Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones

Abstract
:Introduction
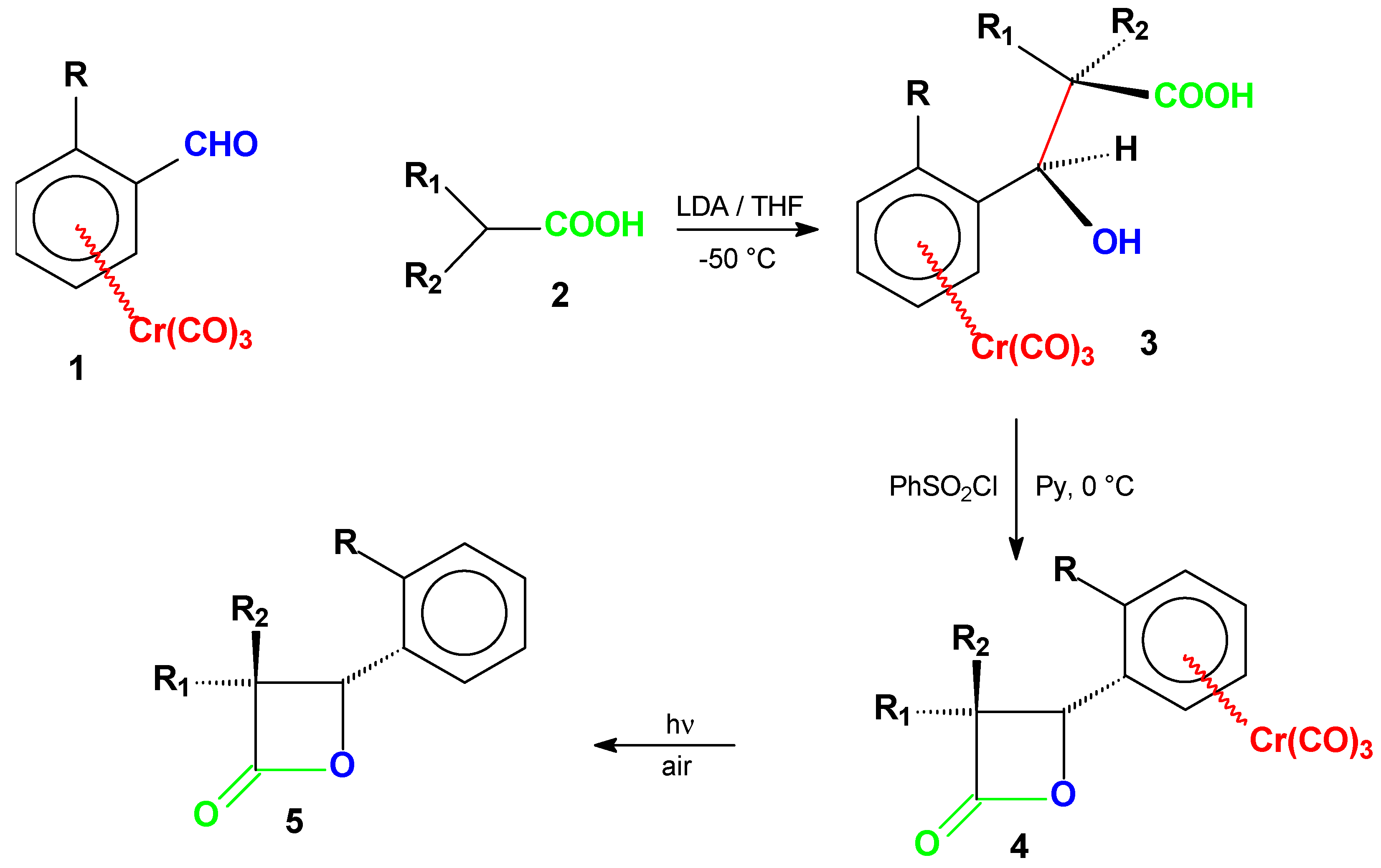

{kind=link}
| Prod. | R | R1 | R2 | Yield % | m.p. °C | [α]D | Absol. Config.a) | Config. of 1 |
|---|---|---|---|---|---|---|---|---|
| 3a | OMe | Me | Me | 95 | 132 | +36.5 | 3R | (-)-1R |
| 3b | Cl | Me | Me | 86 | 142 | +32.1 | 3R | (-)-1R |
| 3c | OMe | Me | H | 95 (1:1) | 98 d | +126 | 2R3R | (-)-1R |
| 3d | OMe | H | Me | 101 d | +127.9 | 2S3R | (-)-1R | |
| 3e | OMe | CMe3 | H | 96 (1:1) | 110 d | -103.5 | 2R3S | (+)-1S |
| 3f | OMe | H | CMe3 | 107 d | -129.7 | 2S3S | (+)-1S | |
| 4a | OMe | Me | Me | 70 | 118 | +42.8 | 4R | (-)-1R |
| 4b | Cl | Me | Me | 65 | 111 | -20.3 | 4R | (-)-1R |
| 4c | OMe | Me | H | 60 | 67 | +67.1 | 3R4R | (-)-1R |
| 4d | OMe | H | Me | -- | -- | -- | -- | (-)-1R |
| 4e | OMe | CMe3 | H | 60 | 110 | -54 | 3R4S | (+)-1S |
| 4f | OMe | H | CMe3 | 60 | 105 | -63.3 | 3S4S | (+)-1S |
| 5a | OMe | Me | Me | 97 | Oil | +94.7 | 4R | (-)-1R |
