Results and Discussion
Brimble
et al. have reported the synthesis of 2-methyl-1-pyrrolo[1,2-a] pyrazinone (
5) from methyl pyrrole-2-carboxylate (
4) and 1,2-dibromoethane (
Scheme 1) [
2]. Based on this scheme, we wished to prepare compounds
2 by replacing the 1,2-dibromoethane with 1,2-dibromo-1-phenylethane, but the N-alkylation of compound
4 failed to take place and 2-bromo-1-phenylethene was separated from the reaction mixture. This revealed that instead of the desired reaction, an elimination of 1,2-dibromo-1- phenylethane took place under the alkaline reaction conditions.
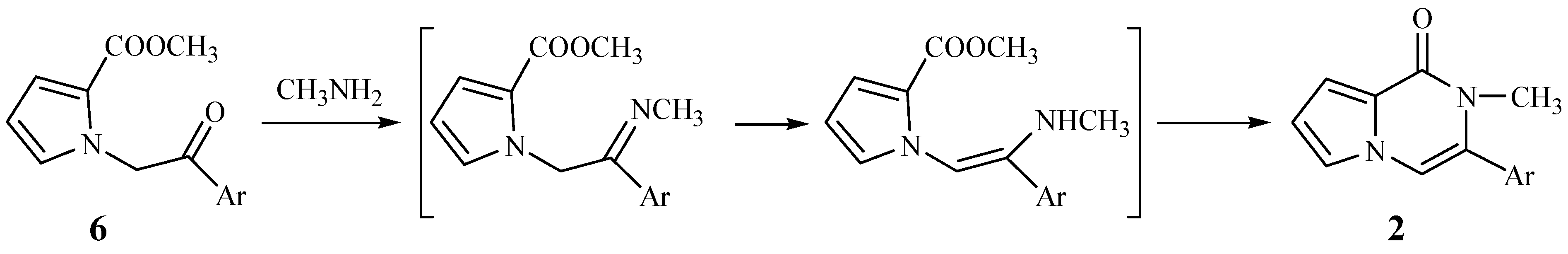
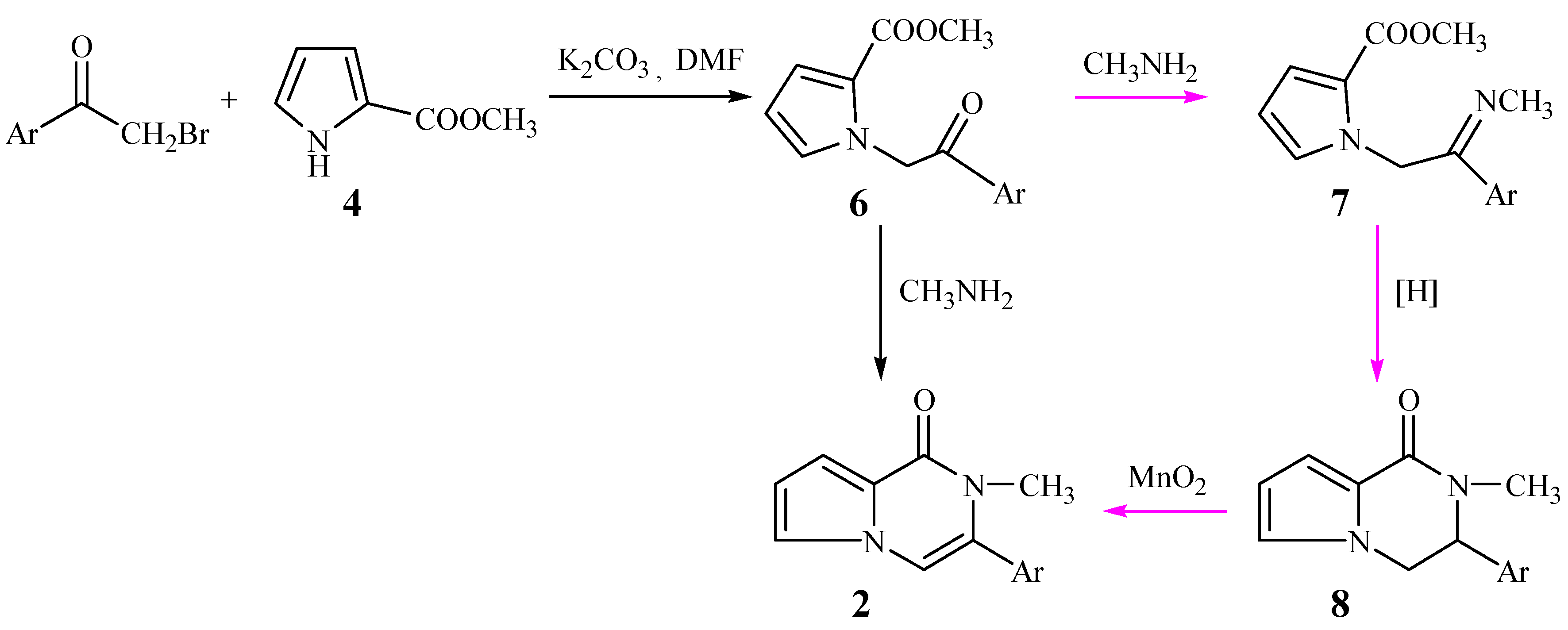
With α-bromoacetophenone replacing 1,2-dibromo-1-phenylethane, the new route shown
Scheme 2 was designed. The N-alkylations of compound
4 took place smoothly to form the intermediates
6. Our original intention was to convert compounds
6 to the title compounds
2 through imidization to
7, reduction to
8, and finally oxidation to compounds
2, but unexpectedly, when
6 reacted with methylamine in absolute methanol, the title 3-aryl-2-methyl-1-pyrrolo[
1,
2,[1,2-a]pyrazinones (
2) were obtained directly.
