianos, AG. Synthesis of inpoly(organophosphazenes) with glycolic acid ester and lactic acid ester side groups: Prototypes for new bioerodible polymers. Macromolecules 1994, 27, 1–4. [Google Scholar]
- Nair, LS; Laurencin, CT. Polymers as biomaterials for tissue engineering and controlled drug delivery. In Tissue Engineering I Advances in Biochemical Engineering/Biotechnology; Lee, K, Kaplan, D, Eds.; Springer: Berlin, Germany, 2006; pp. 47–90. [Google Scholar]
- Sethuraman, S; Nair, LS; El-Amin, S; Farrar, R; Nguyen, MT; Singh, A. In vivo biodegradability and biocompatibility evaluation of novel alanine ester based polyphosphazenes in a rat model. J. Biomed. Mater. Res. A 2006, 77, 679–687. [Google Scholar]
- Nair, LS; Lee, D; Laurencin, CT. Polyphosphazenes as novel biomaterials. In Handbook of Biodegradable Polymeric Materials and Applications; Narasimhan, A, Mallapragada, A, Eds.; American Scientific Publication: Stevenson Ranch, CA, USA, 2004; pp. 277–306. [Google Scholar]
- Nair, LS; Khan, YM; Laurencin, CT. Polyphosphazenes. In An Introduction to Biomaterials; Hollinger, H, Ed.; CRC Publications: Boca Raton, FL, USA, 2006; pp. 273–290. [Google Scholar]
- Laurencin, CT; Nair, LS. Polyphosphazene nanofibers for biomedical applications: Preliminary studies. In Nanoengineered Nanofibrous Materials, NATO-ASI Proceedings; Guceri, S, Gogotsi, YG, Kuznetsov, V, Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; pp. 281–300. [Google Scholar]
- Kumbar, SG; Bhattacharyya, S; Nukavarapu, SP; Khan, YM; Nair, LS; Laurencin, CT. In vitro and in vivo characterization of biodegradable poly(organo phosphazenes) for biomedical applications. J. Inorg. Organomet. Chem 2006, 16, 365–385. [Google Scholar]
- Nair, LS; Lee, DA; Bender, JD; Barrett, EW; Greigh, YE; Brown, PW. Synthesis, characterization and osteocompatibility evaluations of novel alanine based polyphosphazenes. J. Biomed. Mater. Res 2006, 76A, 206–213. [Google Scholar]
- Swaminathan, S; Nair, LS; Anurima, S; Bender, JD; Greish, YE; Brown, PW; Allcock, HR; Laurencin, CT. Development of novel biodegradable amino acid ester based polyphosphazene-hydroxyapatite composites for bone tissue engineering. In Nanoscale Materials Science in Biology and Medicine Boston; Laurencin, CT, Botchwey, E, Eds.; Materials Research Society: Warrendale, PA, USA, 2005. [Google Scholar]
- Greish, YE; Bender, JD; Lakshmi, S; Brown, PW; Allcock, HR; Laurencin, CT. Low temperature formation of hydroxyapatite-poly(alkyl hydroxyl benzoate) phosphazene composites for biomedical applications. Biomaterials 2005, 26, 1–9. [Google Scholar]
- Greish, YE; Bender, JD; Lakshmi, S; Brown, PW; Allcock, HR; Laurencin, CT. Composite formation from hydroxyapatite with sodium and potassium salts of polyphosphazene. J. Mater. Sci. Mater. Med 2005, 16, 613–620. [Google Scholar]
- Greish, YE; Bender, JD; Lakshmi, S; Brown, PW; Allcock, HR; Laurencin, CT. Formation of hydroxyapatite-poly[-bis(calcium carboxylato henoxy) phosphazene] composites at physiologic temperature. J. Biomed. Mater. Res 2006, 77A, 416–425. [Google Scholar]
- Grego, AV; Mingrone, G. Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: An update. Clin. Nutrit 1995, 14, 143–148. [Google Scholar]
