The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries

{kind=link}
{kind=link}
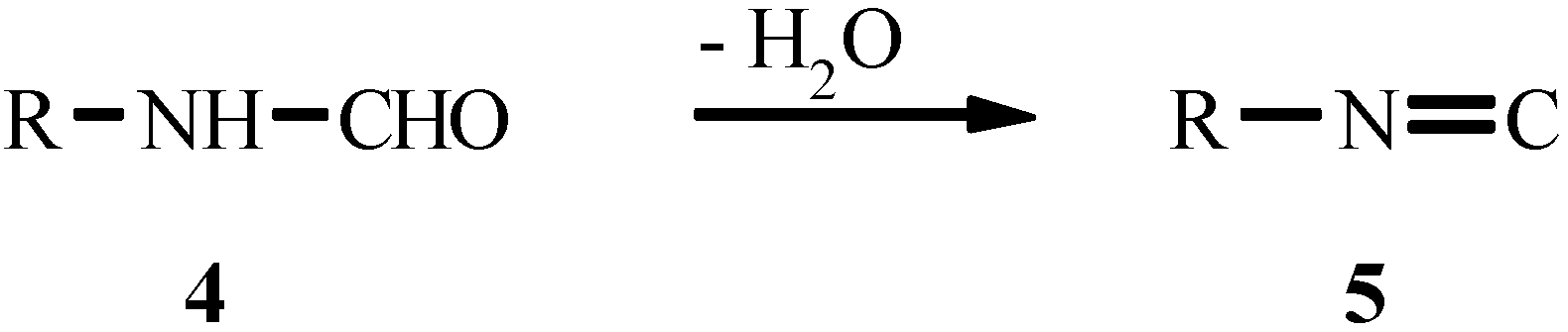{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. SYNTHESES BY ONE-POT REACTIONS
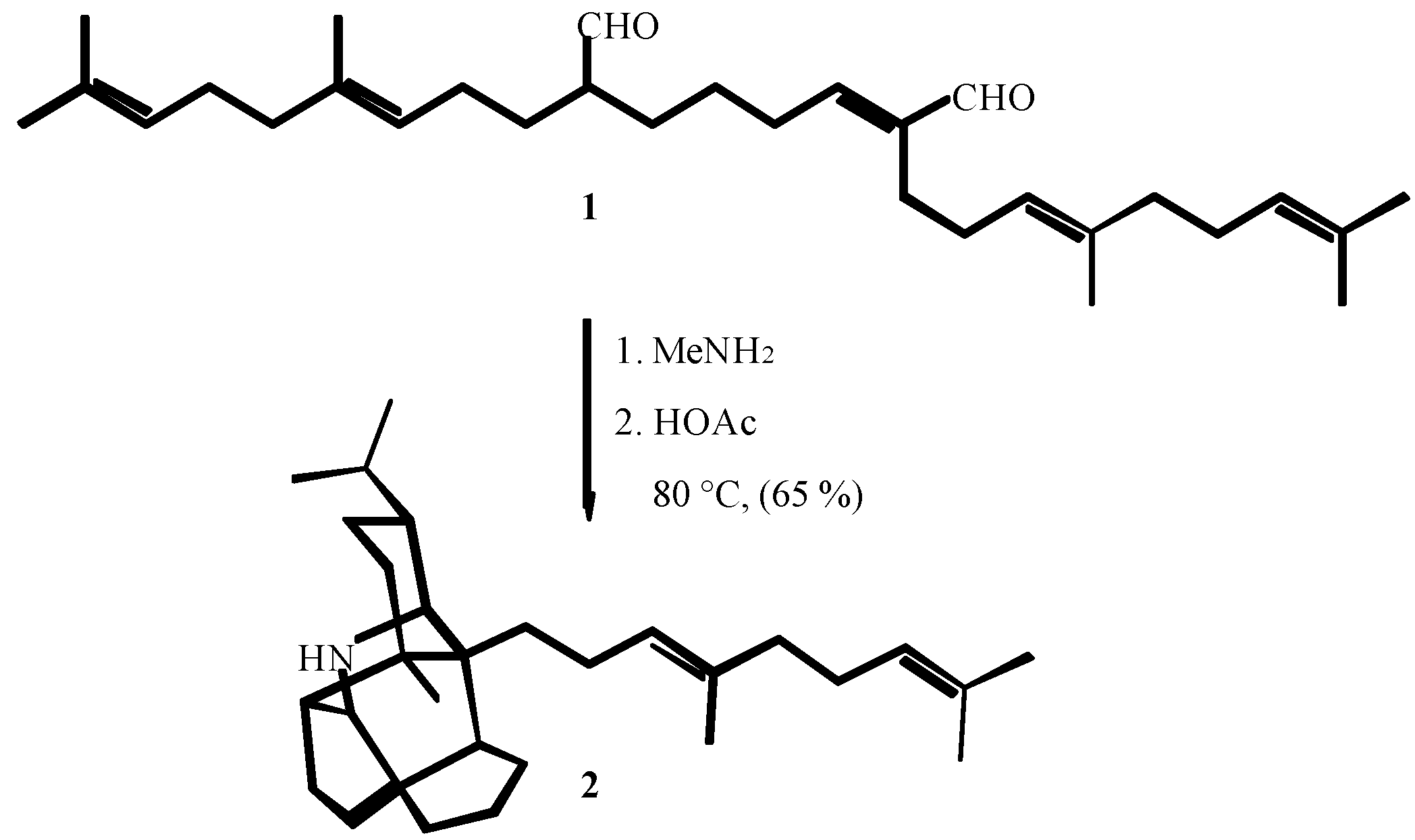
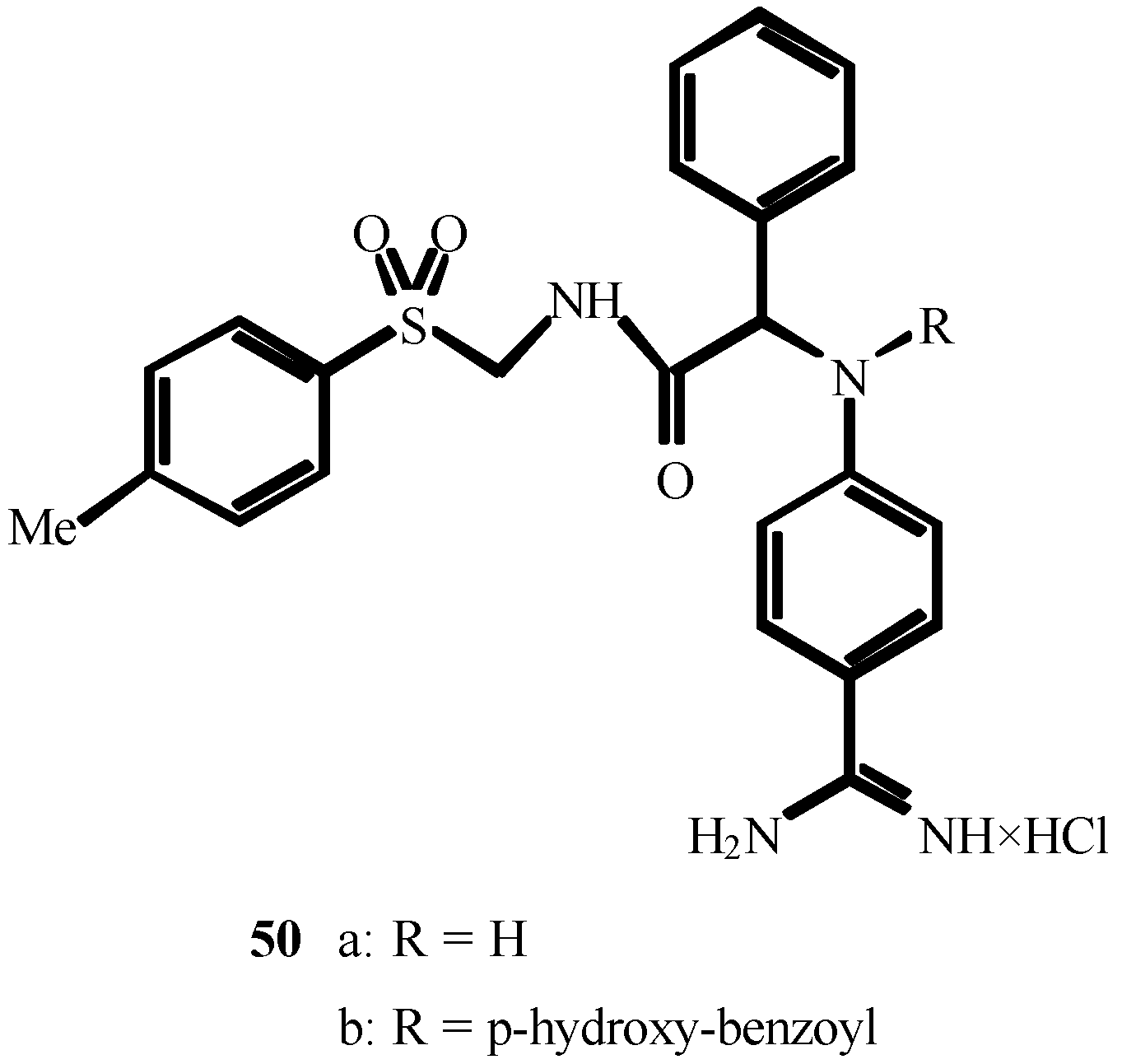
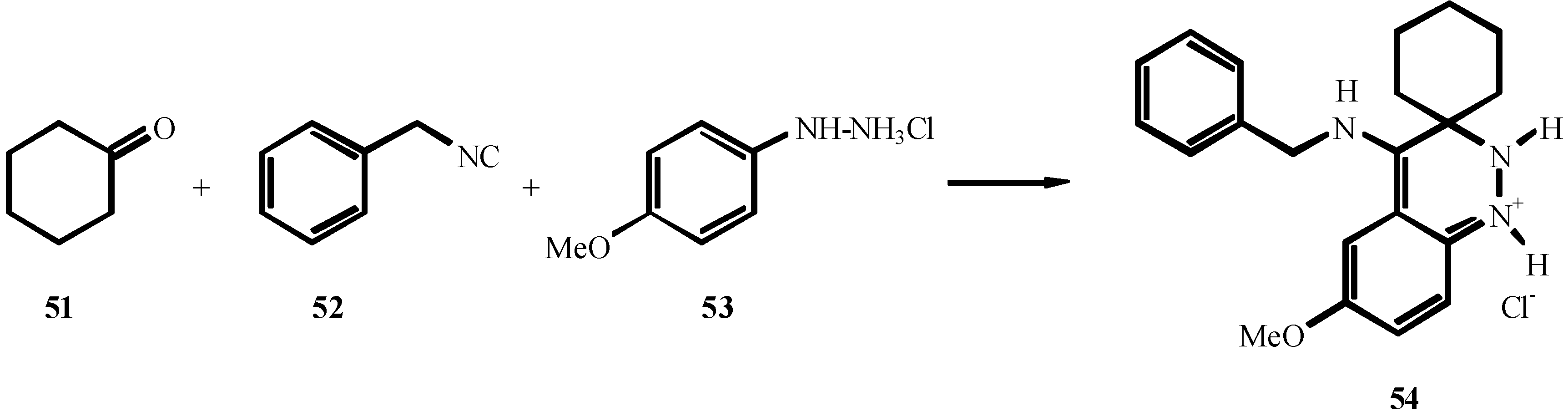
2. THE FIRST CENTURY OF ISOCYANIDE CHEMISTRY
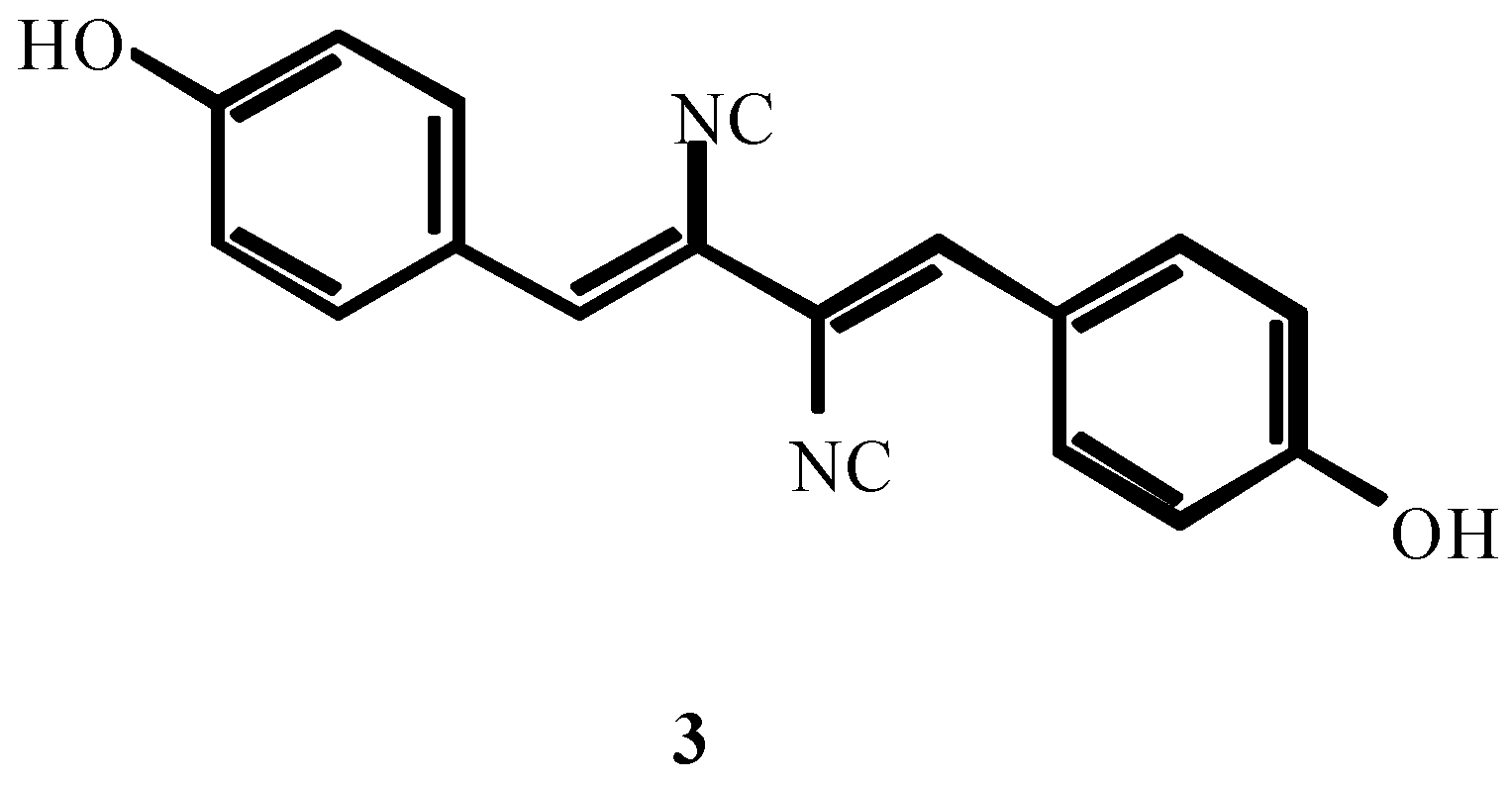
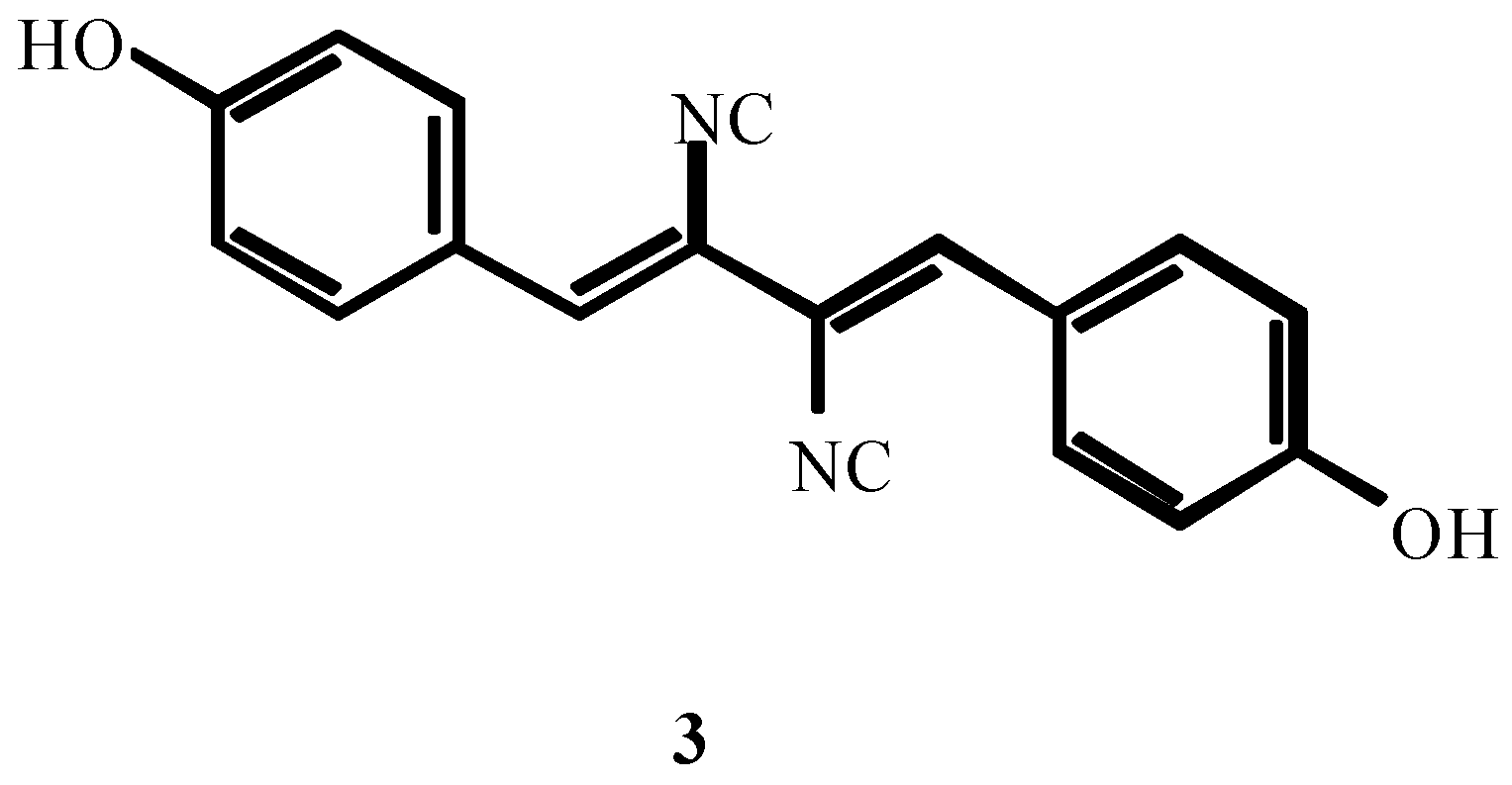
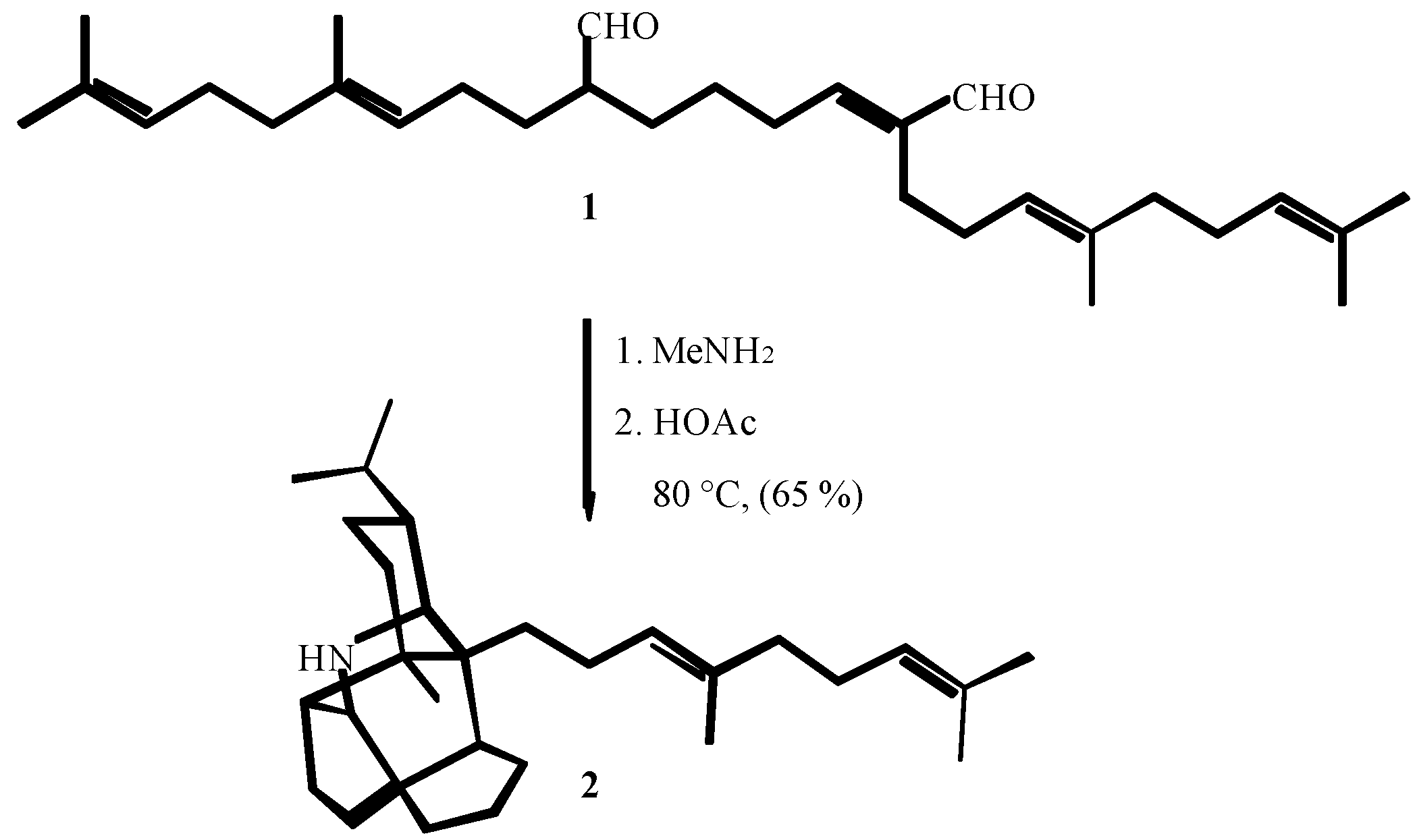
3. THE SECOND PERIOD OF THE ISOCYANIDE CHEMISTRY

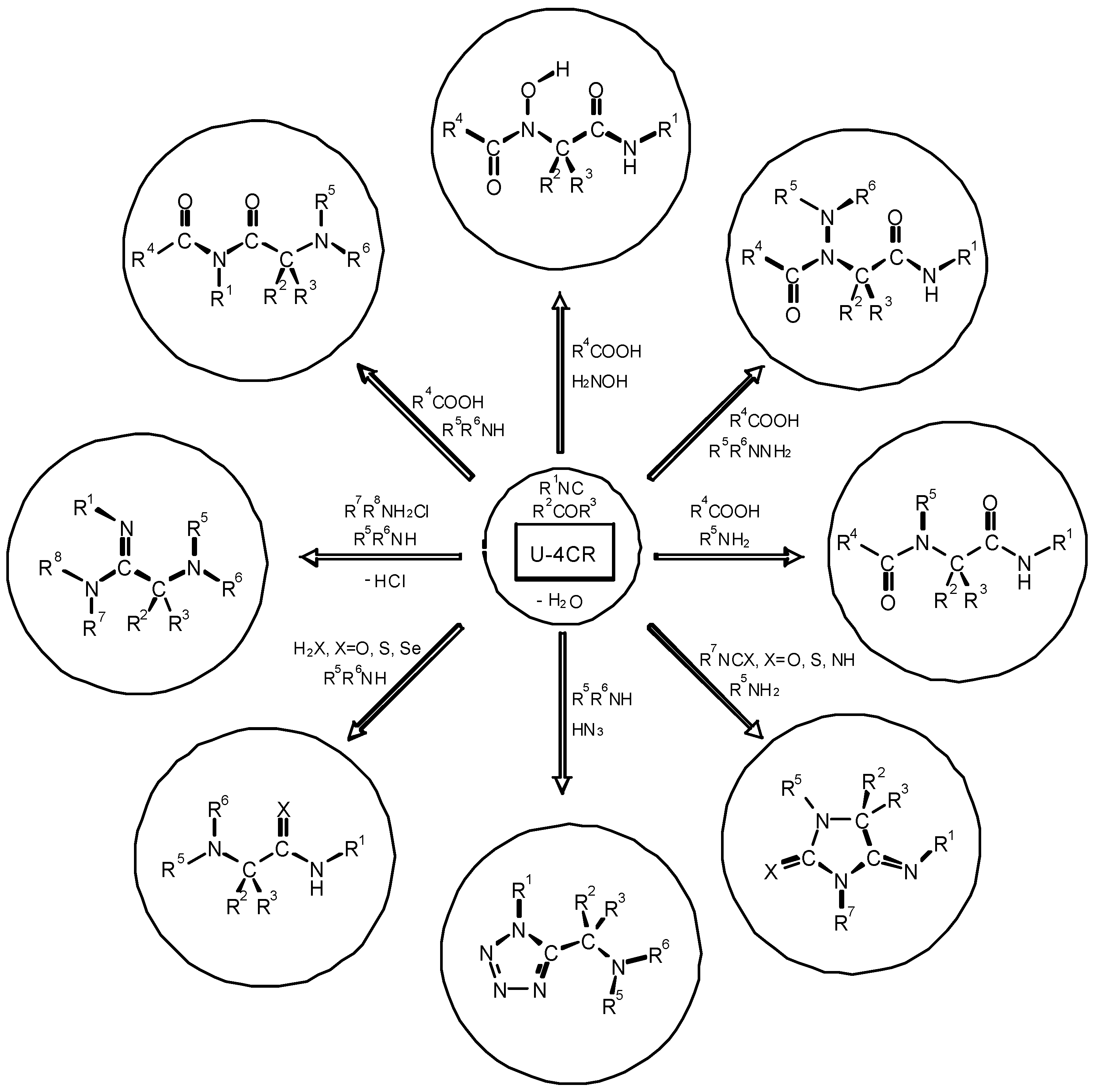

