Preparation and Crystal Structure of (1S, 5S, 7S, 8R)-8-Hydroxy-7-phenyl-2,6-dioxabicyclo[3.3.0]octan-3-one

Abstract
:Introduction
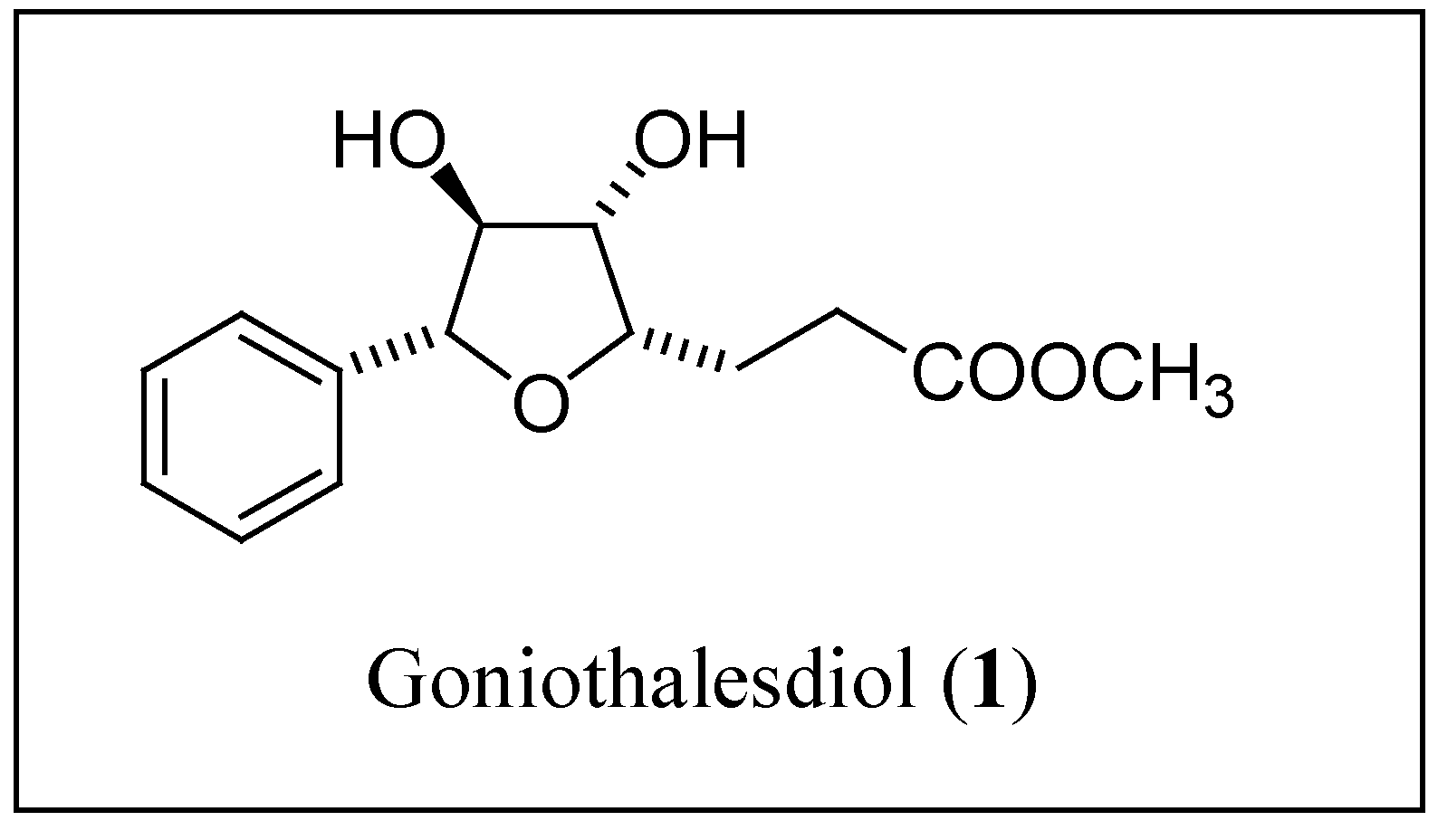
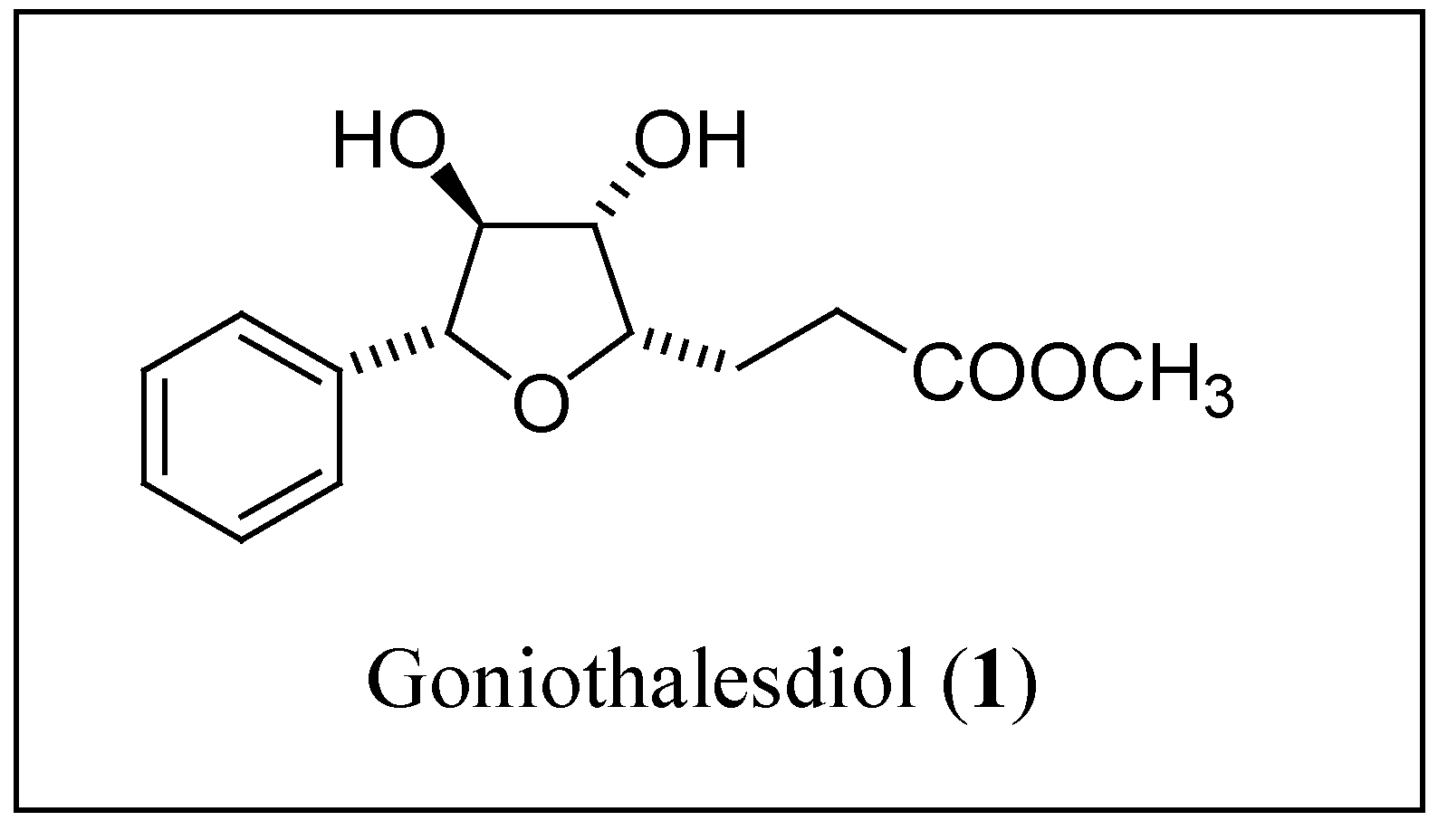
Results and Discussion
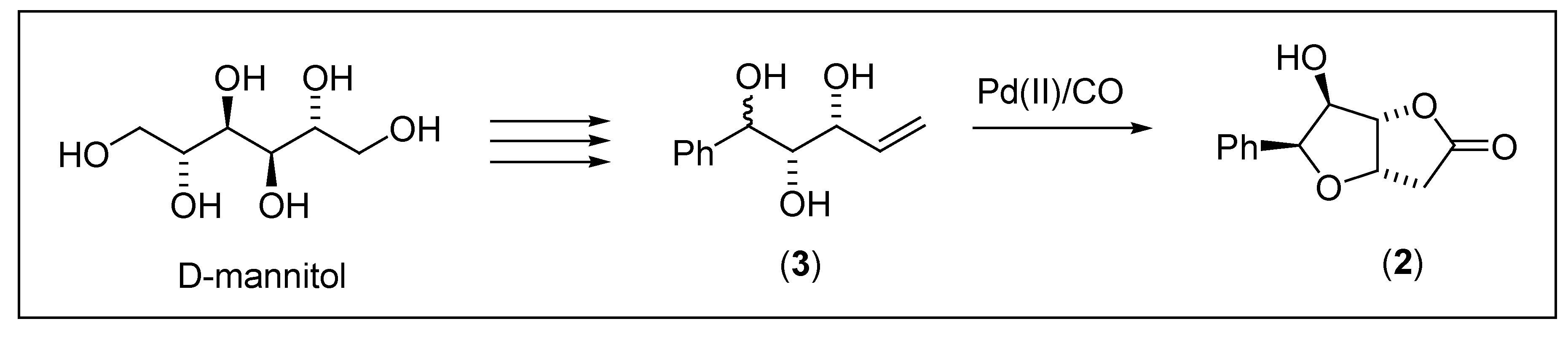
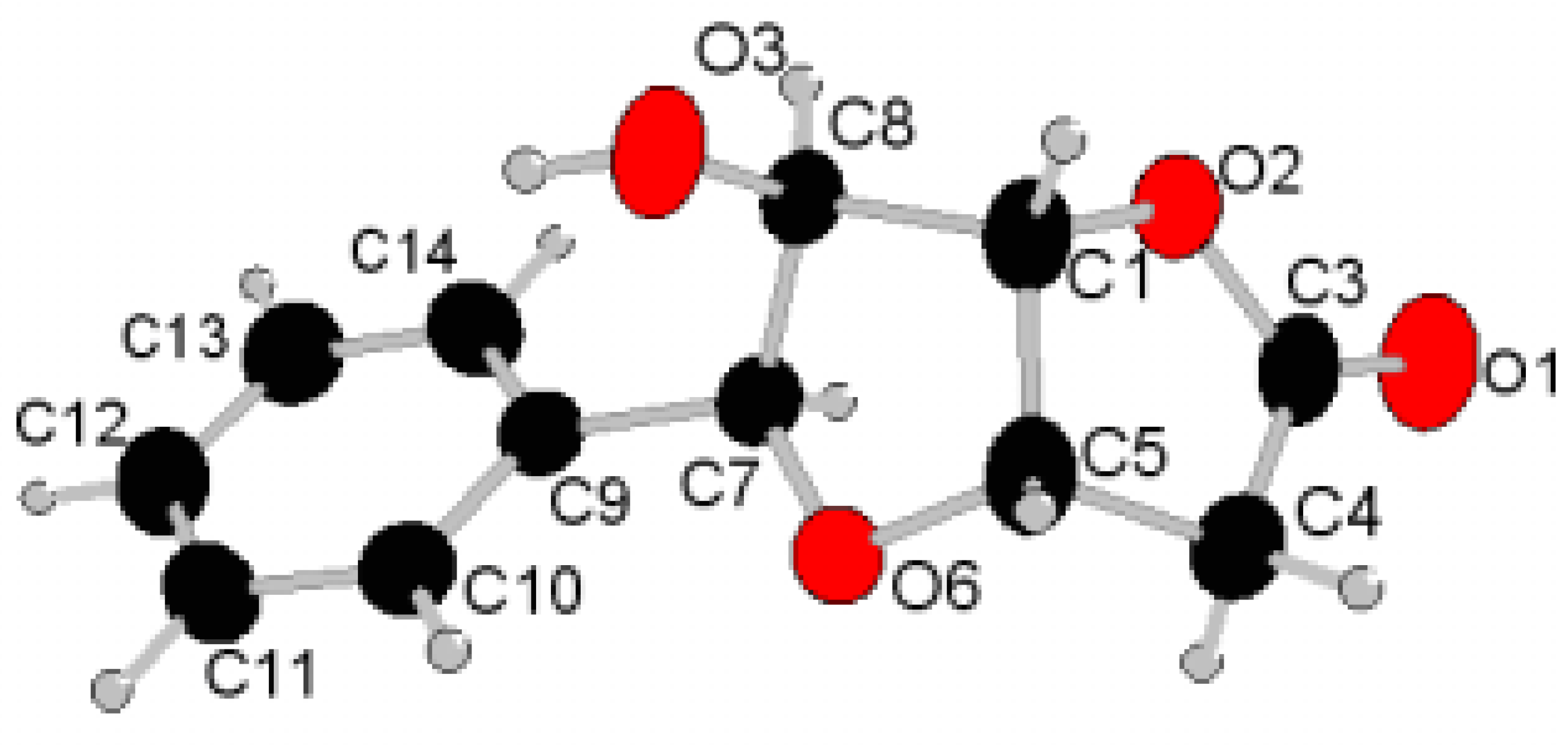

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C12H12O4 |
| Formula weight | 220.22 |
| Temperature, T (K) | 183(2) |
| Wavelength, λ (Å) | 0.71073 |
| Crystal system | orthorhombic |
| Space group | P212121 |
| Unit cell dimensions (Å) | a = 5.7069(1) |
| b = 8.4010(1) | |
