Introduction
The asymmetric reduction of prochiral ketones with simple chiral borane complexes resulted in disappointingly low
ee (1.5 – 3.5%) [
1]. However, the N-BH
3 coordinated oxazaborolidines pioneered by Itsuno [
2] and developed by Corey [
3] gave excellent results. The success of oxazaborolidines could be due to the operation of two center catalysis enumerated by Helmchen
et al. involving a Lewis acidic center and a Lewis basic center [
4]. Obviously the absence of such two centered reaction in the case of amino acid ester complex could be the reason for the low
ee. Hence we opted to design compounds in which the ester moiety of the amino acid is retained and construct the oxazaborolidine ring as shown below (
Figure 1).
Figure 1.
Where R=iso-propyl (1a), iso-butyl (1b), sec-butyl (1c)
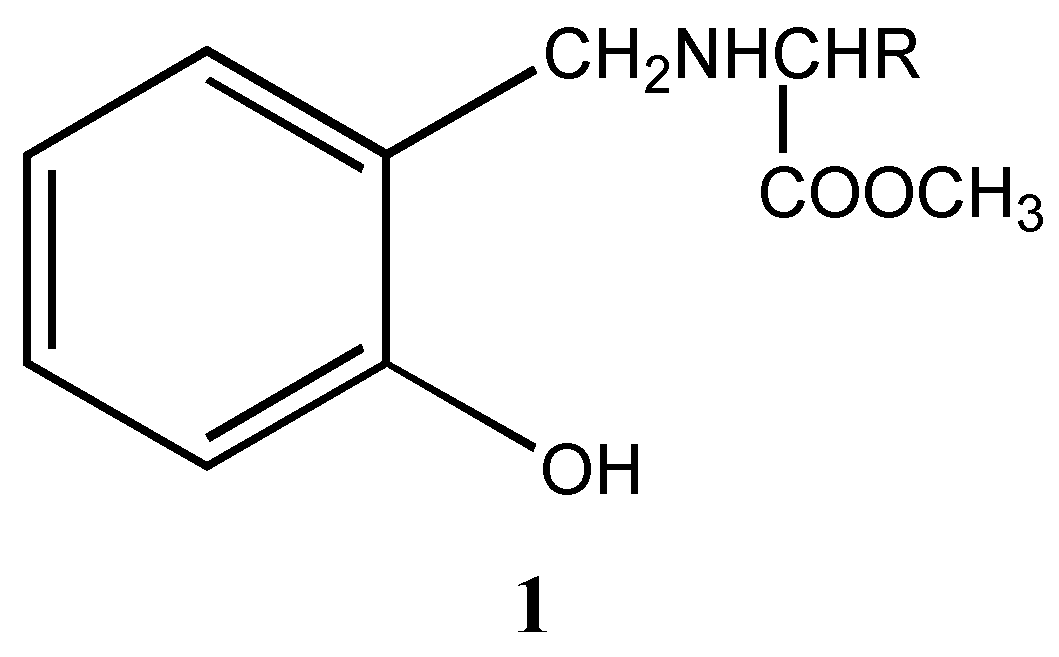
Indeed we found that these ligands can induce chiral switching in the reduction of prochiral ketones, which is described here. The chiral ligands (
1a-c) were synthesized using salicylaldehyde and various amino acid esters as starting materials through the formation of the corresponding imine esters and selective reduction of the imine esters to amine esters using tetrabutylammonium borohydride [
5,
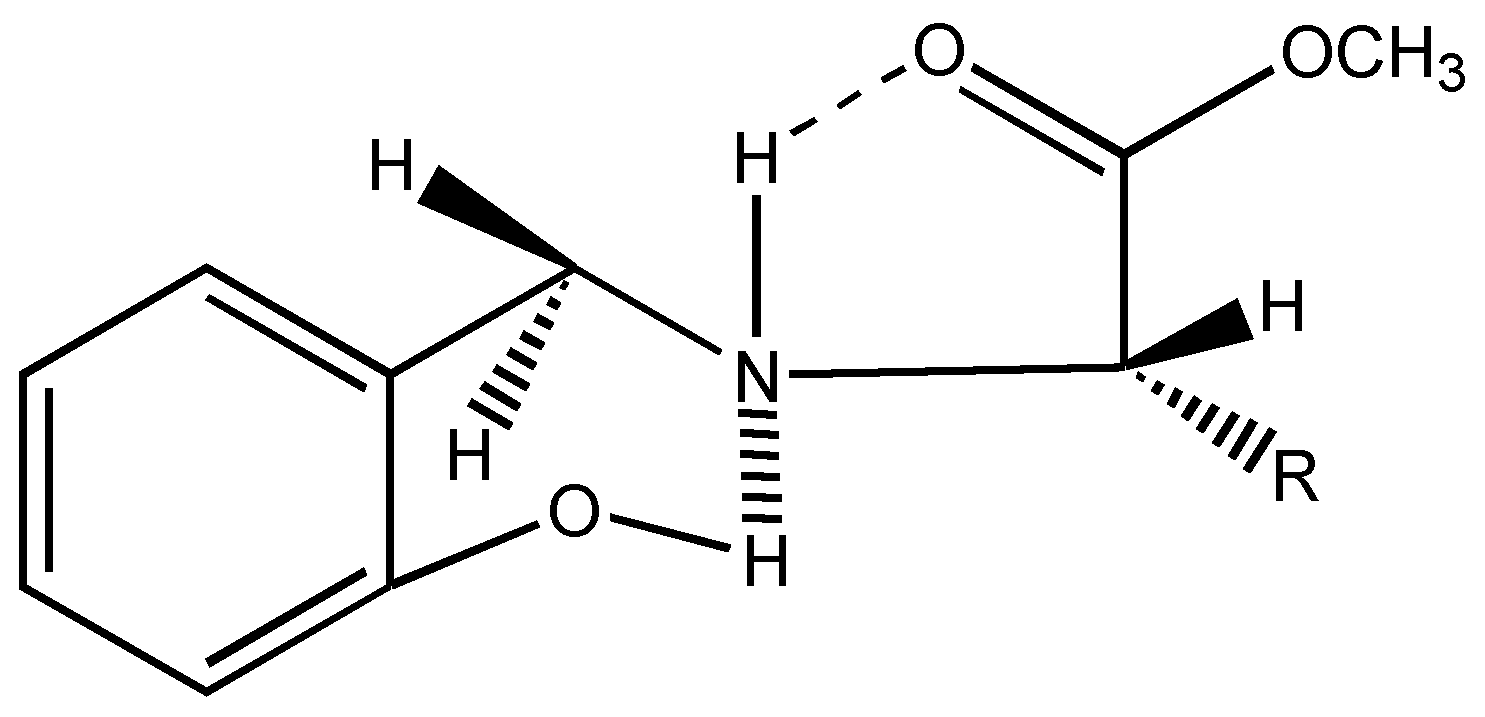
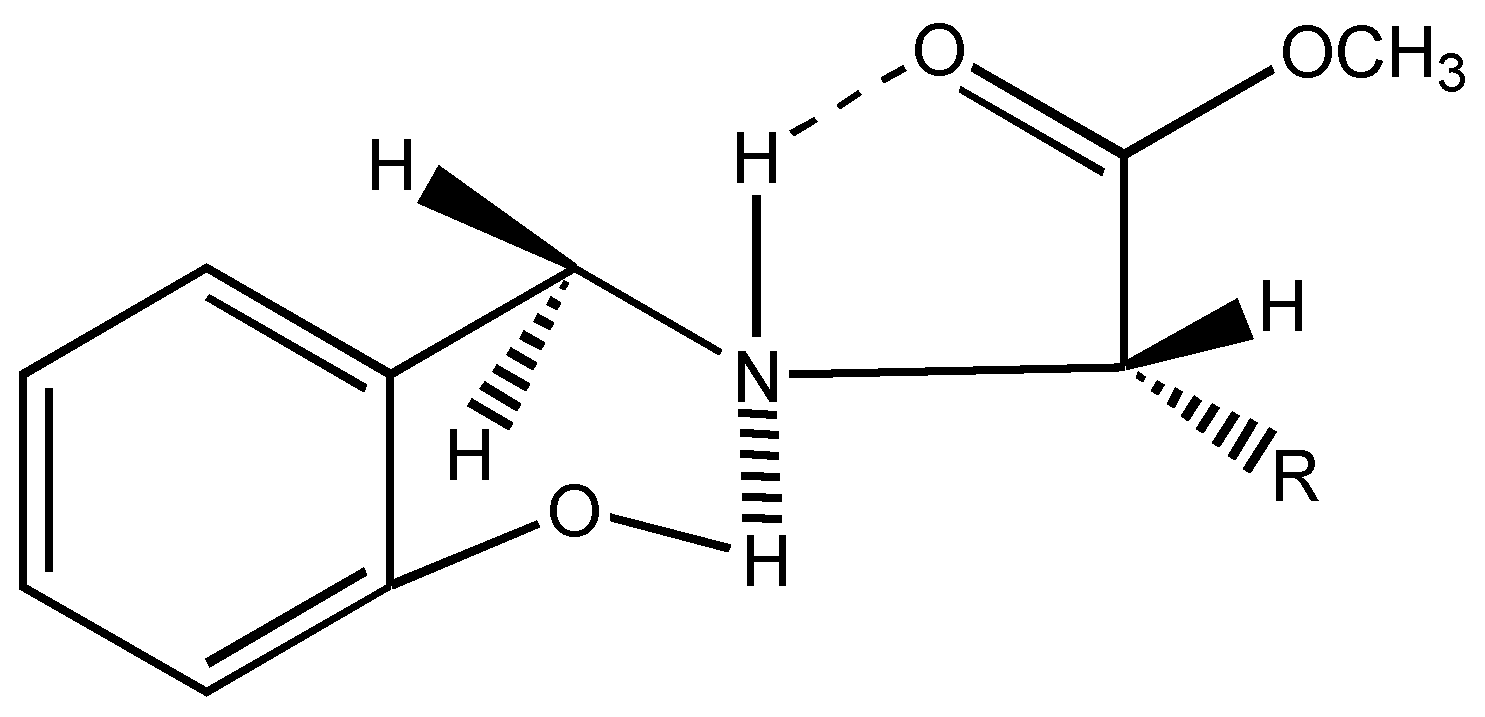
6]. Interestingly in all these chiral ligands, the two benzylic protons remain stereochemically non equivalent as shown by two doublets at δ = 4.1 ppm and δ = 3.6 ppm, J = 13.5 Hz in their
1H-NMR spectra. This can be attributed to the conformational locking achieved through hydrogen bonding as seen below (
Figure 2).