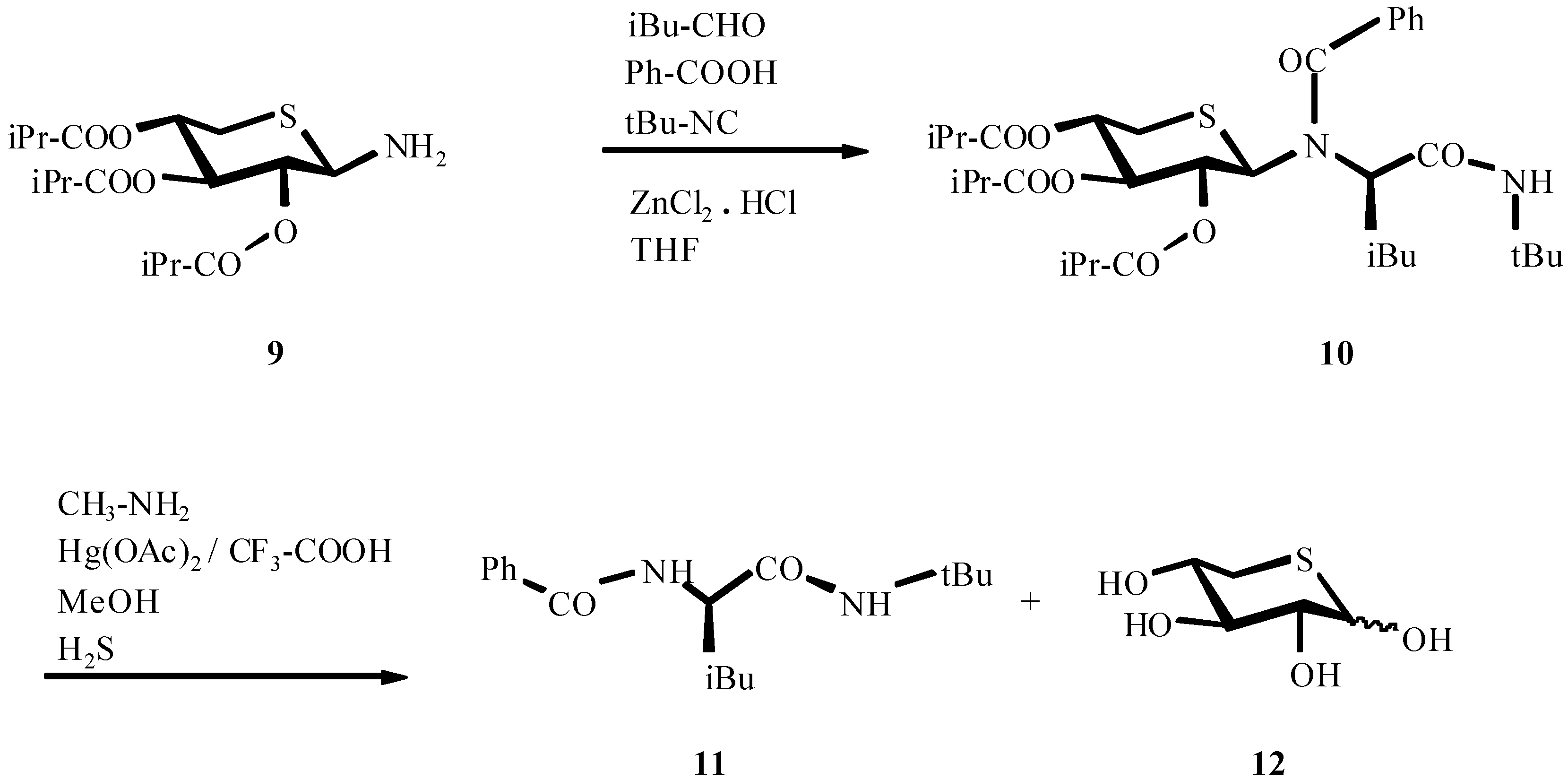
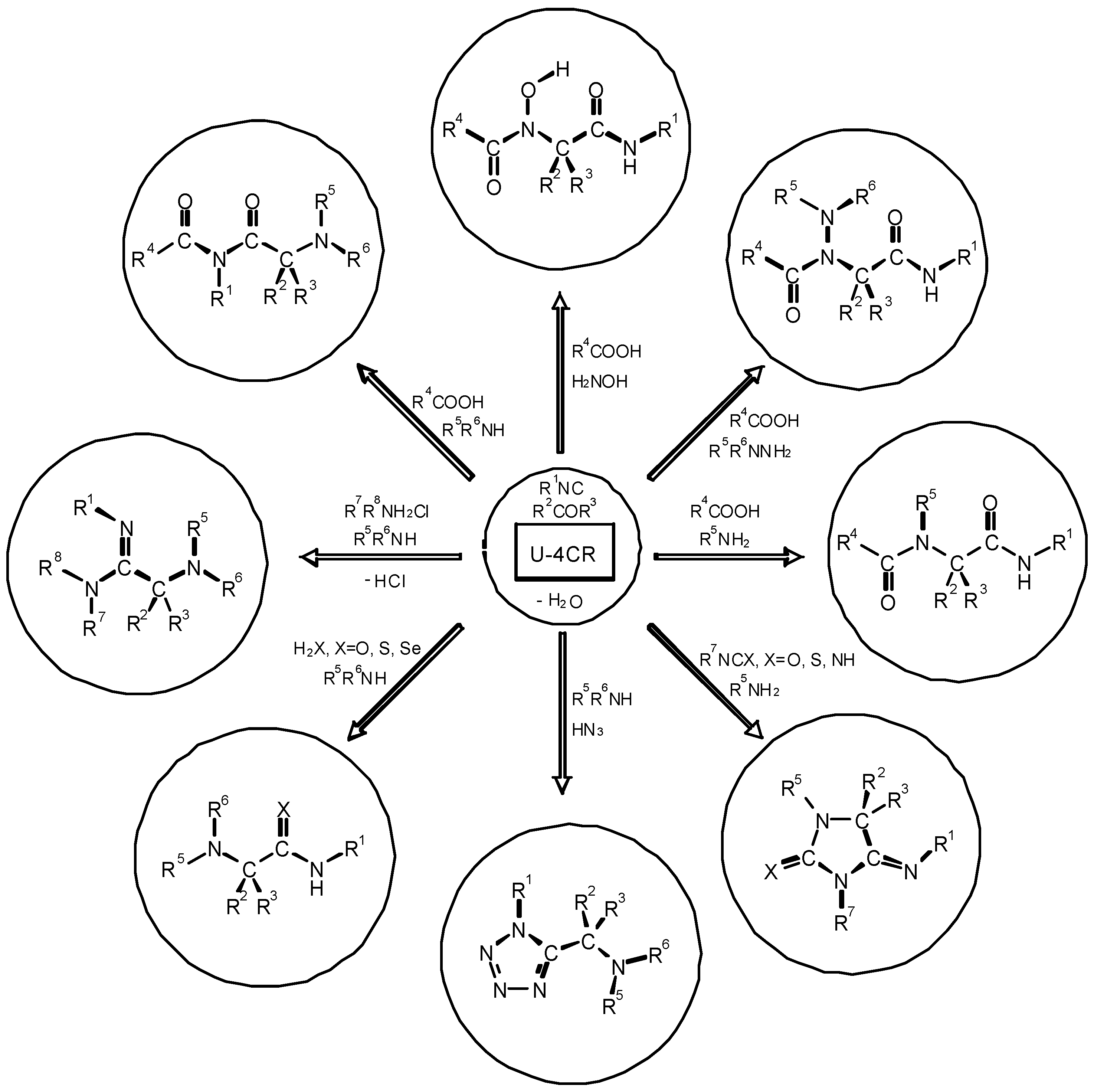
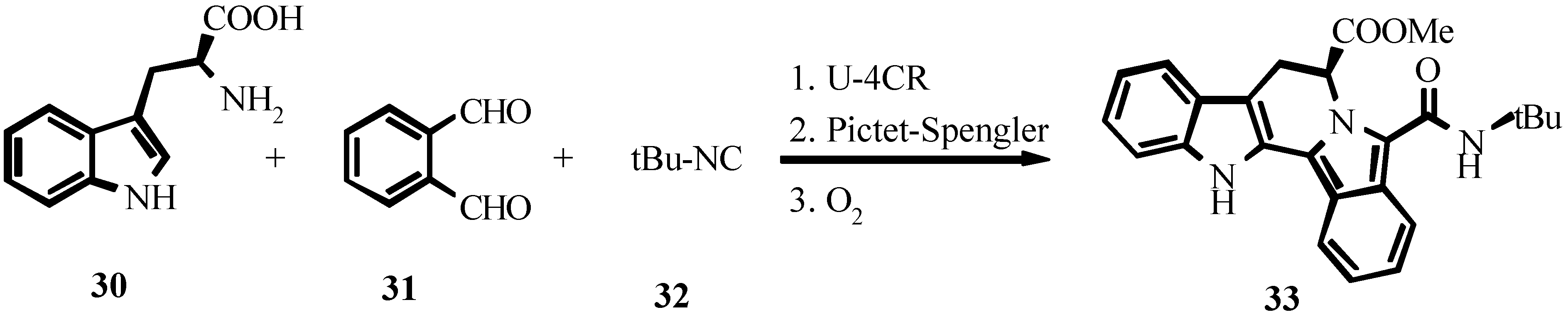
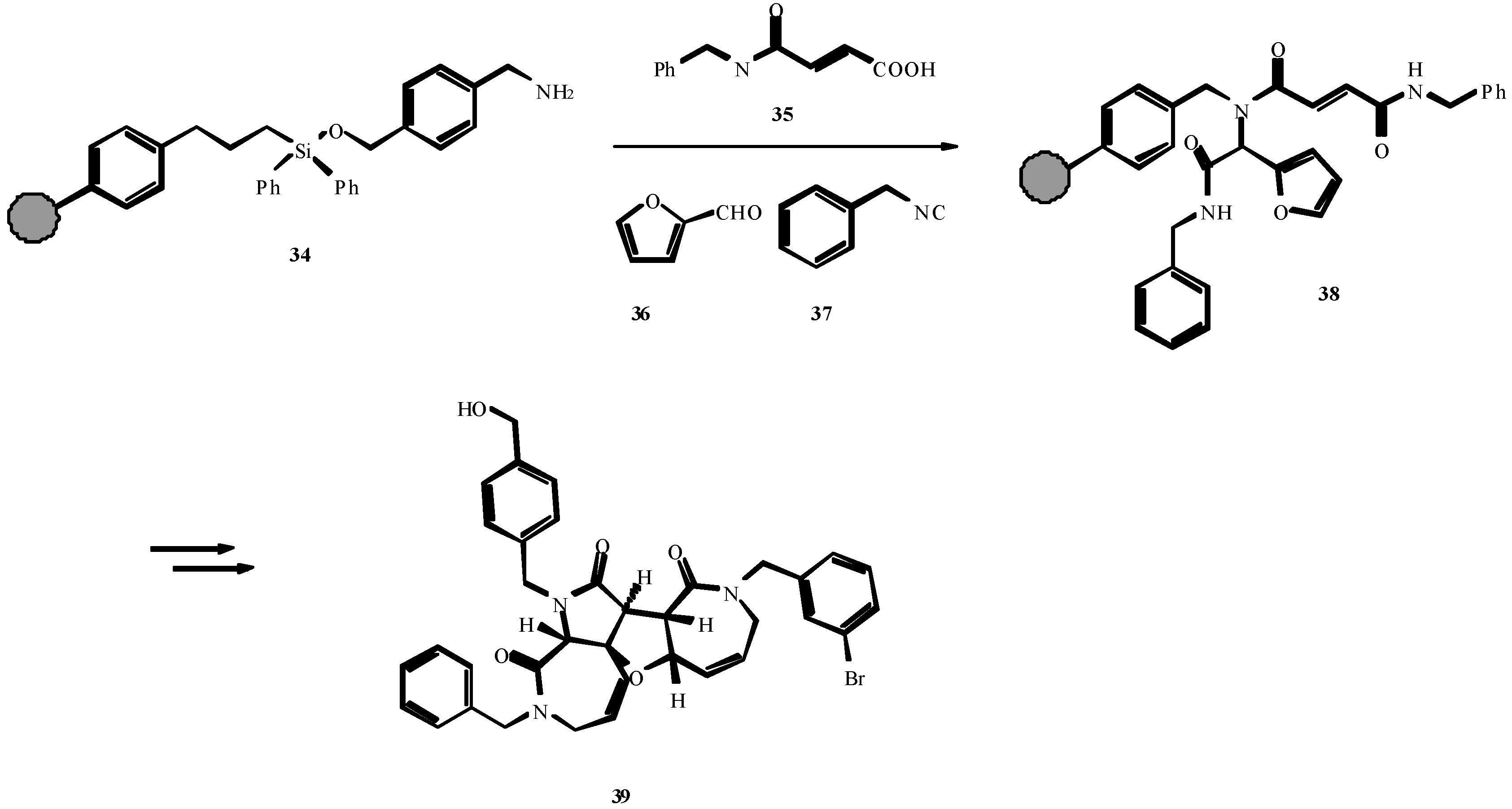
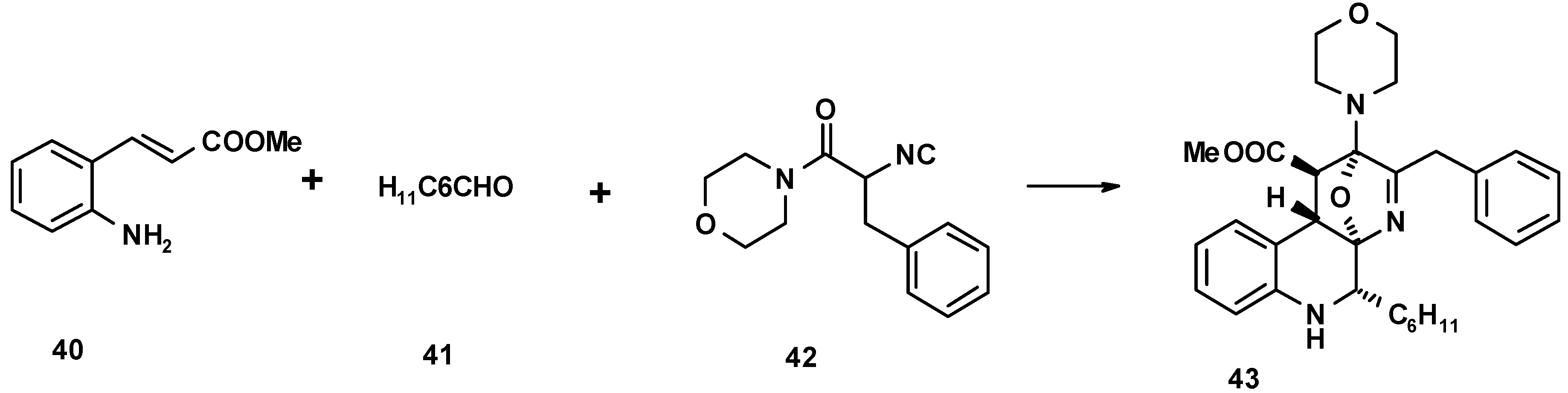
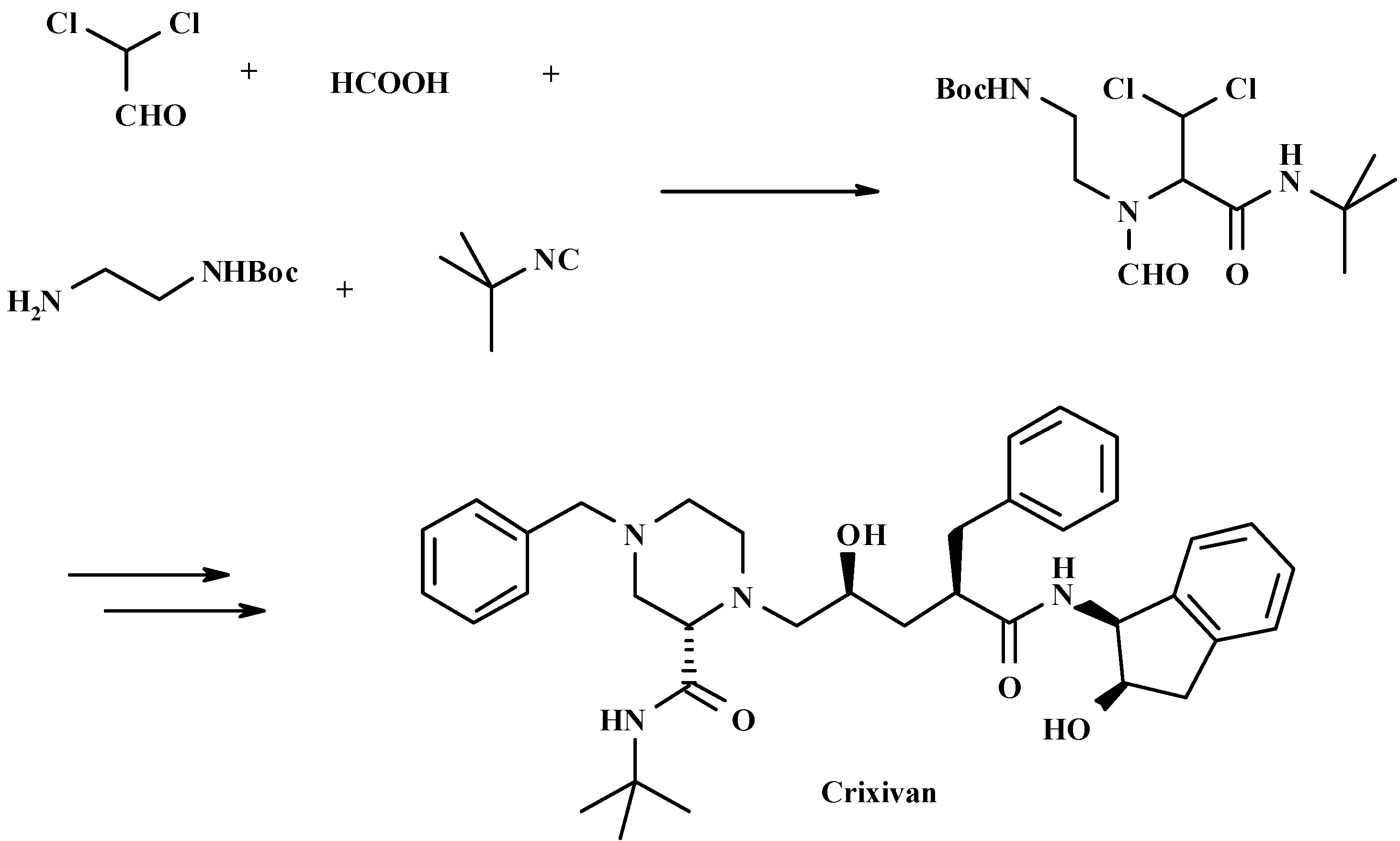
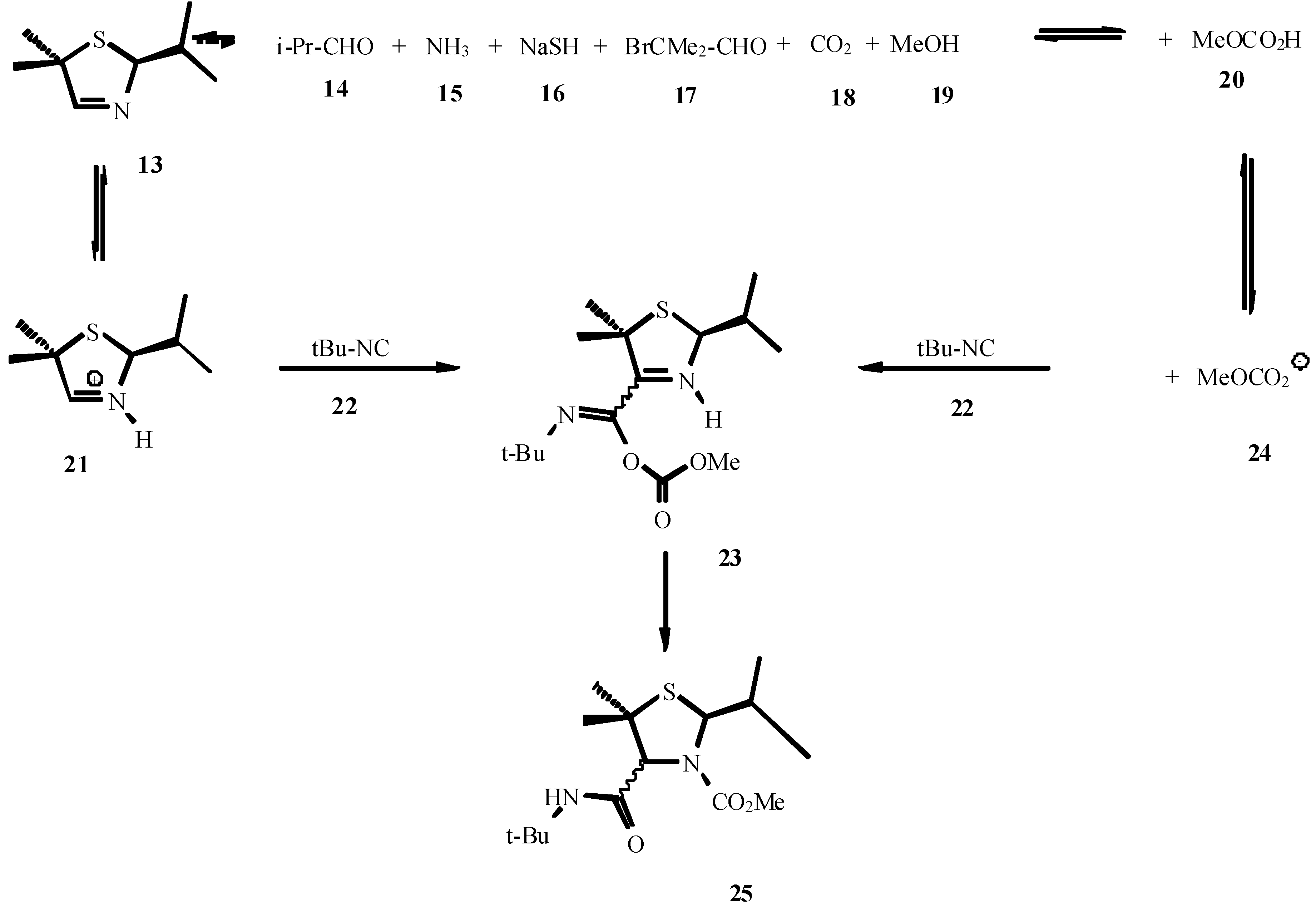
4. THE LAST PERIOD OF ISOCYANIDE CHEMISTRY

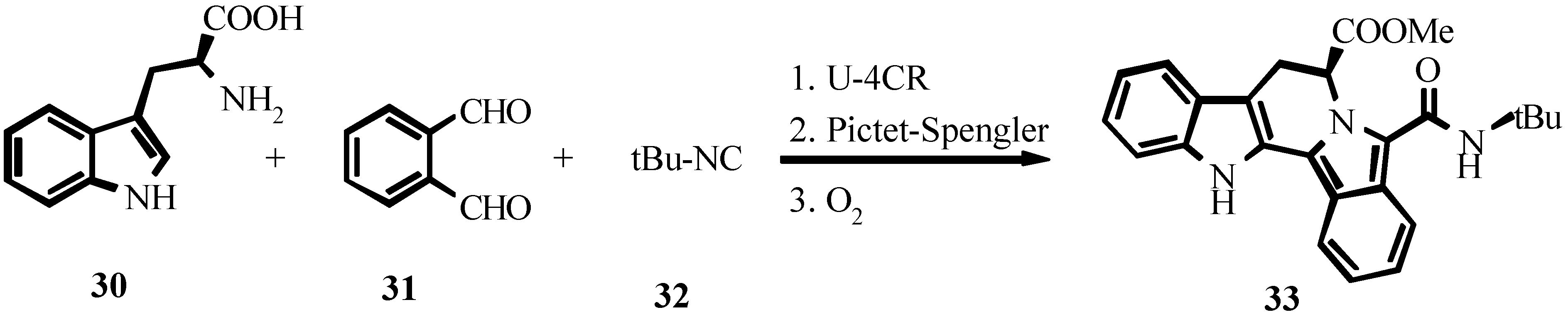
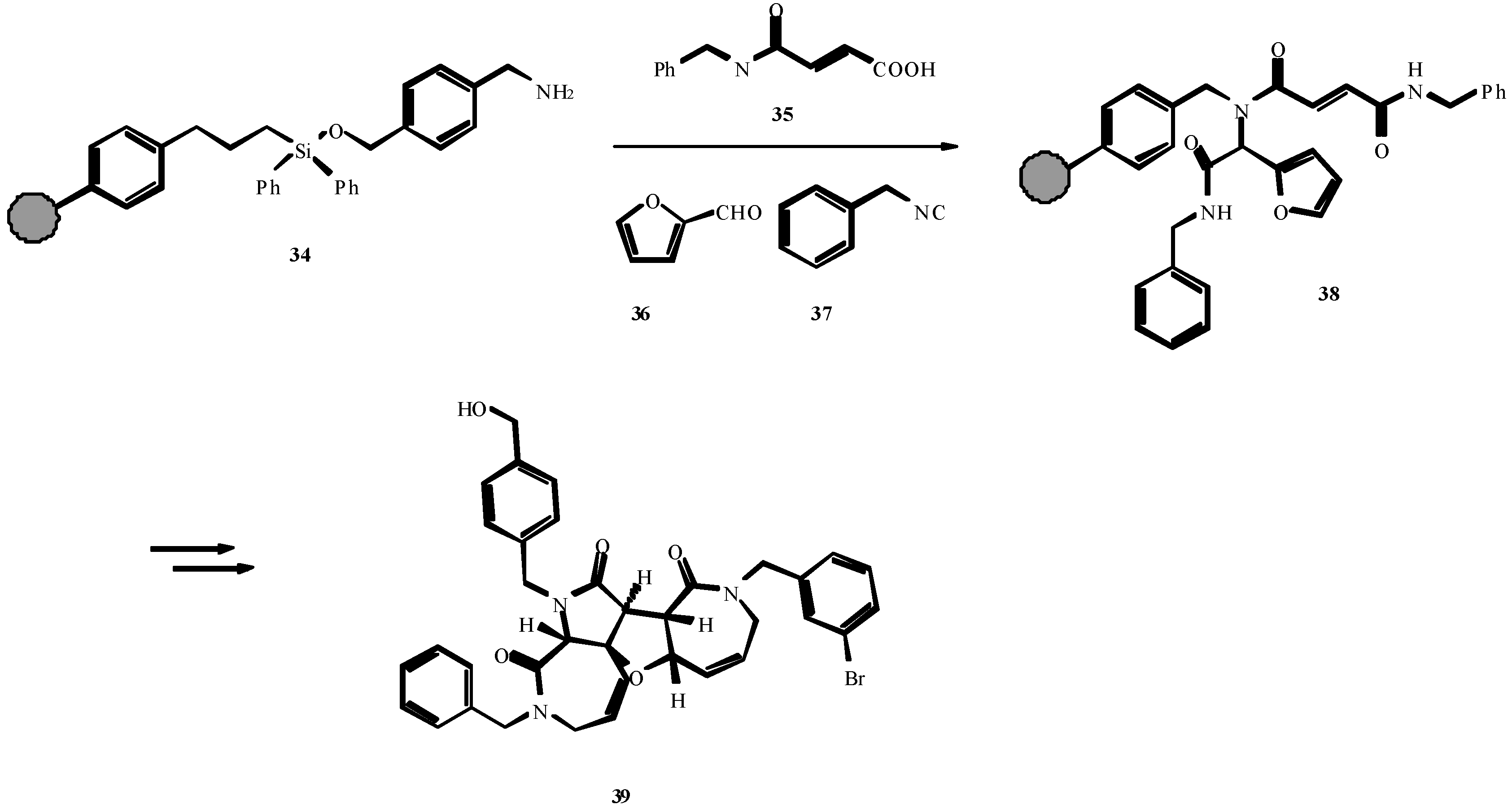
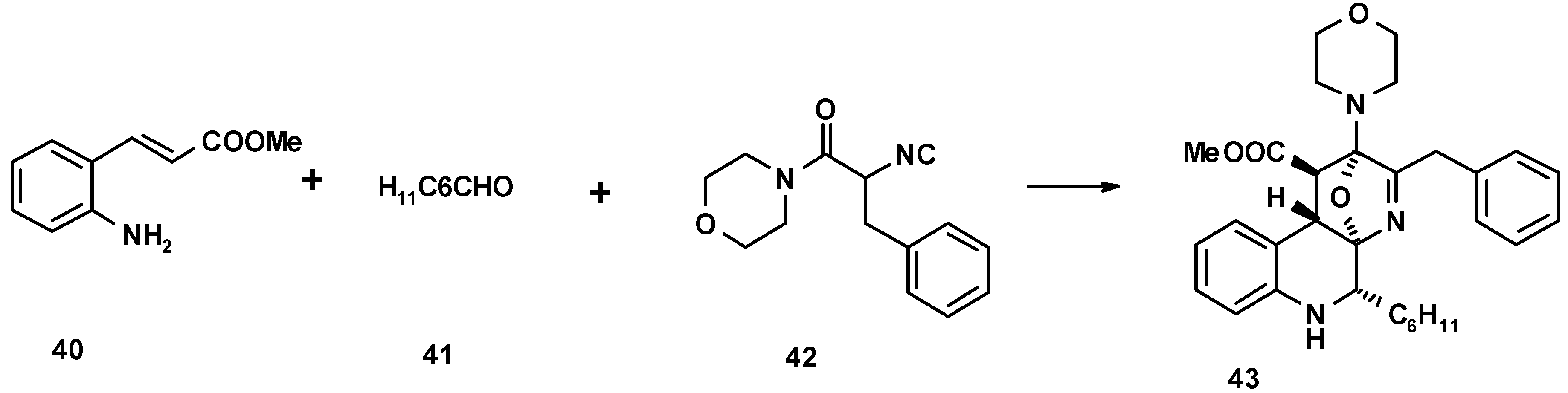

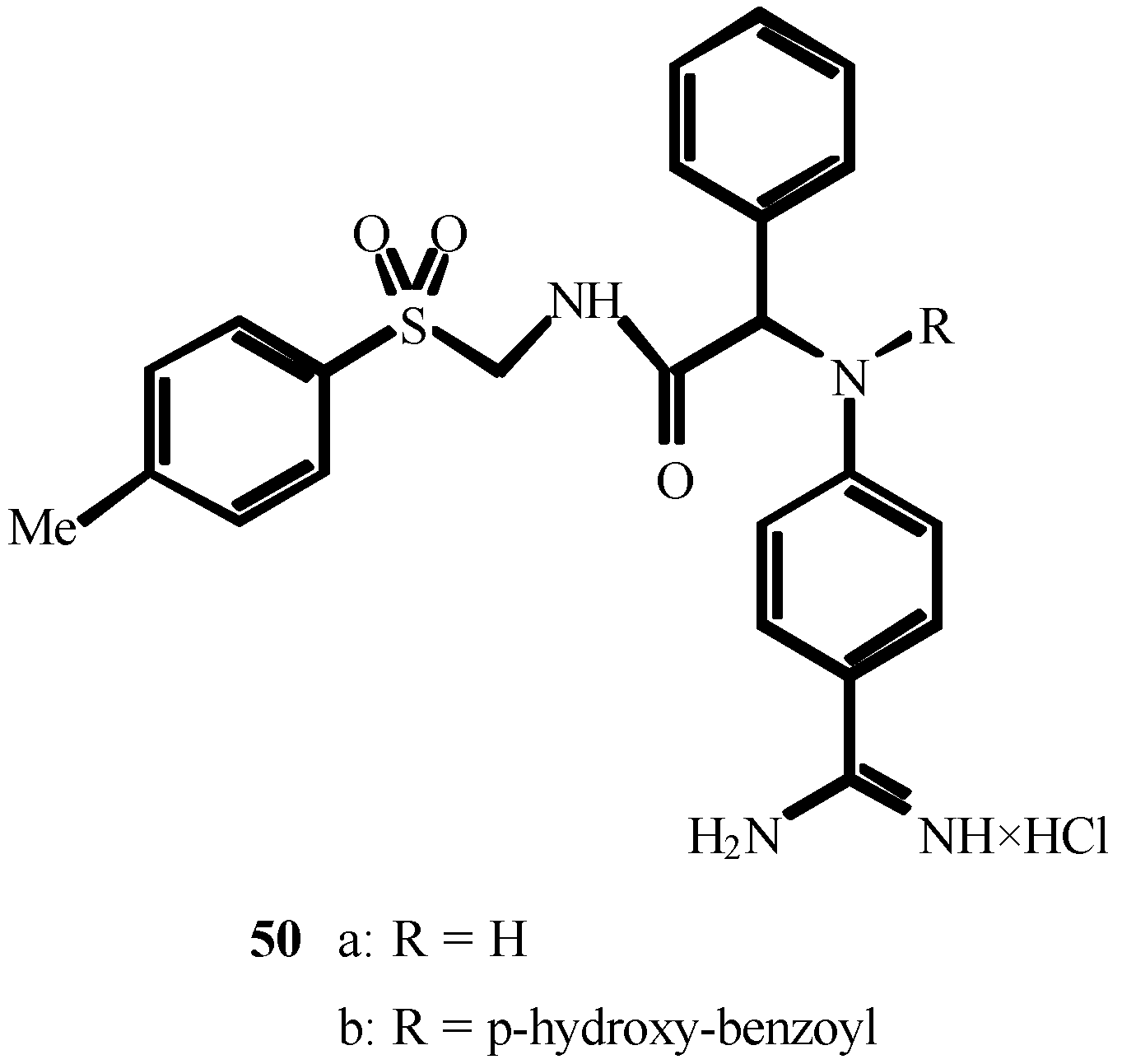
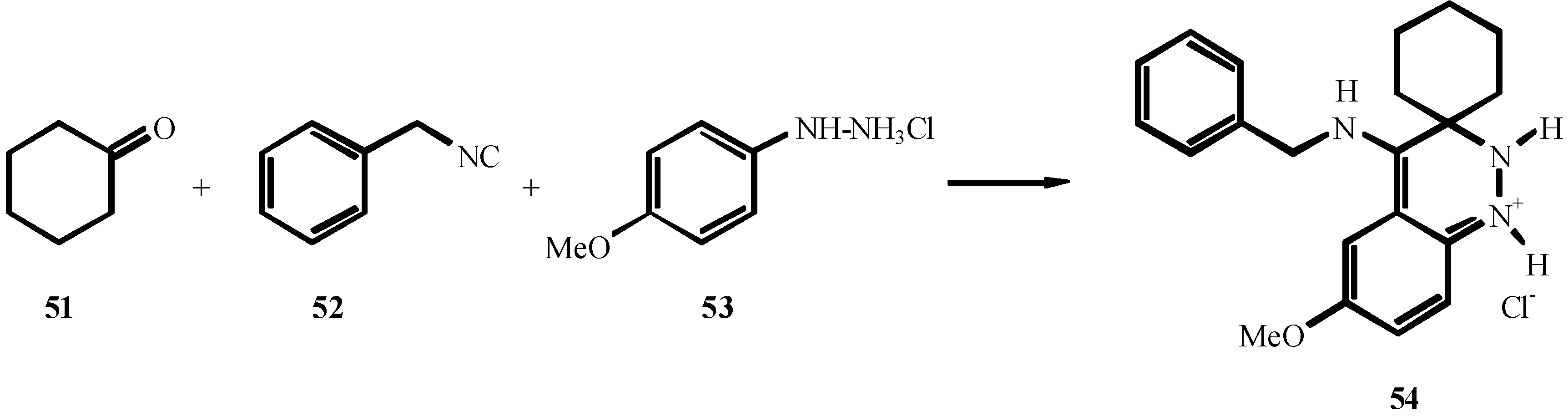
References
- Janezic, D.; Hodoscek, M.; Ugi, I. Internet Electr. J. Mol. Design 2002, 1, 293.