| c = 21.2427(4) | |
| Unit cell volume, V (Å3) | 1018.45(3) |
| Formula units per unit cell Z | 4 |
| Dcalcd (g/cm3) | 1.436 |
| Absorption coefficient, μ (mm-1) | 0.108 |
| F(000) | 464 |
| Crystal size (mm) | 0.70 × 0.54 × 0.44 |
| Diffractometer | Siemens SMART CCD |
| θ Range (°) | 2.61–28.25 |
| Range of h | –7→7 |
| Range of k | –11→11 |
| Range of l | –27→27 |
| Reflections | 11525 |
| Independent reflections | 2371 (Rint = 0.0328) |
| Completeness to θ = 28.25 (%) | 95.7 |
| Absorption correction | multi-scan |
| Max. and min. transmission | 0.9539 and 0.9281 |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 2371 / 0 / 158 |
| Goodness-of-fit on F2 | 1.002 |
| Final R indices [I>2σ(I)] | R1 = 0.0336, wR2 = 0.0798 |
| R indices (all data) | R1 = 0.0400, wR2 = 0.0846 |
| Largest difference peak and hole (e/Å3) | 0.195 and –0.209 |
| O(1)-C(3) | 1.2065(17) | C(5)-C(1)-C(8) | 104.37(11) | |
| O(2)-C(3) | 1.3533(18) | O(1)-C(3)-O(2) | 120.39(15) | |
| O(2)-C(1) | 1.4474(16) | O(1)-C(3)-C(4) | 128.97(14) | |
| O(3)-C(8) | 1.4211(15) | O(2)-C(3)-C(4) | 110.63(12) | |
| O(6)-C(7) | 1.4379(17) | C(3)-C(4)-C(5) | 105.48(12) | |
| O(6)-C(5) | 1.4382(17) | O(6)-C(5)-C(1) | 106.39(11) | |