The corresponding dihydrooxazaborins
2 can be prepared by the method of Brown and Corey [
7]. The reaction resulted in the initial liberation of hydrogen at room temperature. Thus, 1 mmol of
1 liberates 2 mmol of H
2 upon treatment with 1 mmol of borane in THF at room temperature. After the liberation of H
2 ceased, the reaction mixture was refluxed for 1.5h and the formation of
2 was confirmed using
11B-NMR (δ-24ppm).
Scheme 1.
Where R=iso-propyl (a), iso-butyl (b), sec-butyl (c)
The chiral reducing agents 3 were prepared in situ by refluxing 2 and BH3/THF in a 1:1 ratio for one hour. Interestingly, the ester group remained intact as evidenced by the peak at 1735 cm-1 in the IR spectrum. Also, the ligand isolated after hydrolysis did not exhibit any change in the functional groups.
Results and Discussion
Reduction of acetophenone with these chiral reducing agents
3a-3c was representative. The reductions were instantaneous resulting in optically active 1-phenyl ethanol. Interestingly, the dihydrooxazaborins derived from valine methyl ester,
3a showed good enantioselectivity (
ee of 75% of the R –enantiomer). However,
3b and
3c showed poor to moderate enantioselectivity (15-55%) (
Table 1, entries 1-3). The instantaneous reduction indicates that the reduction is effected by nitrogen-coordinated borane (N-BH
3), which is more nucleophilic. The predominant formation of the
R-enantiomer may be due to the more selective transfer of hydride from the reagent to the
Si face of the ketone. Hence, Corey’s mechanism involving the hydride transfer from the reagent to the
Si face of the ketone
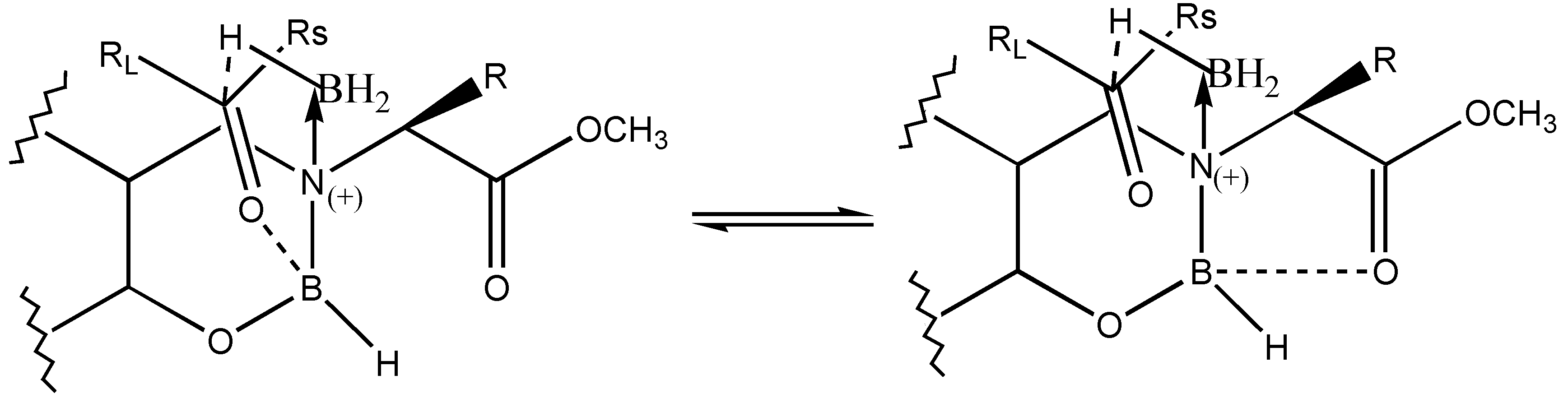
via the formation of a six membered cyclic transition state is expected for the reduction. However, the lower observed
ee for the reduction may be due to the competition of the carbonyl group of the ketone for coordination with boron from ester carbonyl of the reagent (
Figure 3).
The variation of
ee with respect to the nature of alkyl group of the dihydrooxazaborin may be due to the different steric influence offered by the
R group towards stereo selective reduction. The reductions were also carried out at lower temperatures to find out if
ee could be improved. But, when the reactions were performed at 0°C, the
ee was decreased which could be due to the formation of a dimer of dihydrooxazaborin as suggested by Corey [
8]. Moreover, at low temperature (0°C), the equilibrium could be shifted more towards the intramolecular ester carbonyl coordination with boron resulting in a less selective reduction of ketone accounting for lesser
ee. At room temperature, (25°C) the equilibrium would be shifted more towards the ketone carbonyl coordination with boron, which helps in more selective reduction of ketone and is thus responsible for fairly high
ee. The results are listed in
Table 1.
Table 1.
Reduction of acetophenone using dihydrooxazaborins at different temperatures
Table 1.