- Nef, I. Justus Liebigs Ann. Chem. 1892, 270, 267, 1899, 309, 126.
- Ugi, I. Isonitrile Chemistry; Academic Press: New York, 1971. [Google Scholar]
- Dömling, A.; Ugi, I. Angew. Chem. 2000, 112, 3300, [Angew. Chem. Int. Ed. Engl. 2000, 39, 3168].
- Ugi, I. J. Prakt. Chem. 1997, 339, 499.
- Strecker, A. Ann. Chem. 1850, 75, 27.
- Mannich, C.; Krötsche, I. Arch. Pharm. 1912, 250, 647. Organic Reaction; Blick, F. F.; Adams, R. (Eds.) John Wiley & Sons: New York; Vol. 1, 1942; p. 303.
- Hantzsch, A. Justus Liebigs Ann. Chem. 1892, 215, 1, ibid. 1892, 219, 1; Ber. Dtsch. Chem. Ges. 1890, 23, 1474; see also: . Böttinger, C. Justus Liebigs Ann. Chem. 1981, 208, 122. Eisener, U.; Kuthan, J. Chem. Rev. 1972, 72, 1.
- Radziszewski, B. Ber. Dtsch. Chem. Ges. 1882, 15, 1499, 2706.
- Biginelli, P. Ber. Dtsch. Chem. Ges. 1891, 24, 1317, 2962, ibid. 1893, 26, 447.
- Bergs, H. Ger. Pat. 566094 (1929) [Chem. Abstr. 1933, 27, 1001]. Bucherer, H.T.; Steiner, W. J. Prakt. Chem. 1934, 140, 291, Bucherer, H.T., ibid. 1934, 141, 5.
- Asinger, F. Angew. Chem. 1956, 68, 413. Asinger, F.; Thiel, M.; Pallas, E. Liebigs Ann. Chem. 1957, 602, 37. Asinger, F.; Thiel, M. Angew. Chem. 1967, 79, 953. Asinger, F.; Leuchtenberg, W.; Offermanns, H. Chem. Zeitung 1974, 94, 6105. Asinger, F.; Gluzek, K.H. Monats. Chem. 1983, 114, 47.