| C(1)-C(5) | 1.540(2) | O(6)-C(5)-C(4) | 112.97(13) | |
| C(1)-C(8) | 1.5206(18) | C(1)-C(5)-C(4) | 103.71(12) | |
| C(3)-C(4) | 1.499(2) | O(6)-C(7)-C(9) | 112.15(12) | |
| C(4)-C(5) | 1.524(2) | O(6)-C(7)-C(8) | 104.29(11) | |
| C(7)-C(9) | 1.5076(19) | C(9)-C(7)-C(8) | 114.36(11) | |
| C(7)-C(8) | 1.5315(19) | O(3)-C(8)-C(1) | 105.62(11) | |
| C(3)-O(2)-C(1) | 111.22(11) | O(3)-C(8)-C(7) | 112.58(12) | |
| C(7)-O(6)-C(5) | 108.55(11) | C(1)-C(8)-C(7) | 101.00(11) | |
| O(2)-C(1)-C(5) | 106.51(11) | C(10)-C(9)-C(7) | 123.04(13) | |
| O(2)-C(1)-C(8) | 109.74(11) | C(14)-C(9)-C(7) | 117.78(13) |
| O(2)-C(1)-C(5)-C(4) | 15.69(14) | O(2)-C(1)-C(5)-O(6) | –103.68(12) | |
| C(1)-C(5)-C(4)-C(3) | –12.53(15) | C(8)-C(1)-C(5)-C(4) | 131.77(11) | |
| C(5)-C(4)-C(3)-O(2) | 5.14(16) | C(3)-C(4)-C(5)-O(6) | 102.24(14) | |
| C(4)-C(3)-O(2)-C(1) | 5.32(16) | C(5)-O(6)-C(7)-C(9) | –157.52(11) | |
| C(3)-O(2)-C(1)-C(5) | –13.49(14) | O(2)-C(1)-C(8)-O(3) | –159.61(11) | |
| O(6)-C(5)-C(1)-C(8) | 12.40(15) | C(5)-C(1)-C(8)-O(3) | 86.59(13) | |
| C(5)-C(1)-C(8)-C(7) | –30.83(13) | O(2)-C(1)-C(8)-C(7) | 82.97(13) | |
| C(1)-C(8)-C(7)-O(6) | 39.33(13) | O(6)-C(7)-C(8)-O(3) | –72.88(14) | |
| C(8)-C(7)-O(6)-C(5) | –33.23(15) | C(9)-C(7)-C(8)-O(3) | 49.98(16) | |