Obviously, this represents a very facile route for the synthesis of compounds
2. Since primary amines readily react with ketones to yield imines, the mechanism in
Figure 1 would appear reasonable.
Figure 1.
Proposed mechanism of the first cyclization
Figure 1.
Proposed mechanism of the first cyclization
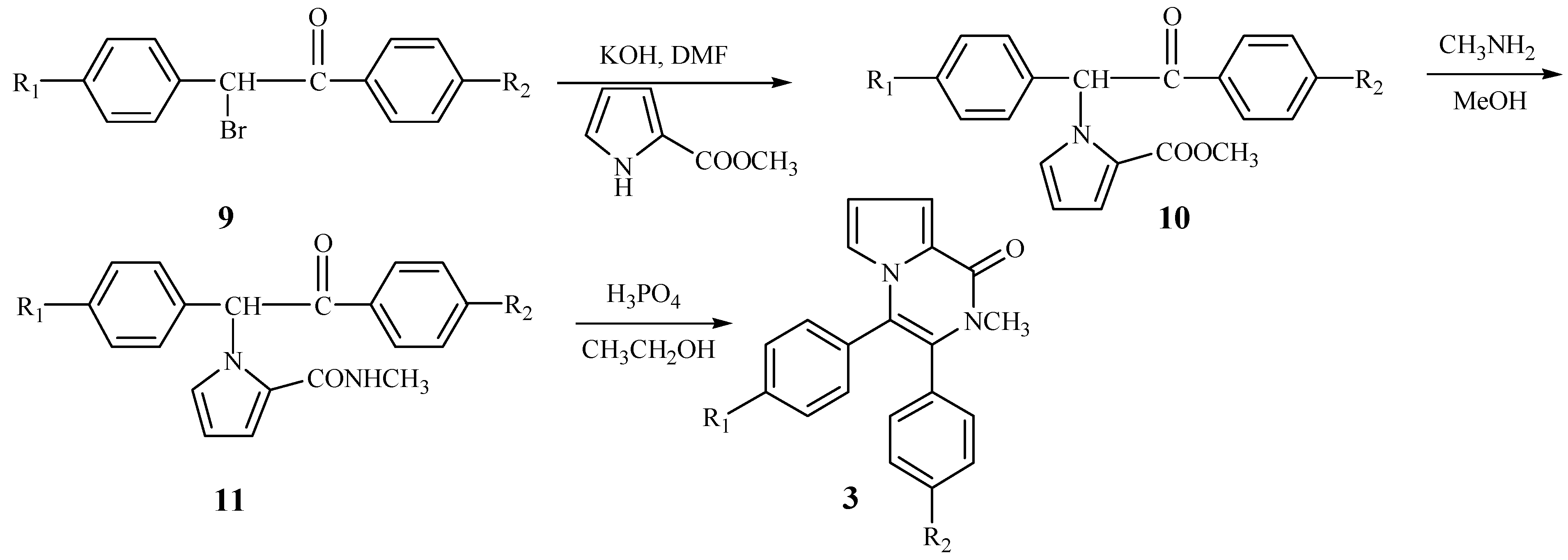
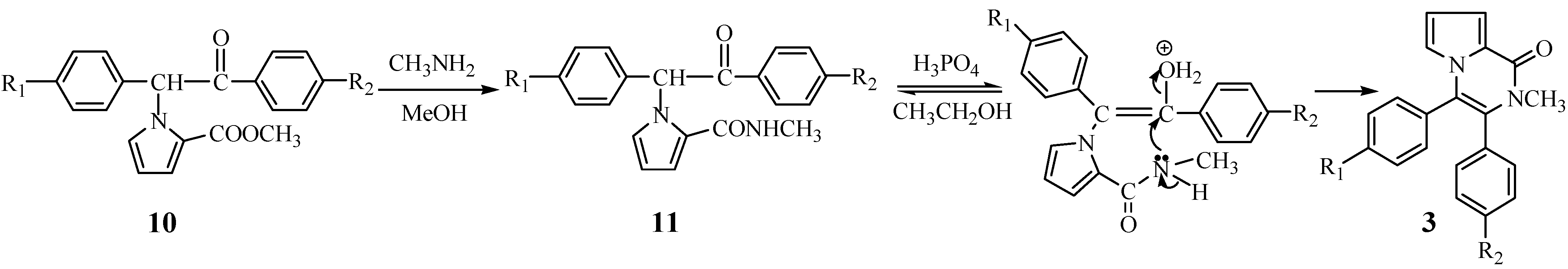
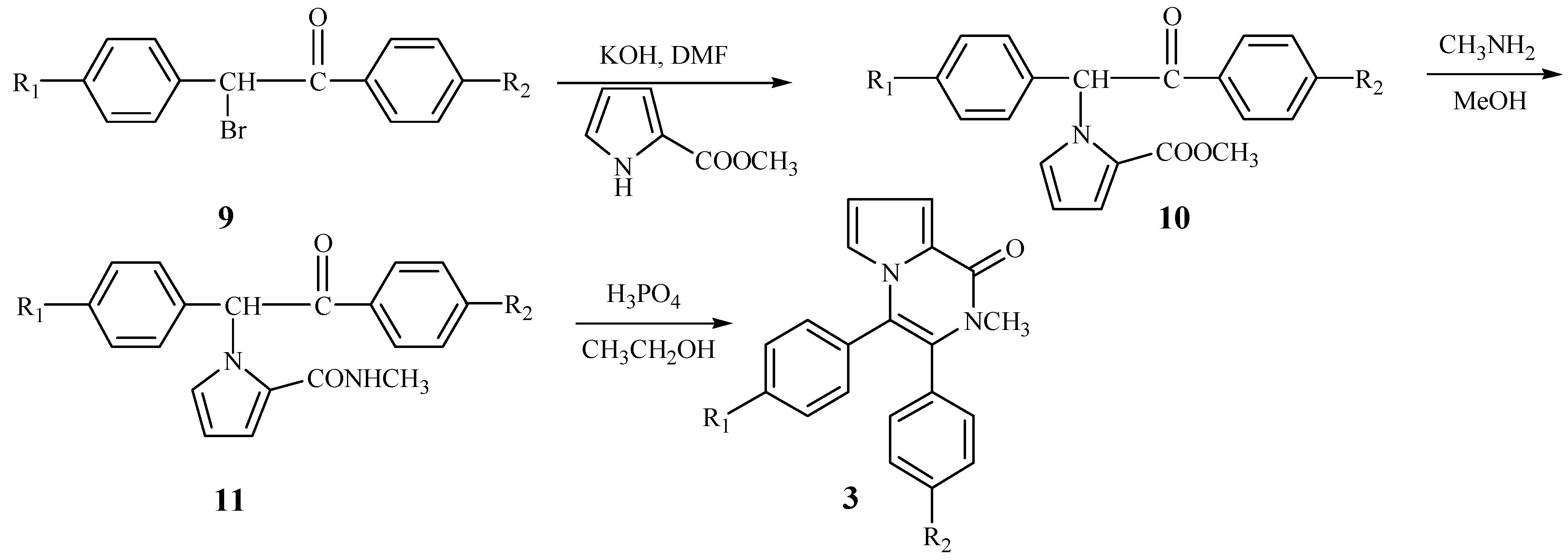
According to
Scheme 2, we wished to prepare 3,4-diaryl-2-methyl-1-pyrrolo[1,2-a]pyrazinones
3 from 1-bromo-1,2-diarylethanones. The N-alkylation of compound