- Mortensen, PB. C6–C10-dicarboxylic aciduria in starved, fat-fed and diabetic rats receiving decanoic acid or medium-chain triacylglycerol. An in vivo measure of the rate of beta-oxidation of fatty acids. Biochim. Biophys. Acta 1981, 664, 349–355. [Google Scholar]
- Fu, J; Fiegel, J; Krauland, E; Hanes, J. New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials 2002, 23, 4425–4433. [Google Scholar]
- Sundback, CA; Shyu, JY; Wang, YD; Faquin, WC; Langer, RS; Vacanti, JP; Hadlock, TA. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials 2005, 26, 5454–5464. [Google Scholar]
- Bettinger, CJ; Orrick, B; Misra, A; Langer, R; Borenstein, JT. Microfabrication of poly(glycerol–sebacate) for contact guidance applications. Biomaterials 2006, 27, 2558–2565. [Google Scholar]
- Motlagh, D; Yang, J; Lui, KY; Webb, AR; Ameer, GA. Hemocompatibility evaluation of poly(glycerol-sebacate) in vitro for vascular tissue engineering. Biomaterials 2006, 27, 4315–4324. [Google Scholar]
- Chen, QZ; Bismarck, A; Hansen, U; Junaid, S; Tran, MQ; Harding, SE; Ali, NN; Boccaccini, AR. Characterisation of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue. Biomaterials 2008, 29, 47–57. [Google Scholar]
- Liu, QY; Tian, M; Ding, T; Shi, R; Zhang, LQ. Preparation and characterization of a biodegradable polyester elastomer with thermal processing abilities. J Appl Polym Sci 2005, 98, 2033–2041. [Google Scholar]
- Liu, QY; Tian, M; Ding, T; Shi, R; Feng, YX; Zhang, LQ; Chen, DF; Tian, W. Preparation and characterization of a thermoplastic poly(glycerol-sebacate) elastomer by two-step method. J. Appl. Polym. Sci 2007, 103, 1412–1419. [Google Scholar]
- Liu, QY; Tian, M; Shi, R; Zhang, LQ; Chen, DF; Tian, W. Structure and properties of thermoplastic poly(glycerol sebacate) elastomers originating from prepolymers with different molecular weights. J. Appl. Polym. Sci 2007, 104, 1131–1137. [Google Scholar]
- Bruggeman, JP; de Bruin, BJ; Bettinger, CJ; Langer, R. Biodegradable poly(polyol sebacate) polymers. Biomaterials 2008, 29, 4726–4735. [Google Scholar]
- Qiu, HJ; Yang, J; Kodali, P; Koh, J; Ameer, GA. A citric acid-based hydroxyapatite composite for orthopedic implants. Biomaterials 2006, 27, 5845–5854. [Google Scholar]
- Ameer, G; Webb, AR. Biodegradable nanocomposites with enhanced mechanical properties for soft tissue. US Pat Appl 20070071790 2006. [Google Scholar]
- Lei, LJ; Ding, T; Shi, R; Liu, QY; Zhang, LQ; Chen, DF; Tian, W. Synthesis, characterization and in vitro degradation of a novel degradable poly((1,2-propanediol-sebacate)-citrate) bioelastomer. Polym. Degrad. Stabil 2007, 92, 389–396. [Google Scholar]
- Yang, J; Motlagh, D; Webb, AR; Ameer, GA. Novel biphasic elastomeric scaffold for small-diameter blood vessel. Tissue Eng 2005, 11, 1876–1886. [Google Scholar]
- Hoshi, RA; Behl, S; Ameer, GA. Nanoporous biodegradable elastomers. Adv. Mater 2009, 21, 188–192. [Google Scholar]
- Kang, Y; Yang, J; Khan, S; Anissian, L; Ameer, GA. A new biodegradable polyester elastomer for cartilage tissue engineering. J. Biomed. Mater. Res 2006, 77A, 331–339. [Google Scholar]