- Hellmann, H.; Opitz, G. α-Aminoalkylierung; Verlag Chemie: Weinheim, 1960. [Google Scholar]
- Chattopahyaya, J.; Dömling, A.; Lorenz, K.; Richter, W.; Ugi, I.; Werner, B. Nucleosides & Nuclotides 1977, 16, 843.
- Ho, T.L. Tandem Organic Chemistry; J. Wiley & Sons: New York, 1992. [Google Scholar] see also: Tietze, L.F.; Beifuss, U. Angew. Chem.; 1993; Volume 105, p. 137, [Angew. Chem. Int. Ed. Engl. 1993, 32, 131]. [Google Scholar]
- Eschenmoser, A.; Heusser, H. Experientia 1953, 9, 357. Eschenmoser, A.; Ruzicka, L.; Jeger, J.O.; Arigoni, D. Helv. Chim. Acta 1955, 38, 1890. see also: Barton, D.H.R.; Moss, G.R. Chem. Comm. 1966, 261. Corey, E.J.; Russey, W.E.; Ortiz de Mortellano, R.R. J. Amer. Chem. Soc. 1966, 88, 4750.
- Johnson, W.S. Angew. Chem. 1976, 88, 33, [Angew. Chem. Int. Ed. Engl. 1976, 15, 9].
- Posner, G.H. Chem. Rev. 1986, 86, 831. [Google Scholar]
- Chem. Rev. 1996, 96, 1–600.
- Heathcock, C.H. Angew. Chem. 1992, 104, 6751, [Angew. Chem. Int. Ed. Engl. 1992, 31, 665].
- Lieke, W. Justus Liebigs Ann. Chem. 1859, 112, 316.
- Gautier, A. Justus Liebigs Ann. Chem. 1867, 142, 289, Ann. Chim. (Paris) 1869, 17, 103, 203.