4 also took place smoothly to yield the intermediate
10. When
10 reacted with methylamine, the cyclization did not take place and the product
11 resulting from aminolysis of
10 was obtained. Fortunately, an acid-catalyzed cyclization as shown in
Scheme 3 was discovered. When the intermediate
11 was treated with phosphoric acid in ethanol, it was successfully converted into the title compounds
3 in moderate yield.
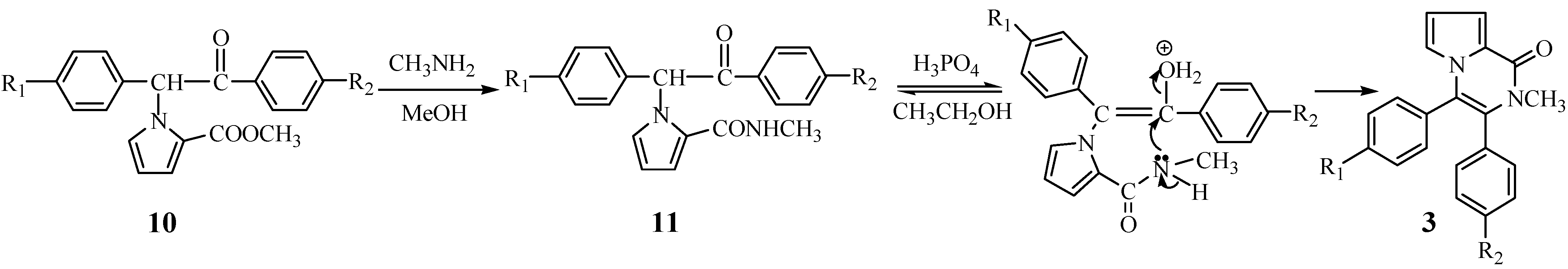
A cyclization mechanism involving a nucleophilic substitution as outlined in
Figure 2 is suggested.