- Lange, R. Biomaterials in drug delivery and tissue engineering: One laboratory’s experience. Acc. Chem. Res 2000, 30, 94–101. [Google Scholar]
- Fakirov, S; Gogeva, T. Poly(ether ester)s based on poly(butylene terephthalate) and poly(ethylene oxide)glycols,1. Makromol. Chem 1990, 191, 603–614. [Google Scholar]
- Fakirov, S; Gogeva, T. Poly(ether ester)s based on poly(butylene terephthalate) and poly(ethylene oxide) glycol, 2. Makromol. Chem 1990, 191, 615–624. [Google Scholar]
- Fakirov, S; Gogeva, T. Poly(ether ester)s based on poly(butylene terephthalate) and poly(ethylene oxide) glycols, 3. Makromol. Chem 1990, 191, 2341–2354. [Google Scholar]
- Fakirov, S; Gogeva, T. Poly(ether ester)s based on poly(butylene terephthalate) and poly(ethylene oxide) glycols, 4. Makromol. Chem 1990, 191, 2355–2365. [Google Scholar]
- Fakirov, S; Apostolov, AA; Boeseke, P; Zachmann, HG. Structure of segmented poly(ether ester)s as revealed by synchrotron radiation. J. Macromol. Sci. Phys 1990, B29, 379–395. [Google Scholar]
- Fakirov, S; Fakirov, C; Fischer, EW; Stamm, M. Deformation behaviour of poly(ether ester) thermoplastic elastomers as revealed by Saxs. Polymer 1991, 32, 1173–1180. [Google Scholar]
- Apostolov, A; Fakirov, S. Effect of the block length on the deformation behavior of polyetheresters as revealed by small-angle X-ray scattering. J. Macromol. XI.—Phys 1992, B31, 329–355. [Google Scholar]
- Fakirov, S; Fakirov, C; Fischer, EW; Stamm, M. Polymer, deformation behaviour of poly(ether ester) thermoplastic elastomers with destroyed and regenerated structure as revealed by small-angle X-ray scattering. Polymer 1992, 33, 3818–3827. [Google Scholar]
- Fakirov, S; Fakirov, C; Fischer, EW; Stamm, M; Apostolov, AA. Reversible morphological changes in poly(ether ester) thermorplastic elastomers during deformation as revealed by small-angle X-ray scattering. Colloid Polym. Sci 1993, 271, 811–823. [Google Scholar]
- Fakirov, S; Denchev, Z; Apostolov, AA; Stamm, M; Fakirov, C. Morphological characterization during deformation of a poly(ether ester) thermoplastic elastomer by small-angle X-ray scattering. Colloid Polym. Sci 1994, 272, 1363–1372. [Google Scholar]
- Stribeck, N; Sapoundjieva, D; Denchev, Z; Apostolov, AA; Zachmann, HG; Stamm, M; Fakirov, S. Deformation behavior of poly(ether ester) copolymer as revealed by small- and wide-angle scattering of X-ray radiation from synchrotron. Macromolecules 1997, 30, 1329–1339. [Google Scholar]
- Grote, JJ. Reconstruction of the middle ear with hydroxylapatite implants: Long-term results. Ann. Otol. Rhinol. Laryngol 1990, 99, 12–16. [Google Scholar]
- Grote, JJ; Bakker, D; Hesseling, SC; van Blitterswijk, CA. New alloplastic tympanic membrane material. Am. J. Otol 1991, 12, 329–335. [Google Scholar]
- Bakkum, EA; Trimbos, JB; Dalmeyer, RAJ; van Blitterswijk, CA. Preventing postoperative intraperitoneal adhesion formation with polyactive, a degradable copolymer acting as a barrier. J. Mater. Sci. Mater. Med 1995, 6, 41–45. [Google Scholar]
- Radder, AM; Leenders, H; van Blitterswijk, CA. Bone-bonding behaviour of PEO/PBT copolymer coatings and bulk implants: A comparative study. Biomaterials 1994, 21, 532–537. [Google Scholar]