- Hofmann, A.W. Ber. Dtsch. Chem. Ges. 1870, 3, 63. see also: Weber, W.P.; Gokel, G.W.; Ugi, I. Angew. Chem. 1972, 84, 587, [Angew. Chem. Int. Ed. Engl. 1972, 11, 530].
- Oliveri-Mandala, E.; Alagna, B. Gazz. Chim. Ital. 1910, 40 (II), 441. Zimmerman, B.N.; Olafson, R.A. Tetrahedron Lett. 1969, 5081.
- Passerini, M. Gazz. Chim. Ital. 1921, 51(II), 126, 181, Passerini, M., Ragni, G., ibid., 1931, 61, 964.
- Rothe, W. Pharmazie 1950, 5, 190.
- Hagedorn, I.; Tönjes, H. Pharmazie 1956, 11, 409, 1957, 12, 567. Hagedorn, I.; Eholzer, U.; Lüttringhaus, A. Chem. Ber. 1960, 93, 1584.
- Ugi, I.; Meyr, R. Angew. Chem. 1958, 70, 702. Ugi, I.; Meyr, R. Chem. Ber. 1960, 93, 239. Ugi, I.; Fetzer, U.; Eholzer, U.; Knupfer, H.; Offermann, K. Angew. Chem. 1965, 77, 492, [Angew. Chem. Int. Ed. Engl. 1965, 4, 452]. Obrecht, R.; Herrmann, R.; Ugi, I. Synthesis 1985, 400.
- Edenborough, M.S.; Herbert, R.B. Nat. Prod. Rep. 1988, 55, 229.
- Scheuer, P.J. Acc. Chem. Res. 1992, 25, 433. Chang, C.W.J.; Scheuer, P.J. Top. Curr. Chem. 1993, 167, 33.
- Marconi, G.G.; Molloy, B.B.; Nagarajan, R.; Martin, J.R.; Deeter, J.B.; Okolowitz, J.L. J. Antibiot. 1978, 31, 27. Baldwin, J.E.; Baansai, H.S.; Chondrogianni, H.S.; Field, L.D.; Taha, A.A.; Thaller, V.; Brewer, D.; Taylor, A. Tetrahedron 1985, 41, 1931. Bornemann, V.; Patterson, G.M.L.; Moore, R.E. J. Am. Chem. Soc. 1988, 110, 2339. Itoh, J.; Takeuchi, Y.; Gomi, S.; Inouye, S.; Mikawa Yoshikawa, T.N.; Okhishi, H. J. Antibiot. 1990, 43, 456.
- Ugi, I. Angew. Chem. 1962, 74, 9, [Angew. Chem. Int. Ed. Engl. 1962, 1, 8].
- Ugi, I.; Steinbrückner, C. Chem. Ber. 1961, 94, a) 734; b) 2802.
- Ugi, I.; Lohberger, S.; Karl, R. Comprehensive Organic Synthesis: Selectivity for Synthetic Efficiency; vol. 2, chap. 4.6; Trost, B.M., Heathcock, C. H., Eds.; Pergamon: Oxford, 1991; pp. 1083; 1090. [Google Scholar]
- Bowers, M.M.; Caroll, P.; Joullié, M.M. J. Chem. Soc. Perkin Trans. I 1989, 857.
- Dömling, A.; Ugi, I. Angew. Chem. 2000, 112, 3300, [Angew. Chem. Int. Ed. Engl. 2000, 39, 3168]. Dömling, A.; Ugi, I.; Herdtweck, E. Acta Chem. Scand. 1998, 52, 107.
- Ugi, I.; Marquarding, D.; Urban, R. Chemistry and Biochemistry of Amino Acids, Peptides and Proteins; Vol. 6, Weinstein, B., Ed.; Marcel Dekker: New York, 1982; p. 245. [Google Scholar]
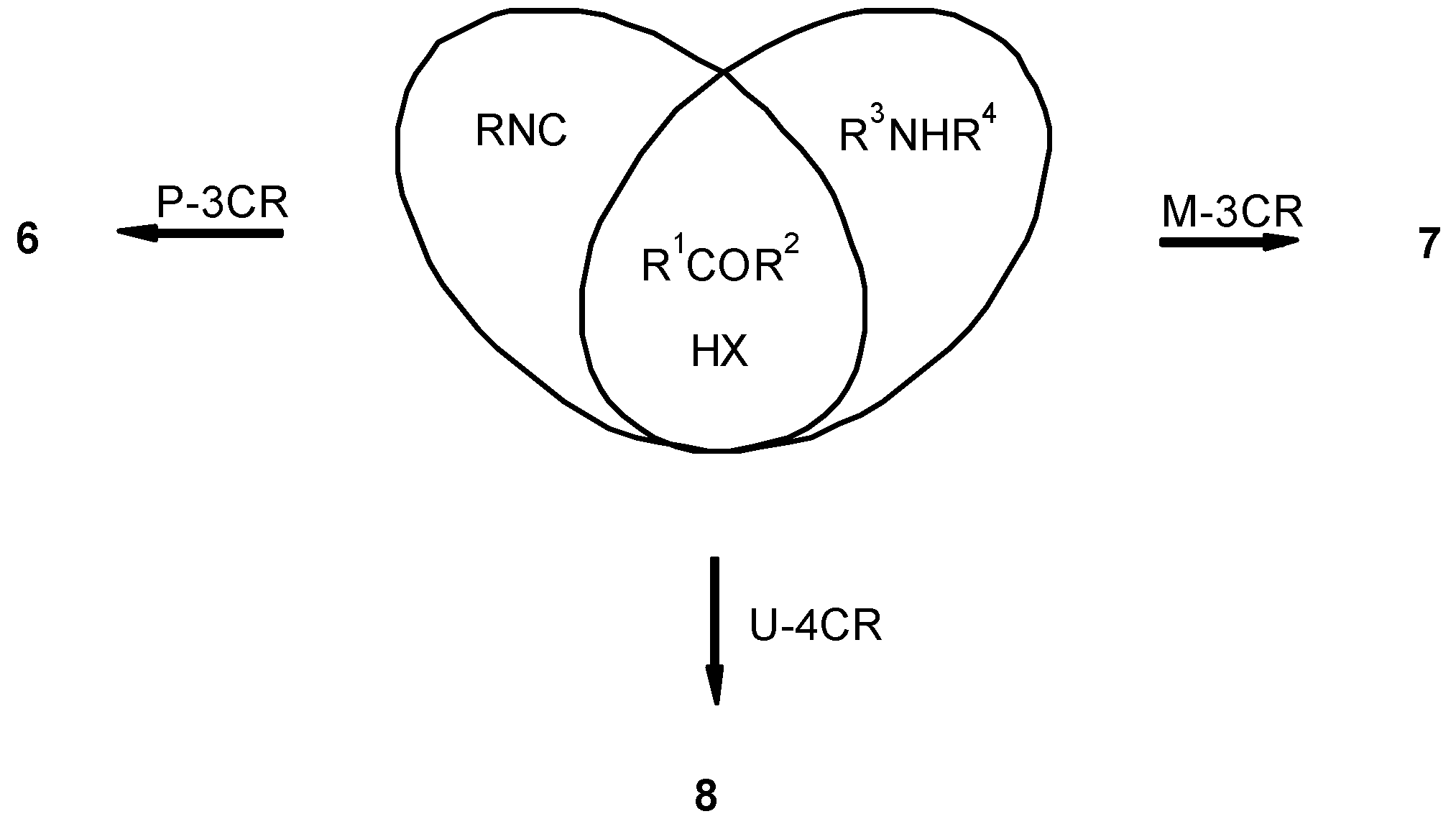
- Math. Unions: Mac Lane, S.; Birkhoff, G. Algebra; MacMillan Company: New York, 1967; p. 3. [Google Scholar] Given sets of R and S have the intersection R ∩ S with the common elements R and S. This means R ∩ S = {x | x ⊂ R and x ⊂ S}, where as a union R ∪ S is R ∪ S = {x | x ⊂ R or x ⊂ S}
- Ugi, I.; Dömling, A.; Hörl, W. Endeavour 1994, 18, 115, GIT Fachzeitschrift für das Laboratorium 1994, 38, 430.