Figure 2.
Proposed mechanism of the second cyclization
Figure 2.
Proposed mechanism of the second cyclization
From above-mentioned two proposed mechanisms, it can be found that the intermediates 6 and 10 are similar, but the cyclization of 10 does not happen under the conditions of intermediates 6. After preliminary studies, we assume that it may be the steric influence that has resulted in the two proposed mechanism of cyclization.
Pharmacological tests
Male Kunming mice were used to test the anti-inflammatory and analgesic activities. These tests were carried out by administering the compounds intragastrically (i.g.) at a dose of 200mg/kg, and each compound was tested on 10 mice. The anti-inflammatory activities were evaluated by the xylene- induced ear edema model, and the analgesic activities were evaluated by acetic acid-induced writhing method. The results show that compounds
2b,
2f,
3a and
3c have remarkable activities. The anti- inflammatory activities of
2f and
3c are more potent than that of ibuprofen, the positive control (see
Table 1).
Table 1.
Inhibition on xylene-induced mice ear edema and acetic acid-induced mice writhing
Table 1.
Inhibition on xylene-induced mice ear edema and acetic acid-induced mice writhing
| Compd. | Xylene-induced mice ear edema | Acetic acid-induced mice writhing |
| Dose/mg.kg-1 | Mice/n | Inhb.rate/% | Dose/mg.kg-1 | Mice/n | Inhb.rate/% |
| CMC | 200 | 10 | ----- | 200 | 10 | ----- |
| ibuprofen | 200 | 10 | 36.9 | 200 | 10 | 98.3 |
| 2a | 200 | 10 | 6.1 | 200 | 10 | 80.0* |
| 2b | 200 | 10 | 30.7* | 200 | 10 | 85.7* |
| 2c | 200 | 10 | 13.1 | 200 | 10 | 82.8* |
| 2d | 200 | 10 | 14.6 | 200 | 10 | 46.0 |
| 2e | 200 | 10 | 29.5 | 200 | 10 | 93.5** |
| 2f | 200 | 10 | 71.6** | 200 | 10 | 96.0** |
| 3a | 200 | 10 | 26.8* | 200 | 10 | 78.5** |
| 3b | 200 | 10 | 16.4 | 200 | 10 | 27.6 |
| 3c | 200 | 10 | 53.9** | 200 | 10 | 62.6* |
| 3d | 200 | 10 | 25.4 | 200 | 10 | 50.2 |
| 3e | 200 | 10 | 16.1 | 200 | 10 | 35.3 |
Experimental
General
Melting points were determined by the capillary tube method, and the thermometer was uncorrected. 1H-NMR spectra were recorded on a Bruker ARX-300 spectrometer. MS were measured with Finnigan LCQ LS/MS instrument. IR spectra were recorded on a Bruker IR-FIS-55 spectrometer.
General Procedure for the Preparation of Methyl 1-(2-aryl-2-oxoethyl)pyrrole-2-carboxylates 6.
The corresponding α-bromoarylethanone (13.6 mmol) was added to a mixture of methyl pyrrole-2- carboxylate (
4, 13.3 mmol) and K
2CO
3 (2.7 g) in DMF (15 mL). After stirring at room temperature for 30 min, the reaction mixture was poured into water. The mixture was filtered, and the solid was washed with water and air-dried to yield the crude compounds
6. The crude products were purified by recrystallization from methanol or column chromatography on silica gel [
3].
Methyl 1-(
2-
oxo-2-
phenylethyl)
pyrrole-
2-
carboxylate (
6a). Yield 62.5%; m.p. 105-107°C (Lit. [
3] 105- 106°C).
Methyl 1-[2-(4-hydroxyphenyl)-2-oxoethyl]pyrrole-2-carboxylate (6d). Yield 80.5%; m.p. 209- 210°C; 1H-NMR (DMSO) δ (ppm): 3.61 (s, 3H, -OCH3), 5.79 (s, 2H, -CH2), 6.17 (br. t, 1H, pyrrole H- 4), 6.90 (br. d, 3H, pyrrole H-3, Ar-H), 7.12 (br. s, 1H, pyrrole H-5), 7.91 (d, 2H, J=8.3Hz, Ar-H), 10.48 (s, 1H, -OH).