Reduction of acetophenone using dihydrooxazaborins at different temperatures
| S.No | Chiral reducing agent | Temp | [α]D23°c=5 (CHCl3)[9] | ee (%) | Yield (%) |
|---|
| 1 | iso-propyl (3a) | 30°C | 31.5 (+42)* | 75 | 99 |
| 2 | iso-butyl (3b) | 30°C | 6.3 | 15 | 92 |
| 3 | sec-butyl (3c) | 30°C | 23.1 | 55 | 90 |
| 4 | iso-propyl (3a) | 0°C | 6.3 | 15 | 94 |
| 5 | sec-butyl (3c) | 0°C | 4.2 | 10 | 93 |
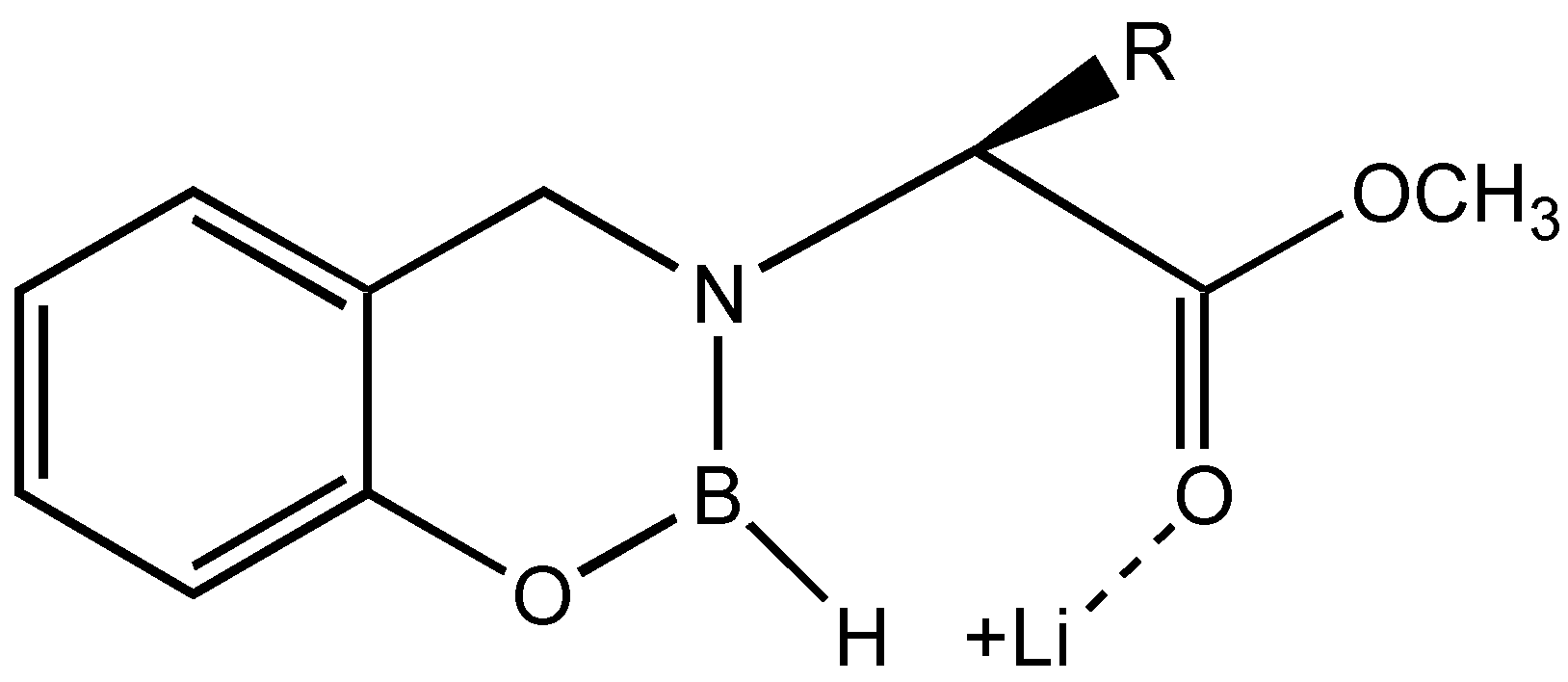
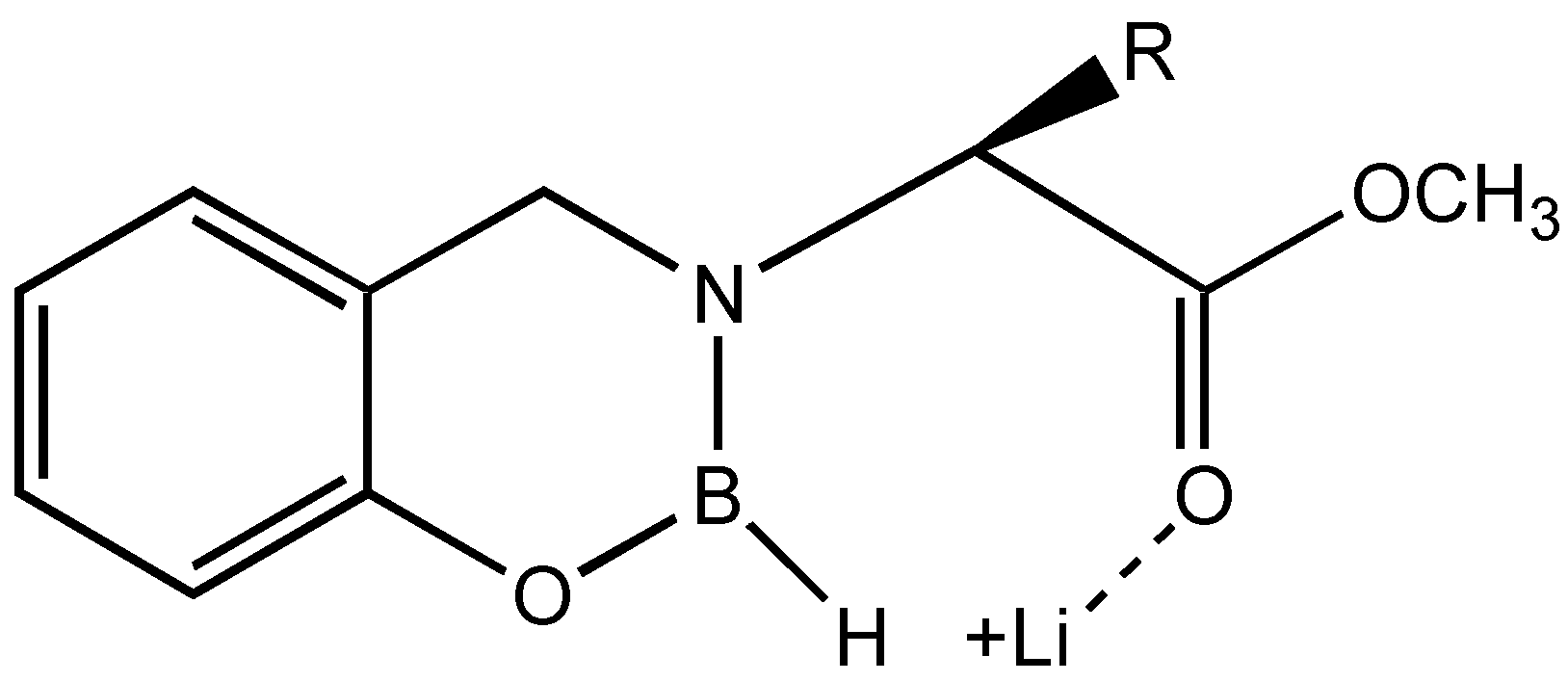
The results suggest that if the equilibrium is shifted more towards the coordination of the ketone carbonyl and the boron, the
ee can be enhanced. This can be achieved by the addition of hard acids such as lithium salts, which can disrupt the coordination between ester and boron without affecting the conformational locking (
Figure 4).
Hence, reductions were carried out with chiral reducing agents derived from valine in presence of stoichiometric quantities of lithium salts. Interestingly, when LiBr was added to a freshly prepared solution of N-borane coordinated dihydrooxazaborin, and stirred for 15 min. at room temperature followed by the addition of 5 mmol of acetophenone increased the