| C(7)-O(6)-C(5)-C(1) | 13.00(15) | C(9)-C(7)-C(8)-C(1) | 162.18(12) | |
| C(3)-O(2)-C(1)-C(8) | –125.91(12) | C(8)-C(7)-C(9)-C(10) | –106.15(16) | |
| C(7)-O(6)-C(5)-C(4) | –100.14(14) | O(6)-C(7)-C(9)-C(14) | –168.01(12) |
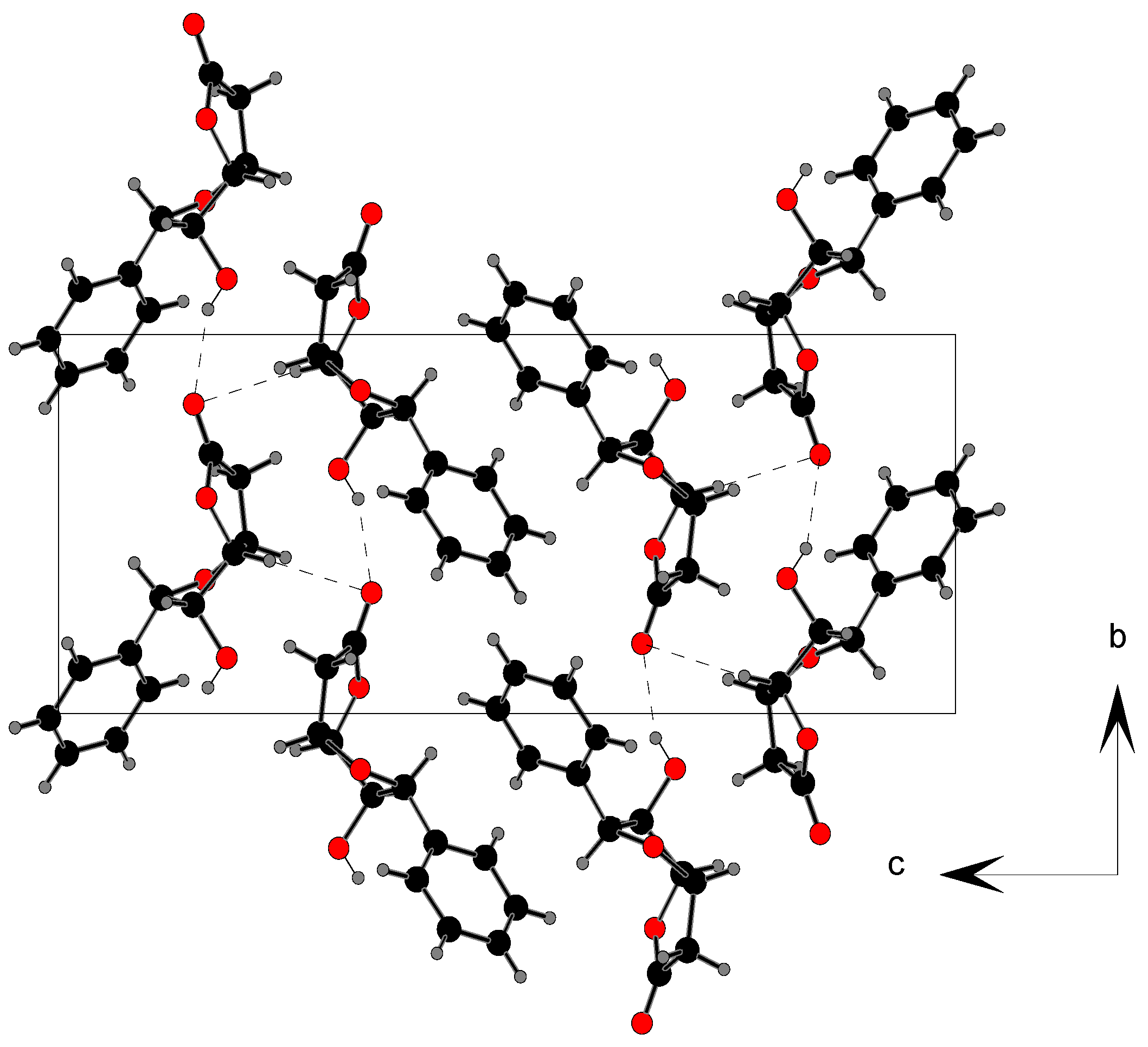
| X–H…Y | Symmetry code | X–H(Å) | H…Y(Å) | X…Y(Å) | X–H…Y(°) |
| O(3)–H(3)…O(1) | x, y+1, z | 0.84 | 2.12 | 2.8885(14) | 151.2 |
| C(1)–H(1)…O(1) | –x, y+0.5, –z+0.5 | 1.00 | 2.55 | 3.4976(19) | 157.3 |
Experimental
General
X-ray techniques
(1S, 5S, 7S, 8R)-8-Hydroxy-7-phenyl-2,6-dioxabicyclo[3.3.0]octan-3-one (2).