- Beumer, GJ; van Blitterswijk, CA; Bakker, D; Ponec, M. A new biodegradable matrix as a part of a cell seeded skin substitute for the treatment of deep skin defects: A physico-chemical characterisation. Clin. Mater 1993, 14, 21–27. [Google Scholar]
- Wang, S; Wan, ACA; Xu, XY; Gao, SJ; Mao, HQ; Leong, KW; Yu, H. A new nerve guide conduit material composed of a biodegradable poly(phosphoester). Biomaterials 2001, 22, 1157–1169. [Google Scholar]
- Zhao, Z; Eong, LKW. Polyphosphoestes in drug and gene delivery. Adv. Drug Deliv. Rev 2003, 55, 483–499. [Google Scholar]
- van Blitterswijk, CA; Bakker, D; Leenders, H; van den Brink, J; Hesseling, SC; Bovell, Y; Radder, AM; Sakkers, RJ; Gaillard, ML; Heinze, PH; Beumer, GJ. Interfacial reactions leading to bone bonding with PEO/PBT copolymers (Polyactive™). In Bonebonding Biomaterials; Ducheyne, P, Kokubo, T, van Blitterswijk, CA, Eds.; Reed Healthcare Communications: Leiderdorp, The Netherlands, 1992; pp. 13–30. [Google Scholar]
- van Blitterswijk, CA; van den Brink, J; Leenders, H; Bakker, D. The effect of PEO ratio on degradation, calcification and bonebonding of PEO/PBT copolymer (Polyactive™). Cell Mater 1993, 3, 23–36. [Google Scholar]
- Beumer, GJ; van Blitterswijk, CA; Bakker, D; Ponec, M. Cell seeding and in vitro biocompatibility evaluation of polymeric matrices of PEO/PBT copolymers and PLLA. Biomaterials 1993, 14, 598–604. [Google Scholar]
- Beumer, GJ; van Blitterswijk, CA; Ponec, M. Biocompatibility of a biodegradable matrix used as a skin substitute: An in vitro evaluation. J. Biomed. Mater. Res 1994, 28, 545–552. [Google Scholar]
- van Loon, JA; Leenders, H; Goedemoed, JH; van Blitterswijk, CA. Tissue reactions during long-term implantation in relation to degradation. In A Study of a Range of PEO/PBT Copolymers; Proceedings of the 20th Annual Meeting of the Society for Biomaterials: Boston, MA, USA, 1994; p. 370. [Google Scholar]
- Sakkers, RJB; Dalmeyer, RAJ; Wijn, JR; Blitterswijk, CA. Use of bone-bonding hydrogel copolymers in bone: An in vitro and in vivo study of expanding PEO-PBT copolymers in goat femora. J. Biomed. Mater. Res 2000, 49, 312–318. [Google Scholar]
- Reed, AM; Gilding, DK. Biodegradable polymers for use in surgery-poly(ethylene oxide) poly(ethylene terephthalate) (PEO/PET) copolymers: 2. in vitro degradation. Polymer 1981, 22, 499–504. [Google Scholar]
- Hayen, H; Deschamps, AA; Grijpma, DW; Feijen, J; Karst, U. Liquid chromatographic–mass spectrometric studies on the in vitro degradation of a poly(ether ester) block copolymer. J. Chromatogr. A 2004, 1029, 29–36. [Google Scholar]
- Stokes, K; Urbanski, P; Upton, J. The in vivo auto-oxidation of polyether polyurethane by metal ions. J. Biomater. Sci. Polym. Ed 1990, 1, 207–230. [Google Scholar]
- Deschamps, AA; van Apeldoorn, AA; Hayen, H; de Bruijn, JD; Karst, U; Grijpma, DW; Feijen, J. In vivo and in vitro degradation of poly(ether ester) block copolymers based on poly(ethylene glycol) and poly(butylene terephthalate). Biomaterials 2004, 25, 247–258. [Google Scholar]