ee of the alcohol to 90%. The results are presented in
Table 2.
Table 2.
Reduction of ketones in presence of lithium salts at room temperature (30°C)
Table 2.
Reduction of ketones in presence of lithium salts at room temperature (30°C)
| Salts | Ketones | Product | [α]D23°c=5 (CHCl3) [9] | ee (%) | Yield (%) |
|---|
| LiBr | Acetophenone | 1-phenylethanol | 37.8 (+42)# | 90 | 90 |
| LiClO4 | Acetophenone | 1-phenylethanol | 33.6 | 80 | 93 |
| LiBr | Phenacyl chloride | 2-chloro-1-phenylethanol | 35.7 (-41.3) | 85 | 90 |
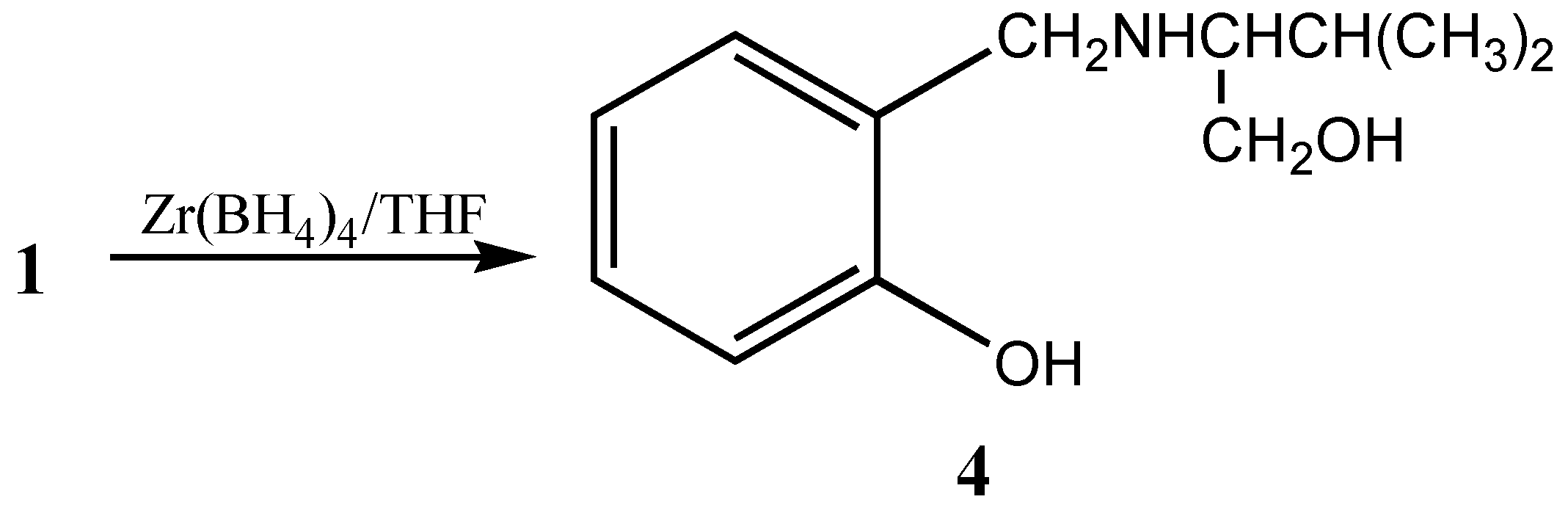
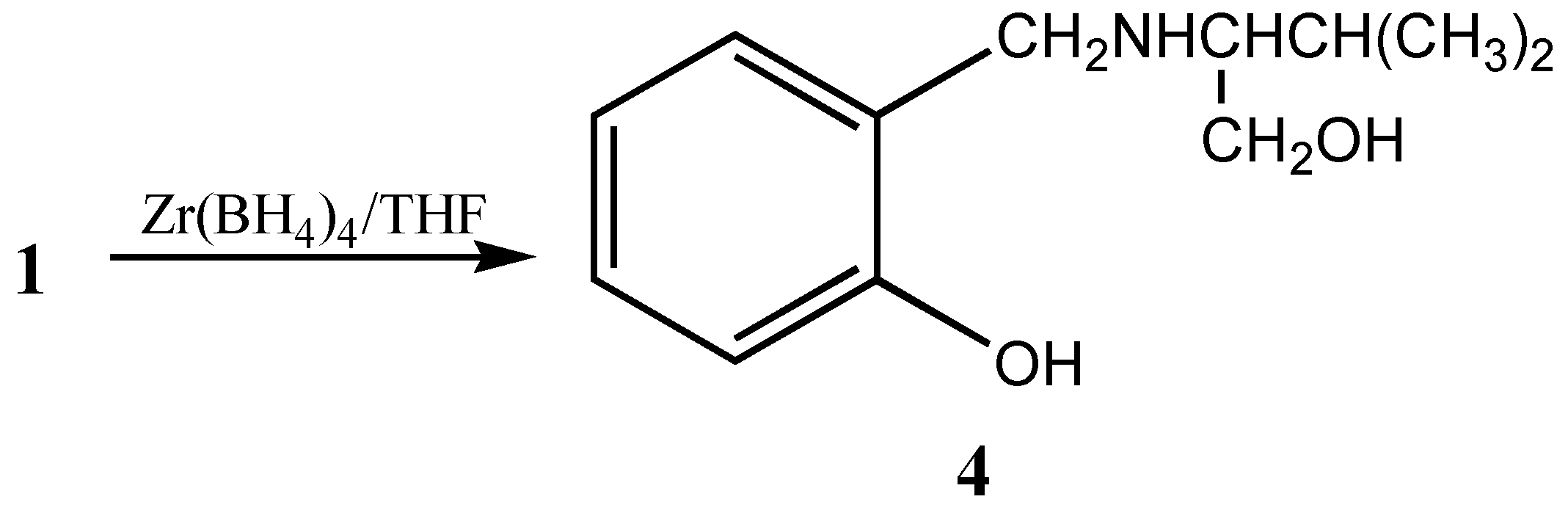
Alternatively, the coordination of boron and ester carbonyl can also be disrupted by reducing the ester moiety by forming a tridentate chiral precursor
4 [
6] (
Scheme 2).
This chiral precursor with a tridentate ligand is considered to be more appropriate for asymmetric reductions than an unidentate or bidentate ligand due to the rigid complex formation,. These tridentate chiral precursors can also be utilized for the formation of heterobimetallic chiral reagent using LiAlH
4 leading to the formation of chiral dioxazaluminium complex, which is another potential chiral reagent for selective reduction of ketones [
10] (
Scheme 3).
Thus, several bicyclic oxazaborolidines (
5) were prepared from the amino acids valine, leucine and
iso-leucine and they were studied for the reduction of prochiral ketones at room temperature (
Table 3).
Table 3.
Reduction of Acetophenone at room temperature with various bicyclic oxazaborolidines
Table 3.
Reduction of Acetophenone at room temperature with various bicyclic oxazaborolidines
| R | [α]D23°c=5 (CHCl3)[9] | ee (%) | Yield (%) |
|---|
| 5a iso-propyl | 37.8 | 90 | 95 |
| 5b iso-butyl | 4.2 | 10 | 89 |
| 5c sec-butyl | 23.1 | 55 | 90 |
This predominant formation of the R enantiomer using 5a follows Corey’s mechanism involving the hydride transfer from the reagent to the Si face of the ketone via the formation of a six membered cyclic transition state.