Acknowledgements
References and Notes
- Gracza, T.; Hasenöhrl, T.; Stahl, U.; Jäger, V. Synthesis of 3,5-Anhydro-2-deoxy-1,4-glyconolactones by Palladium(II)-Catalyzed, Regioselective Oxycarbonylation of C5- and C6-Enitols. ω-Homologation of Aldoses to Produce Intermediates for C-Glycoside/C-Nucleoside Synthesis. Synthesis 1991, 1108–1118. [Google Scholar]
- Gracza, T.; Jäger, V. Palladium(II)-Catalyzed Oxycarbonylation of Unsaturated Polyols: Synthesis of (-)-Goniofufurone and Assignment of Absolute Configuration to the Natural (+)-Enantiomer, a Cytotoxic Styryllactone. Synlett 1992, 191–193. [Google Scholar]
- Gracza, T.; Jäger, V. Synthesis of Natural and Unnatural Enantiomers of Goniofufurone and Its 7-Epimers from D-Glucose. Application of Palladium(II)-Catalyzed Oxycarbonylation of Unsaturated Polyols. Synthesis 1994, 1359–1368. [Google Scholar]
- Dixon, D. J.; Ley, S.V.; Gracza, T.; Szolcsanyi, P. Total synthesis of the polyenoyltetramic acid mycotoxin erythroskyrine. J. Chem. Soc., Perkin Trans. 1 1999, 839–841. [Google Scholar]
- Babjak, M.; Kapitán, P.; Gracza, T. The First Total Synthesis of Goniothalesdiol. Tetrahedron Lett. 2002, 43, 6983–6985. [Google Scholar] [CrossRef]
- Cao, Sh.-G.; Wu, X.-H.; Sim, K.-Y.; Tan, B. K. H.; Pereira, J. T.; Goh, S.-H. Styryl-Lactone Derivatives and Alkaloids from Goniothalamus borneensis (Annonaceae). Tetrahedron 1998, 54, 2143–2148. [Google Scholar] [CrossRef]
- CCDC 194248 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (Fax: +44-1223-336030; e-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
- Cremer, D.; Pople, J. A. A General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1573. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R. E.; Shimoni, L.; Chang, L.-N. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem., Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar]
- Siemens AXS. SMART & SAINT: Madison, WI, USA, 1995.
- Sheldrick, G. M. Program SADABS; University of Göttingen: Germany, 2001. [Google Scholar]
- Blessing, R. H. An Empirical Correction for Absorption Anisotropy. Acta Crystallogr., Sect. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bruker AXS Inc. SHELXTL Version 6.10; Madison, WI, USA, 2001. [Google Scholar]
- Brandenburg, K. DIAMOND: Visual Crystal Structure Information System, Version 2.1d; Crystal Impact GbR: Bonn, Germany, 2000. [Google Scholar]
- Samples Availability: Compound 2 reported in this paper is available from MDPI.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
Langer, V.; Gyepesová, D.; Koman, M.; Kapitán, P.; Babjak, M.; Gracza, T.; Koóš, M. Preparation and Crystal Structure of (1S, 5S, 7S, 8R)-8-Hydroxy-7-phenyl-2,6-dioxabicyclo[3.3.0]octan-3-one. Molecules 2003, 8, 599-606. https://doi.org/10.3390/80700599
Langer V, Gyepesová D, Koman M, Kapitán P, Babjak M, Gracza T, Koóš M. Preparation and Crystal Structure of (1S, 5S, 7S, 8R)-8-Hydroxy-7-phenyl-2,6-dioxabicyclo[3.3.0]octan-3-one. Molecules. 2003; 8(7):599-606. https://doi.org/10.3390/80700599
Chicago/Turabian StyleLanger, Vratislav, Dalma Gyepesová, Marian Koman, Peter Kapitán, Matej Babjak, Tibor Gracza, and Miroslav Koóš. 2003. "Preparation and Crystal Structure of (1S, 5S, 7S, 8R)-8-Hydroxy-7-phenyl-2,6-dioxabicyclo[3.3.0]octan-3-one" Molecules 8, no. 7: 599-606. https://doi.org/10.3390/80700599
APA StyleLanger, V., Gyepesová, D., Koman, M., Kapitán, P., Babjak, M., Gracza, T., & Koóš, M. (2003). Preparation and Crystal Structure of (1S, 5S, 7S, 8R)-8-Hydroxy-7-phenyl-2,6-dioxabicyclo[3.3.0]octan-3-one. Molecules, 8(7), 599-606. https://doi.org/10.3390/80700599