- Ding, T; Liu, QY; Shi, R; Tian, M; Yang, J; Zhang, LQ. Synthesis, characterization and in vitro degradation study of a novel and rapidly degradable elastomer. Polym Degrad Stabil 2006, 91, 733–739. [Google Scholar]
- Hollinger, JO. Preliminary report on the osteogenic potential of a biodegradable copolymer of polyactide (PLA) and polyglycolide (PGA). J. Biomed. Mater. Res 1983, 17, 71–82. [Google Scholar]
- Pitt, CG; Gratzl, MM; Kimmel, GL; Surles, J; Schindler, A. Aliphatic polyesters □.The degradation of poly(dl-lactide), poly(epsilon-caprolactone), and their copolymers in vivo. Biomaterials 1981, 2, 215–220. [Google Scholar]
- Xie, J; Ihara, M; Jung, Y; Kwon, IlK; Kim, SH; Kim, YH; Matsuda, T. Mechano-active scaffold design based on microporous poly(l-lactide-co-ε-caprolactone) for articular cartilage tissue engineering: Dependence of porosity on compression force-applied mechanical behaviors. Tissue Eng 2006, 12, 449–458. [Google Scholar]
- Jeong, SI; Kim, SH; Kim, YH; Jung, Y; Kwon, JH; Kim, BS; Lee, YM. Manufacture of elastic biodegradable PLCL scaffolds for mechano-active vascular tissue engineering. J. Biomater. Sci. Polym. Ed 2004, 15, 645–660. [Google Scholar]
- Jeong, SI; Kwon, JH; Lim, JI; Cho, SW; Jung, Y; Sung, WJ; Kim, SH; Kim, YH; Lee, YM; Kim, BS; Choi, CY; Kim, SJ. Mechano-active tissue engineering of vascular smooth muscle using pulsatile perfusion bioreactors and elastic PLCL scaffolds. Biomaterials 2005, 26, 1405–1411. [Google Scholar]
- Cohn, D; Salomon, AH. Designing biodegradable multiblock PCL/PLA thermoplastic elastomers. Biomaterials 2005, 26, 2297–2305. [Google Scholar]
- Jeong, SI; Kim, BS; Kang, SW; Kwon, JH; Lee, YM; Kim, SH; Kim, YH. In vivo biocompatibilty and degradation behavior of elastic poly(l-lactide-co-ε-caprolactone) scaffolds. Biomaterials 2004, 25, 5939–5946. [Google Scholar]
- Min, CC; Cui, WJ; Bei, JZ; Wang, SG. Biodegradable shape-memory polymer-polylactide-co-poly(glycolide-co-caprolactone) multiblock copolymer. Polym. Adv. Technol 2005, 16, 608–615. [Google Scholar]
- Engelberg, I; Kohn, J. Physio-mechanical properties of degradable polymers used in medical applications: A comparative study. Biomaterials 1991, 12, 292–304. [Google Scholar]
- Pěgo, AP; Poot, AA; Grijpma, DW; Feijen, J. Copolymers of trimethylene carbonate and epsilon-caprolactone for porous nerve guides: Synthesis and properties. J. Biomat. Sci. Polym. Ed 2001, 12, 35–53. [Google Scholar]
- Pêgo, AP; Siebum, B; van Luyn, MJA; van Seijen, XJGY; Poot, AA; Grijpma, DW; Feijen, J. Preparation of degradable porous structures based on 1,3-trimethylene carbonate and d,l-lactide(co)polymers for heart tissue engineering. Tissue Eng 2003, 9, 981–994. [Google Scholar]
- Pêgo, AP; van Luyn, MJA; Brouwer, LA; van Wachem, PB; Poot, AA; Grijpma, DW; Feijen, J. In vitro degradation of trimethylene carbonate based (co)polymers. Macromol. Biosci 2002, 2, 411–419. [Google Scholar]
- Barrows, TH. Degradable implant materials: A review of synthetic absorbable polymers and their applications. Clin. Mater 1986, 1, 233–257. [Google Scholar]
- Barrows, TH; Gibson, SJ; Johnson, JD. Poly(ester-amides): In vivo analysis of degradation and metabolism using radiolabelled polymer. Trans. Soc. Biomater 1984, 7, 210. [Google Scholar]