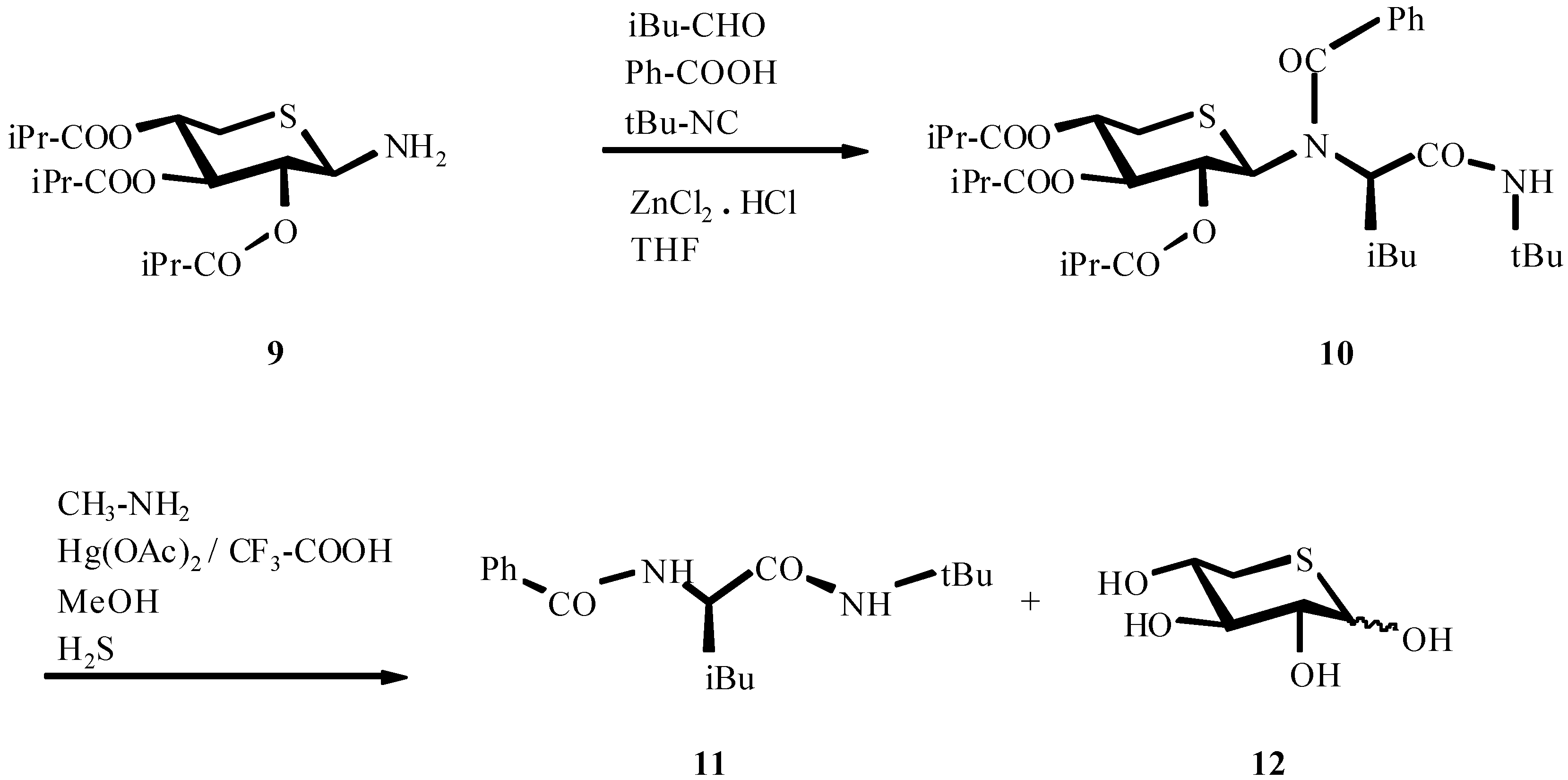
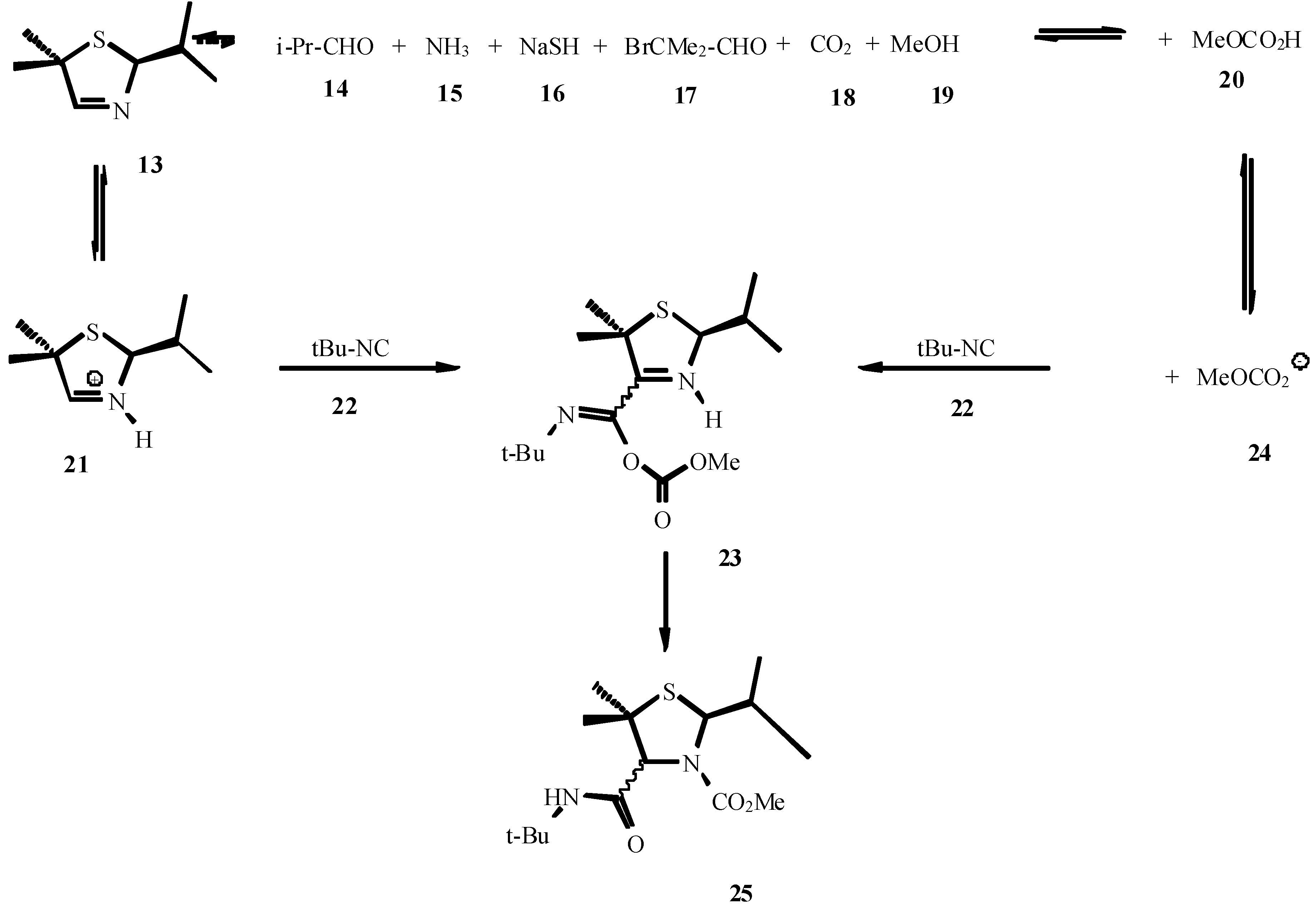
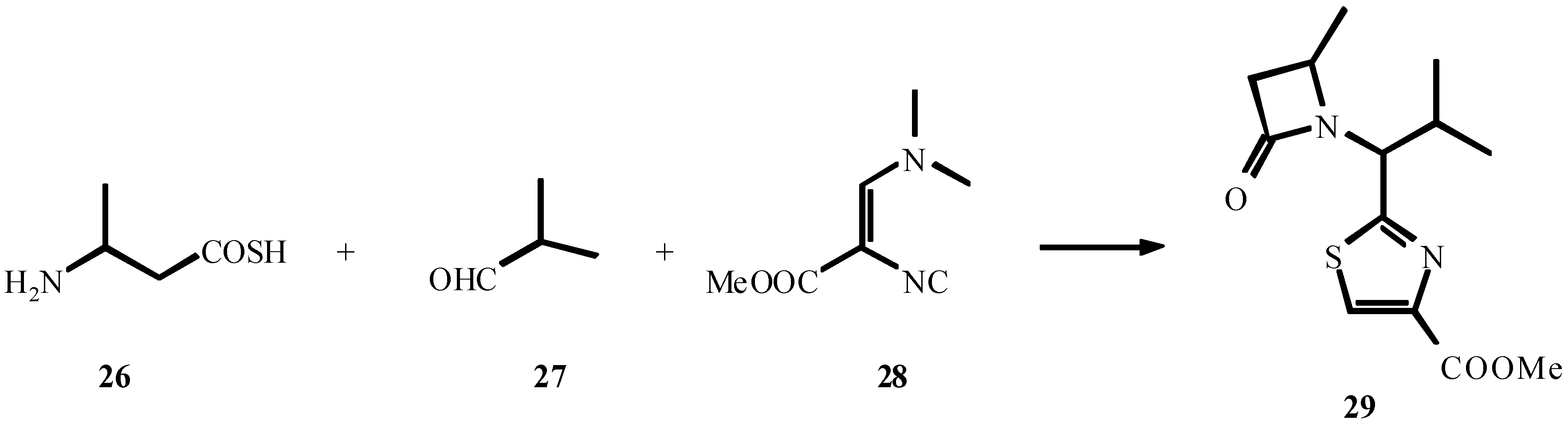
- Roß, G.; Ugi, I. Canad. J. Chem. 2001, 79, 1934. Roß, G.; Herdtweck, E.; Ugi, I. Tetrahedron. in press.
- Bossio, R.; Marcaccini, S.; Pepino, R. Liebigs Ann. Chem. 1990, 935, 1991, 1107. Bossio, R.; Marcaccini, S.; Paoli, P.; Papaleo, P.; Pepino, R. Liebigs Ann. Chem. 1991, 843. Bossio, R.; Marcaccini, S.; Pepino, R.; Torroba, T. Synthesis 1993, 7. Chem. Soc. Perkin Trans. 1 1996, 229. Bossio, R.; Marcaccini, S.; Pepino, R. Liebigs Ann. Chem. 1993, 1229. Bossio, R.; Marcaccini, S.; Papaleo, P.; Pepino, R. J. Heterocycl. Chem. 1994, 31, 297. Bossio, R.; Marcaccini, S.; Pepino, R. Tetrahedron Lett. 1995, 36, 2325. J. Org. Chem. 1996, 61, 2202. Bossio, R.; Marcos, C.F.; Marcaccini, S.; Pepino, R. Synthesis 1997, 1389. Heterocycles 1997, 1589.
- Dömling, A.; Ugi, I. Angew. Chem. 1993, 105, 634, [Angew. Chem. Int. Ed. Engl. 1993, 32, 563].
- Kolb, J.; Beck, B.; Dömling, A. Tetrahedron Lett. 2002, 34, 6897.
- Dömling, A.; Chi, K.-C. unpublished.
- Lee, D.; Sello, J.K.; Schreiber, S.-L. Org. Lett. 2000, 2, 700.
- González-Zamora, E.; Fayol, A.; Bois-Choussy, M.; Chiaroni, A.; Zhi, J. Chem. Comm. 2001, 1684.
- Rossen, K.; Pye, P.J.; DiMichele, L.M.; Volante, R.P.; Reider, P.J. Tetrahedron Lett. 1998, 39, 6823.
- Furka, A. Drug. Dev. Res. 1995, 36, 1.
- Früchtel, J.S.; Jung, G. Angew. Chem. 1996, 108, 19, [Angew. Chem. Int. Ed. Engl. 1996, 35, 17]. Frobel, K.; Krämer, T. Chemie in unserer Zeit 1996, 30, 270. Balkenhohl, F.; Buschen-Hünnefeld, C.v.; Lanshy, A.; Zechel, C. Angew. Chem. 1996, 108, 3436, [Angew. Chem. Int. Ed. Engl. 1996, 35, 2288]. Terrett, N.K. Combinatorial Chemistry; Oxford Univ. Press: Oxford, U.K., 1998. [Google Scholar] Ugi, I.; Dömling, A.; Ebert, B. Combinatorial Chemistry; Jung, G., Ed.; Wiley-VCH: Weinheim, 2000; pp. 125–165. [Google Scholar]
- Armstrong, R.W. Combinatorial Libraries Related to Natural Products. In presented at the ACS National Meeting in Anaheim, Calif; 2 April 1995. [Google Scholar] J. Am. Chem. Soc. 1995, 117, 7842. Stocker, A.M.; Keating, T.A.; Tempest, P.A.; Armstrong, R.W. Tetrahedron Lett. 1996, 37, 1149. Keating, T.A.; Armstrong, R.W.; Tempest, P.A.; Armstrong, R.W. J. Org. Chem. 1998, 63, 867.
- Weber, L.; Waltbaum, S.; Broger, C.; Gubernator, K. Angew. Chem. 1995, 107, 2452, [Angew. Chem. Int. Ed. Engl. 1995, 34, 2280]. Weber, L. Drug Discovery Today 2002, 7, 143.
- Lack, O.; Weber, L. Chimia 1996, 50, 445. Weber, L. Current Opinion in Chemical Biology 1998, 2, 381.
- Weber, L.; Illgen, K.; Almstetter, M. Synlett 1999, 366. Weber, L. Drug Discovery Today 2002, No 5 Suppl, 7.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ugi, I.; Werner, B.; Dömling, A. The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries. Molecules 2003, 8, 53-66. https://doi.org/10.3390/80100053
Ugi I, Werner B, Dömling A. The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries. Molecules. 2003; 8(1):53-66. https://doi.org/10.3390/80100053
Chicago/Turabian StyleUgi, I., B. Werner, and A. Dömling. 2003. "The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries" Molecules 8, no. 1: 53-66. https://doi.org/10.3390/80100053
APA StyleUgi, I., Werner, B., & Dömling, A. (2003). The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries. Molecules, 8(1), 53-66. https://doi.org/10.3390/80100053