Methyl 1-[2-(4-methylthiophenyl)-2-oxoethyl]pyrrole-2-carboxylate (6f). Yield 73.2%; m.p. 136- 138°C; 1H-NMR (CDCl3) δ (ppm): 2.53 (s, 3H, -SCH3), 3.72 (s, 3H, -OCH3), 5.72 (s, 2H, -CH2), 6.25 (br. t, 1H, pyrrole H-4), 6.84 (br. s, 1H, pyrrole H-3), 7.04 (dd, 1H, J=1.5Hz, 3.9Hz, pyrrole H-5), 7.30 (d, 2H, J=8.4Hz, Ar-H), 7.92 (d, 2H, J=8.4Hz, Ar-H).
Methyl 1-[2-(2,4-dimethylphenyl)-2-oxoethyl]pyrrole-2-carboxylate (6b), methyl 1-[2-naphthyl-2-oxo- ethyl]pyrrole-2-carboxylate (6c) and methyl 1-[2-(4-fluorophenyl)-2-oxoethyl]pyrrole-2-carboxylate (6e) were prepared by the same procedure used for 6a.
General Procedure for the Preparation of 3-Aryl-2-methyl-1-pyrrolo[1,2-a]pyrazinones 2a-f.
A solution of the appropriate compound 6 (4.5 mmol) in anhydrous methanol (15 mL) was heated to reflux. A stream of methylamine was passed through this solution for 3-4 hr, and then the solution was refluxed for four days. Removal of solvent under reduced pressure afforded a brown, sticky oil which was purified by column chromatography on silica gel using 4:1 petroleum ether/ethyl acetate as eluant to give the corresponding compounds 2.
2-Methyl-3-phenyl-1-pyrrolo[1,2-a]pyrazinone (2a). Yield 62.5%; overall yield 22.7%; m.p. 141- 142°C; 1H-NMR (CDCl3) δ (ppm): 3.28 (s, 3H, N-CH3), 6.60 (br. t, 1H, H-7), 6.93 (s, 1H, H-4), 7.10 (br. d, 1H, H-8), 7.16 (br. d, 1H, H-6), 7.39-7.49 (m, 5H, Ar-H); IR (KBr) ν(cm-1): 1621.4 (amide, C=O); MS (m/z): 225 (M++1).
3-(2,4-Dimethylphenyl)-2-methyl-1-pyrrolo[1,2-a]pyrazinone (2b). Yield 59.4%, overall yield 26.5%; m.p. 163-164°C; 1H-NMR (CDCl3) δ (ppm): 2.20 (s, 3H, Ar-CH3), 2.39 (s, 3H, Ar-CH3), 3.12 (s, 3H, N-CH3), 6.59 (br. t, 1H, H-7), 6.86 (s, 1H, H-4), 7.07-7.16 (m, 5H, H-8, H-6, Ar-H); IR (KBr) ν(cm-1): 1625.9 (amide, C=O); MS (m/z): 253 (M++1).
2-Methyl-3-naphthyl-1-pyrrolo[1,2-a]pyrazinone (2c). Yield 36.8%, overall yield 10.2%; m.p. 161- 163°C; 1H-NMR (CDCl3) δ (ppm): 3.09 (s, 3H, N-CH3), 6.64 (br. t, 1H, H-7), 7.04 (s, 1H, H-4), 7.12 (br. s, 1H, H-8), 7.21 (br. d, 1H, H-6), 7.53-7.58 (m, 4H, Ar-H), 7.72 (br. d, 1H, Ar-H), 7.92-8.00 (m, 2H, Ar-H); IR (KBr) ν(cm-1): 1621.0 (amide, C=O); MS (m/z): 275 (M++1).
3-(4-Hydroxyphenyl)-2-methyl-1-pyrrolo[1,2-a]pyrazinone (2d). Yield 89.9%, overall yield 40.8%; m.p. 240-241°C; 1H-NMR (DMSO) δ (ppm): 3.13 (s, 3H, N-CH3), 6.55 (br. s, 1H, H-7), 6.86 (br. d, 3H, H-4, Ar-H), 7.31 (br. d, 3H, H-8, Ar-H), 7.39 (br. s, 1H, H-6), 9.82 (s, 1H, -OH); IR (KBr) ν(cm-1): 2991.6 (O-H), 1628.4 (amide, C=O); MS (m/z): 241 (M++1).
3-(4-Fluorophenyl)-2-methyl-1-pyrrolo[1,2-a]pyrazinone (2e). Yield 70.1%, overall yield 34.3%; m.p. 133-135°C; 1H-NMR (CDCl3) δ (ppm): 3.25 (s, 3H, N-CH3), 6.59 (br. t, 1H, H-7), 6.89 (s, 1H, H-4), 7.08-7.19 (m, 4H, H-8, H-6, Ar-H), 7.36-7.40 (m, 2H, Ar-H); IR (KBr) ν(cm-1): 1623.5 (amide, C=O); MS (m/z): 243 (M++1).