- Jamiolkowski, DD; Shalaby, SW. Polyesteramides derived from bis-(oxamidodiols) and dicarboxylic acids. US Pat 4209607 1980. [Google Scholar]
- Katsarava, R; Kharadze, D; Kirmelashvili, L; Medzmariashvili, N; Goguadze, T; Tsitlanadze, G. Polyamides from 2,2’-P-phenylenebis (Δ2-5-oxazolone)s and N,N’-bistrimethyl-silylated diamines-synthesis of polyamides containing dipeptide links in the main chains. Makromol. Chem. Phys 1993, 194, 143–150. [Google Scholar]
- Arabuli, N; Tsitlanadze, G; Edilashvili, L; Kharadze, D; Goguadze, T; Beridze, V. Heterochain polymers based on natural aminoacids—synthesis and enzymatic-hydrolysis of regular poly(ester amide)s based on bis(l-phenylalanine) alpha, omega-alkylene diesters and adipic acid. Macromol. Chem. Phys 1994, 195, 2279–2289. [Google Scholar]
- Katsarava, R; Beridze, V; Arabuli, N; Kharadze, D; Chu, CC; Won, CY. Amino acid-based bioanalogous polymers. Synthesis, and study of regular poly(ester amide)s based on bis(alpha-amino acid) alpha, omega-alkylene diesters, and aliphatic dicarboxylic acids. J. Polym. Sci. Polym. Chem 1999, 37, 391–407. [Google Scholar]
- Paredes, N; Rodriguez-Galan, A; Puiggali, J. Synthesis and characterization of a family of biodegradable poly(ester amide)s derived from glycine. J. Polym. Sci., Polym. Chem 1998, 36, 1271–1282. [Google Scholar]
- Paredes, N; Rodriguez-Galan, A; Puiggali, J; Peraire, C. Studies on the biodegradation and biocompatibility of a new poly(esteramide) derived from l-alanine. J. Appl. Polym. Sci 1998, 69, 1537–1549. [Google Scholar]
- Pivsa-Art, S; Nakayama, A; Kawasaki, N; Yamamoto, N; Aiba, S. Biodegradability study of copolyesteramides based on diacid chlorides, diamines, and diols. J. Appl. Polym. Sci 2002, 85, 774–784. [Google Scholar]
- Martinez, MB; Pinilla, IM; Mata, FZ; Perez, JAG. Hydrolytic degradation of poly(ester amides) derived from carbohydrates. Macromolecules 1997, 30, 3197–3203. [Google Scholar]
- Qian, ZY; Li, S; He, Y; Zhang, HL; Liu, XB. Hydrolytic degradation study of biodegradable polyesteramide copolymers based on epsiloncaprolactone and 11-aminoundecanoic acid. Biomaterials 2004, 25, 1975–1981. [Google Scholar]
- Tsitlanadze, G; Machaidze, M; Kviria, T; Djavakhishvili, N; Chu, CC; Katsarava, R. Biodegradation of amino-acid-based poly(ester amide)s: In vitro weight loss and preliminary in vivo studies. J. Biomater. Sci. Polym. Ed 2004, 15, 1–24. [Google Scholar]
- Tsitlanadze, G; Kviria, T; Katsarava, R; Chu, CC. In vitro enzymatic biodegradation of amino acid based poly(ester amide)s biomaterials. J. Mater. Sci. Mater. Med 2004, 15, 185–190. [Google Scholar]
- Okada, M. Chemical syntheses of biodegradable polymers. Prog. Polym. Sci 2002, 27, 87–133. [Google Scholar]
- Ho, L-H; Huang, SJ. Poly(amide–ester)s derived from a-amino acids. Polym. Prepr 1992, 33, 94–95. [Google Scholar]
- Paredes, NA; Rodríguez-Galán, J. Puiggalí, synthesis and characterization of a family of biodegradable poly(ester amide)s derived from glycine. J. Polym. Sci. Part A: Polym. Chem 1998, 36, 1271–1282. [Google Scholar]
- Alla, A; Rodriguez-Galán, A; Martinez de Ilarduya, A; Munöz-Guerra, S. Degradable poly(ester–amide)s based on l-tartaric acid. Polymer 1997, 38, 4935–4944. [Google Scholar]