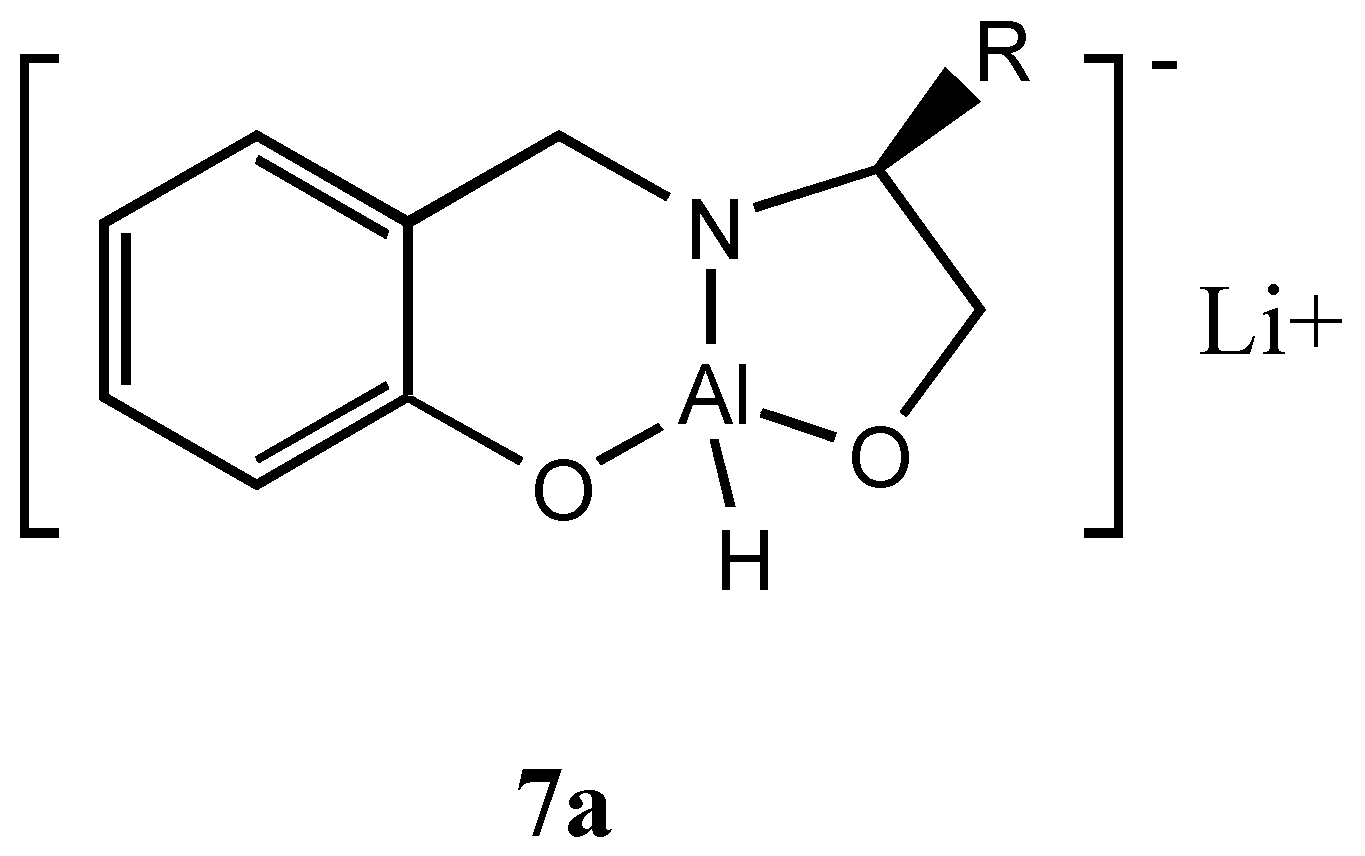
Interestingly, when the reduction was carried out with the heterobimetallic complex
6a the reduction proceeded with reversal of enantioselectivity
albeit with lesser
ee. Probably in the oxazaluminium complex the mechanism could involve the hydride transfer from the reagent to the
Re face of the ketone through aluminium. This could arise due to the exchange of hydride from boron to aluminium. Therefore, we prepared a complex with LiAlH
4 (
7a, Figure 5) without quenching the hydride and studied the asymmetric reduction of acetophenone.
Indeed we obtained the opposite isomer in fairly good
ee. The results are given in
Table 4. Binding of electrophilic carbonyl oxygen with lithium and the additional
pi stacking interaction existing between the phenyl ring of acetophenone and the phenyl ring of the reagent are mainly responsible for more selective transfer of hydride from aluminium to the
Re face of the ketone
via a six membered cyclic transition state thus accounting for the formation of
S - alcohol and the observed
ee.
Table 4.
Reduction of ketones with bicyclic oxazaborolidine and dioxazaluminium complex derived from valine
Table 4.
Reduction of ketones with bicyclic oxazaborolidine and dioxazaluminium complex derived from valine
| Ketones | Reagent/
Temp | Product | Yield
(%) | ee
(%) | [α]D23°
c=5 (CHCl3) [9]a |
|---|
| Acetophenone | 5a/30°C | 1-phenylethanol | 95 | 90(R) | +37.8 (+42) |
| Phenacyl chloride | 5a/30°C | 2-chloro-1-phenylethanol | 90 | 85(S) | +35.7 (+43) |
| Acetophenone | 6a/30°C | 1-phenylethanol | 90 | 30(S) | -12.6 (-41.3) |
| Acetophenone | 7a/0°C | 1-phenylethanol | 90 | 60(S) | -24.7 |
| Phenacyl chloride | 7a/0°C | 2-chloro-1-phenylethanol | 90 | 55(R) | -26.4 (-48) |
Thus, the chiral reducing agents derived from same precursor showed opposite enantioselectivity, which is a unique phenomenon. In conclusion, we have developed novel chiral reagents, which are useful in the syntheses of optically active secondary alcohols of opposite configuration depending upon the nature of the metal complex, which is not possible with other reagents.
Experimental
General
The solvents and chemicals used in this work were obtained commercially and were purified using conventional methods. The NMR spectra were recorded on a BRUKER 200 MHz instrument. Optical rotations were recorded using a Autopol polarimeter using CHCl3 as solvent at 23°C using the sodium D-line monochromator (2580 Å).