2-Methyl-3-(4-methylsulfinylphenyl)-1-pyrrolo[1,2-a]pyrazinone (2f). Yield 70.8%, overall yield 22.7%; m.p. 213-214°C; 1H-NMR (CDCl3) δ (ppm): 2.81 (s, 3H, -SOCH3), 3.28 (s, 3H, N-CH3), 6.61 (br. t, 1H, H-7), 6.93 (s, 1H, H-4), 7.11 (br. s, 1H, H-8), 7.16 (br. d, 1H, H-6), 7.59 (d, 2H, J=8.2Hz, Ar-H), 7.77 (d, 2H, J=8.2Hz, Ar-H); IR (KBr) ν(cm-1): 1049.2 (S=O), 1618.7 (amide, C=O); MS (m/z): 287 (M++1).
General Procedure for the Preparation of 1-Bromo-1,2-diarylethanones 9.
A solution of the 1,2-diarylethanone (2.6 mmol) in ethyl acetate (5 mL) and chloroform (5 mL) was heated to reflux. Powdered cupric bromide (5.1 mmol) was added in small portions over a 3.5-hr period, with the reaction mixture maintained at 75-80°C while a constant stream of nitrogen gas was bubbled through the reaction solution. After the addition was complete, stirring was continued for 1.5 hr until the green color and dark cupric bromide had disappeared, then the reaction mixture was cooled, filtered and the colorless solid cuprous bromide was washed with chloroform (25 mL). The combined filtrate and washings were rotary evaporated and an oily residue was obtained. The residue was redissolved in diethyl ether (200 mL), washed with water, 5% sodium bicarbonate and brine and then dried over sodium sulfate. After removing the solvent, an opaque product was isolated [
4].
1- Bromo-
1-
phenylacetophenone (
9a). Yield 97%; m.p. 54-56°C (Lit. [
4] 55-56°C).
General Procedure for the Preparation of Methyl 1-(1, 2-diaryl-2-oxoethyl)pyrrole-2-carboxylates 10.
The corresponding 1-bromo-1, 2-diarylethanones (13.6 mmol) were added to a mixture of methyl pyrrole-2-carboxylate (
4, 13.3 mmol) and KOH (1.3 g) in DMF (15 mL). After stirring at room temperature for 30 min, the reaction mixture was poured into water. The mixture was filtered, and the solid was washed with water and air-dried to yield the crude products title compounds
10, which were purified by recrystallization from methanol or column chromatography on silica gel [
3].
Methyl 1-[2-(4-methylphenyl)-2-oxo-1-phenylethyl]pyrrole-2-carboxylate (10c). Yield 35.6%; m.p. 126-128°C; 1H-NMR (DMSO) δ (ppm): 2.29 (s, 3H, Ar-CH3), 3.64 (s, 3H, -COOCH3), 6.09 (br. t, 1H, pyrrole H-4), 6.42 (br. t, 1H, pyrrole H-3), 6.97 (dd, 1H, J=1.7Hz, 3.9Hz, pyrrole H-5), 7.28 (d, 2H, J=8.9Hz, Ar-H), 7.42-7.44 (m, 5H, Ar-H), 7.86 (s, 1H, -CH), 7.95 (d, 2H, J=8.9Hz, Ar-H).
General Procedure for the Preparation of N-methyl-1-(1, 2-diaryl-2-oxoethyl)pyrrole-2-amides 11.
A solution of compounds 10 (4.5 mmol) in anhydrous methanol (15 mL) was heated to reflux. A stream of methylamine was passed through this solution for 3-4 hr and then the solution was refluxed for four days. Removal of solvent under reduced pressure afforded a brown sticky oil, which was purified by column chromatography on silica gel using petroleum ether/ethyl acetate (4:1) as eluant to give the title compounds 11.
N-methyl-1-[2-(4-methylphenyl)-2-oxo-1-phenylethyl]pyrrole-2-amide (11c). Yield 70.2%; m.p. 177- 179°C; 1H-NMR (DMSO) δ (ppm): 2.32 (s, 3H, Ar-CH3), 2.64 (d, 3H, J=5.1Hz, -NH-CH3), 6.06 (br. t, 1H, pyrrole H-4), 6.45 (br. s, 1H, pyrrole H-3), 6.77 (br. d, 1H, pyrrole H-5), 7.09 (q, 1H, J=5.1Hz, -NH-CH3), 7.27 (d, 2H, J=7.8Hz, Ar-H), 7.35-7.38 (m, 5H, Ar-H), 7.87 (d, 2H, J=7.8Hz, Ar- H), 8.06 (s, 1H, -CH); IR (KBr) ν(cm-1): 1701.3 (C=O), 1616.4 (amide, C=O); MS (m/z): 333 (M++1).
General Procedure for the Preparation of 3,4-Diaryl-2-methyl-1-pyrrolo[1,2-a]pyrazinones 3.