- Koyama, E; Sanda, F; Endo, T. Syntheses of poly(ester–amide)s derived from optically active amino alcohols. Macromol. Symp 1997, 122, 275–280. [Google Scholar]
- Bettinger, CJ; Bruggeman, JP; Borenstein, JT; Langer, RS. Amino alcohol–based degradable poly(ester amide) elastomers. Biomaterials 2008, 29, 2315–2325. [Google Scholar]
- Kricheldorf, HR; Wollheim, T; Koning, CE; Werumeus-Buning, HG; Altstädt, V. Thermoplastic elastomers 1. Poly(ether–ester–imide)s based on 1,4-diaminobutane, trimellitic anhydride, 1,4-dihydroxybutane and poly(tetra methylene oxide) diols. Polymer 2001, 42, 6699–6708. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
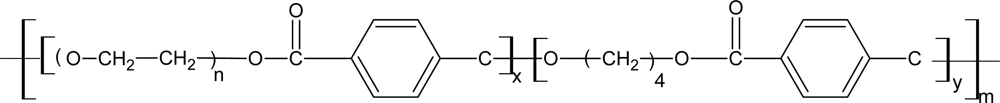{kind=link}
{kind=link}
{kind=link}
| Synthetic Bioelastomers | Tg (°C) | TS (MPa) | EB(%) | Approx DT | Degradation Products | Referrences | |
|---|---|---|---|---|---|---|---|
| PUs | −116 – − 41 | 4–60 | 100–950 | wide range | time | α-hydroxy acids, urethane, urea fragments, lysine (for lysine-derived polyisocyanates) | [15] [16] |
| PNs | − 105 −91 | Wide range | Wide range | wide range | time | phosphate, ammonium salts, amino acids, and ethanol | [17] [18] |
| PGSa | −7 – 46 | >0.5 | >267 | 1 | Glycerol, sebacate | [19,20] | |
| POCb | −5 – 10 | Up to 6.7 | 265 ± 10 | variable | Octanediol, citric acid | [21] | |
| PDCc | −5 – 10 | Up to 3.14 ± 0.5 | 322 ± 20 | variable | 1,10-decanediol Citric acid | [22] | |
| Poly(diol citrates) | − 5 10 | Up to 11.2 | Up to 502% | variable | Citric acid; polyols | [22] | |
| PEG/PBT | – | 8 to 23 | 500% to 1300% | wide range | time | PEG and PBT segment | [23,24] |
| PGCLd | – | <1 | Up to 250 | >1.5 | Short chain oligomers; glycolic acid; 6-hydroxyhexanoic | [24,25], | |
| TMC-DLLA (dry) 50:50 e | 11 | 10 | 570% | <11 | Short chain oligomers; d,l-lactic acid; TMC monomers | [26–28] | |
| TMC-DLLA (dry) 20:80f | 33 | 51 | 7 | <11 | Short chain oligomers; d,l-lactic acid; TMC monomers | [26–28] | |
| TMC-CL 10:90g | >−17 | 23 | – | >24 | Short chain oligomers; Caproic acid | [26–28] | |
| PEAs | variable | variable | variable | wide range | time | Short chain oligomers; diamines,dicarboxylic acids | [29] |
| Diisocyanates | Name and Abbreviation |
|---|---|
 | Butane diisocyanate (BDI) |
 | Hexamethylene diisocyanate (HDI) |
 | Isophorone diisocyanate (IPD) |
 | Lysine diisocyanate (LDI) |
 | Trimethylhexamethylene diisocyanate (TMDI) |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shi, R.; Chen, D.; Liu, Q.; Wu, Y.; Xu, X.; Zhang, L.; Tian, W. Recent Advances in Synthetic Bioelastomers. Int. J. Mol. Sci. 2009, 10, 4223-4256. https://doi.org/10.3390/ijms10104223
Shi R, Chen D, Liu Q, Wu Y, Xu X, Zhang L, Tian W. Recent Advances in Synthetic Bioelastomers. International Journal of Molecular Sciences. 2009; 10(10):4223-4256. https://doi.org/10.3390/ijms10104223
Chicago/Turabian StyleShi, Rui, Dafu Chen, Quanyong Liu, Yan Wu, Xiaochuan Xu, Liqun Zhang, and Wei Tian. 2009. "Recent Advances in Synthetic Bioelastomers" International Journal of Molecular Sciences 10, no. 10: 4223-4256. https://doi.org/10.3390/ijms10104223