Preparation of dihydrooxazaborin 2: BH3/THF solution (5mL, 1M) were added to 5 mmol of the chiral precursor. The formation of 2 was monitored through hydrogen evolution by connecting the system to the gas burette. After the liberation of H2 was complete the reaction mixture was refluxed for 1 to 1.5h.
Reduction of acetophenone using 2a: 1M borane-THF (5 mmol) was added at room temperature to a freshly prepared solution of 5mmol of dihydrooxazaborin, derived from valine methyl ester and salicylaldehyde (5mL) and the resulting mixture was and stirred for 30min. Then acetophenone (0.6mL, 5mmol) was added, the mixture stirred for 10 min at room temperature and then quenched with methanol. The reaction mixture was extracted with hexane and the hexane extract was washed with dil.HCl followed by water, dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. Purification was achieved using column chromatography with hexane: ethyl acetate 98:2 as eluant to give (R)-(+)-1-phenylethanol in 99% chemical yield with ee of 75%; [α]D23 + 31.5°.
Reduction in the presence of lithium salts: LiBr, 0.4g (5mmol) in THF (5mL) was added to a freshly prepared solution of N-borane coordinated dihydrooxazaborin (5mmol) and the mixture stirred for 15min. at room temperature followed by the addition of acetophenone (0,6 mL, 5mmol). The reaction mixture was stirred for 10min. at room temperature and the quenched with methanol. The product was extracted and purified as described above. The alcohol (R)-(+)-1-phenylethanol was obtained in 90% chemical yield and with ee of 90% [α]D23 + 37.8°.
Preparation of bicyclic oxazaborolidine 5a: Borane-THF solution (5mL, 1M, 5 mmol) was added to 5 mmol of 4, and the formation of oxazaborolidine was monitored through the quantitative evolution of hydrogen by connecting the system to gas burette. After the hydrogen liberation was complete the reaction mixture was refluxed for 1 to 1.5h
Reduction of acetophenone using 5a: Borane THF solution (5mL, 1M, 5 mmol) was added to a freshly prepared solution of 5 mmol of 5a, and the mixture stirred at room temperature for 30 min. Then acetophenone (0.6 mL, 5mmol) was added and stirring continued for 10 min at room temperature, then the reaction wasquenched with methanol. The reaction mixture was extracted with hexane and the hexane extract was washed with dil. HCL followed by water, dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. Purification is achieved using column chromatography (hexane: ethyl acetate 98:2 as eluant) to give 0.56g of (R) –(+)-1-phenylethanol (95% yield) with ee of 90%.
Preparation of dioxazaluminium complex 7a: LiAlH4 (0.19g, 5 mmol) was added to 5mmol of 4 in THF and the mixture was stirred at room temperature for 1 to 1.5 h. The formation of dioxazaluminium complex was monitored through the quantitative evolution of hydrogen by connecting the system to a gas burette.
Reduction of acetophenone using 7a: Acetophenone (0.6 mL, 5 mmol) was added to a freshly prepared solution of 5 mmol of dioxazaluminium complex and the resulting mixture was stirred for 1h at 0°C. The reaction was then quenched with dil. HCL. The reaction mixture was extracted with chloroform and washed with water. The chloroform layer was dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. Purification using CC with hexane: ethyl acetate (98:2) as eluant gave 0.54g of the alcohol with 90% chemical yield and with ee of 60% (S)-(-) 1-phenylethanol; [α]D23 –24.7°



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
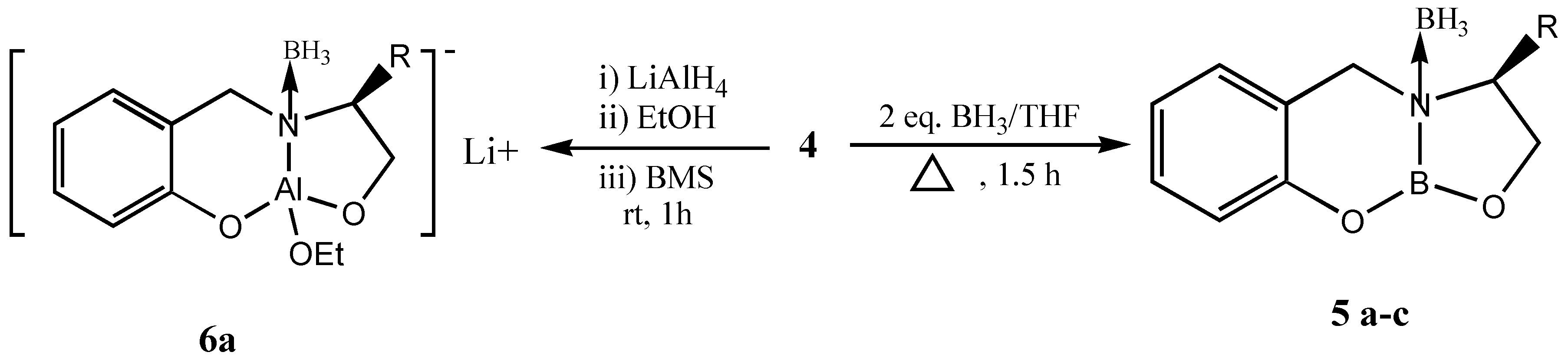{kind=link}