To a solution of compounds 11 (1.4 mmol) in anhydrous ethanol (3 mL) was added 85% phosphoric acid (6 mL). The reaction solution was refluxed for 1 hr. After cooling to room temperature, the reaction solution was poured into water and filtered, giving the crude product as a solid. The crude product could be purified by recrystallization from anhydrous methanol to provide the title compounds 3.
3,4-Diphenyl-2-methyl-1-pyrrolo[1,2-a]pyrazinone (3a, R1=H, R2=H). Yield 65.6%; overall yield 13.4%; m.p. 195-196°C; 1H-NMR (DMSO) δ (ppm): 3.05 (s, 3H, N-CH3), 6.53 (br. t, 1H, H-7), 6.64 (br. s, 1H, H-8), 7.03 (br. d, 1H, H-6), 7.28-7.35 (m, 10H, Ar-H); IR (KBr) ν(cm-1): 1627.9 (amide, C=O); MS (m/z): 301 (M++1).
4-(4-Chlorophenyl)-2-methyl-3-phenyl-1-pyrrolo[1,2-a]pyrazinone (3b, R1=Cl, R2=H). Yield 60.2%, overall yield 10.9%; m.p. 202-204°C; 1H-NMR (DMSO) δ (ppm): 3.05 (s, 3H, N-CH3), 6.56 (br. t, 1H, H-7), 6.70 (br. d, 1H, H-8), 7.05 (dd, 1H, J=1.2Hz, 3.9Hz, H-6), 7.30-7.37 (m, 9H, Ar-H); IR (KBr) ν(cm-1): 1628.6 (amide, C=O); MS (m/z): 335 (M++1).
2-Methyl-3-(4-methylphenyl)-4-phenyl-1-pyrrolo[1,2-a]pyrazinone (3c, R1=H, R2=CH3). Yield 67.8%, overall yield 16.6%; m.p. 204-206°C; 1H-NMR (DMSO) δ (ppm): 2.24 (s, 3H, Ar-CH3), 3.04 (s, 3H, N-CH3), 6.54 (br. d, 1H, H-7), 6.61 (br. s, 1H, H-8), 7.02 (br. d, 1H, H-6), 7.10 (d, 2H, J=7.9Hz, Ar-H), 7.22 (d, 2H, J=7.9Hz, Ar-H), 7.31 (s, 5H, Ar-H); IR (KBr) ν(cm-1): 1631.9 (amide, C=O); MS (m/z): 315 (M++1).
2-Methyl-4-(-methoxyphenyl)-3-(-methylphenyl)-1-pyrrolo[1,2-a]pyrazinone (3d, R1=OCH3, R2=CH3). Yield 94.7%, overall yield 28.8%; m.p. 219-221°C; 1H-NMR (DMSO) δ(ppm): 2.30 (s, 3H, Ar-CH3), 3.07 (s, 3H, N-CH3), 3.75 (s, 3H, -OCH3), 6.42 (dd, 1H, J=2.7Hz, 3.9Hz, H-7), 6.62 (dd, 1H, J=1.5Hz, 2.7Hz, H-8), 6.79 (d, 2H, J=9.0Hz, Ar-H), 6.96 (dd, 1H, J=1.5Hz, 3.9Hz, H-6), 7.05-7.15 (m, 6H, Ar-H); IR (KBr) ν(cm-1): 1633.8 (amide, C=O); MS (m/z): 345 (M++1).
4-(4-Chlorophenyl)-2-methyl-3-(4-methylsulfonylphenyl)-1-pyrrolo[1,2-a]pyrazinone (3e, R1=Cl, R2=SO2CH3). Yield 18.5%; overall yield 6.7%; m.p. 235-237°C; 1H-NMR (DMSO) δ (ppm): 3.04 (s, 3H, N-CH3), 3.22 (s, 3H, -SO2CH3), 6.57 (dd, 1H, J=2.7Hz, 3.9Hz, H-7), 6.72 (dd, 1H, J=1.6Hz, 2.7Hz, H-8), 7.07 (dd, 1H, J=1.6Hz, 3.9Hz, H-6), 7.36 (d, 2H, J=8.7Hz, Ar-H), 7.42 (d, 2H, J=8.7Hz, Ar-H), 7.67 (d, 2H, J=8.4Hz, Ar-H), 7.87 (d, 2H, J=8.4Hz, Ar-H); IR (KBr) ν(cm-1): 1305.2, 1149.8 (O=S=O), 1630.5 (amide, C=O); MS (m/z): 413 (M++1).

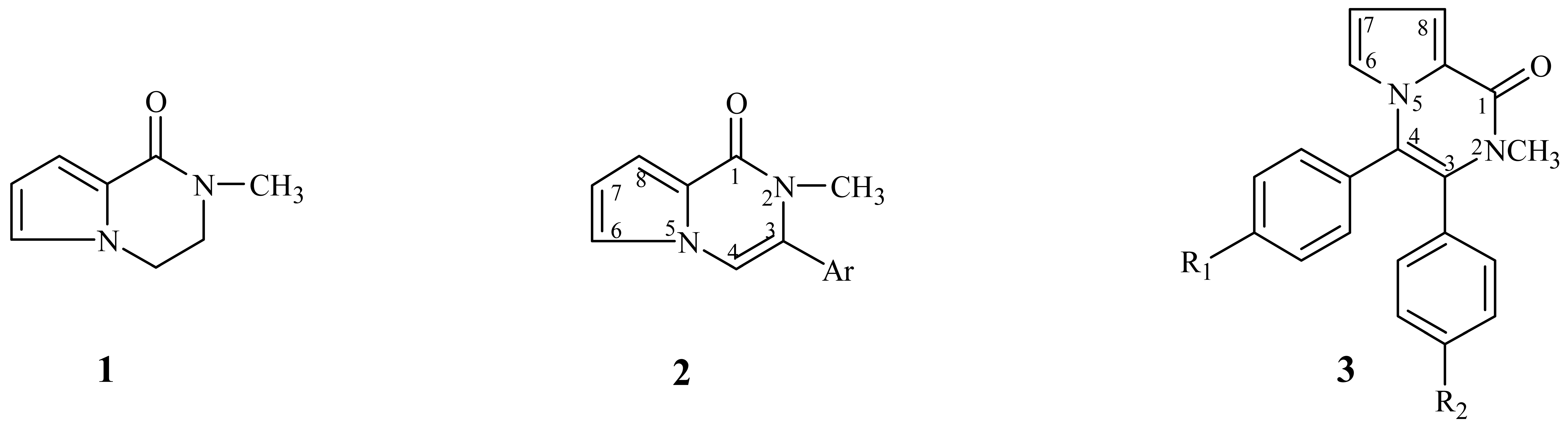 Their structure-activity relationships (SAR) were studied and the results suggested that a large hydrophobic substituent around the 3-position of the pyrazinone ring could result in an additional contribution to the activity. This encouraged us to design and synthesize new aryl derivatives of 2-methyl-1-pyrrolo[1,2-a] pyrazinone (2, 3).
Their structure-activity relationships (SAR) were studied and the results suggested that a large hydrophobic substituent around the 3-position of the pyrazinone ring could result in an additional contribution to the activity. This encouraged us to design and synthesize new aryl derivatives of 2-methyl-1-pyrrolo[1,2-a] pyrazinone (2, 3).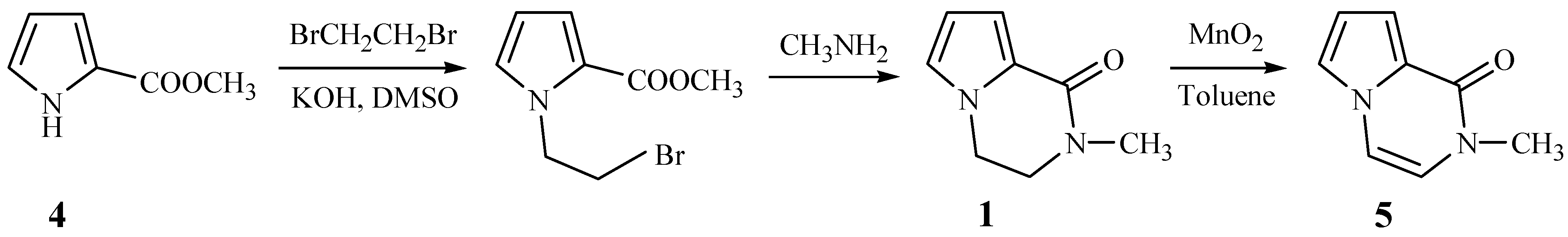



{kind=link}
{kind=link}
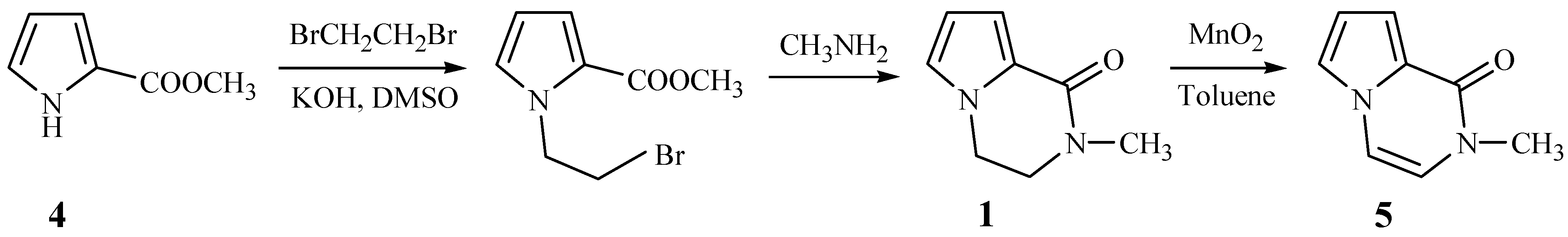{kind=link}
{kind=link}
{kind=link}