| 5b | Cl | Me | Me | 98 | Oil | +74.7 | 4R | (-)-1R |
| 5c | OMe | Me | H | 97 | Oil | +67.1 | 3S4R | (-)-1R |
| 5d | OMe | H | Me | -- | -- | -- | -- | (-)-1R |
| 5e | OMe | CMe3 | H | 98 | Oil | -60.4 | 3R4S | (+)-1S |
| 5f | OMe | H | CMe3 | 98 | Oil | -75.2 | 3S4S | (+)-1S |
Results and Discussion
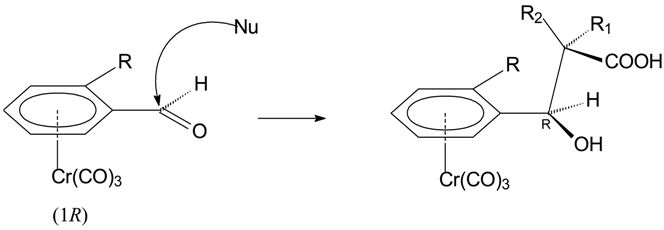
Conclusions
Experimental Section
General
| Prod. | 1H NMR (ppm) | Prod. | 1H NMR (ppm) |
|---|---|---|---|
| 3aa | 1.18 (s, 3H); 1.22 (s, 3H); 3.75 (s, 3H); 4.93 (t, 1H, J=6.1 Hz); 5.01 (d, 1H, J=6.5 Hz); 5.19 (s, 1H); 5.57 (t, 1H, J=6.1 Hz); 5.84 (d, 1H, J=6.1 Hz) | 4cc | 0.8 (d, 3H, J=7.8 Hz); 3.75 (s, 3H); 4.0 (dq, 1H, J=7.8 and 6.1 Hz); 4.92 (t, 1H, J=6.2 Hz); 5.55 (d, 1H, J=6.3 Hz); 5.56 (d, 1H, J=6.1 Hz); 5.8 (t, 1H, J=6.3 Hz); 6.8 (d, 1H, J=6.2 Hz) |
| 3ba | 1.18 (s, 3H); 1.21 (s, 3H); 5.09 (t, 1H, J=6.2 Hz); 5.19 (s, 1H); 5.38 (d, 1H, J=6.3 Hz); 5.47 (t, 1H, J=6.3 Hz); 5.79 (d, 1H, J=6.2 Hz) | 4ec | 0.95 (s, 9H); 3.75 (d, 1H, J=6.6 Hz); 3.79 (s, 3H); 4.91 (t, 1H, J=6.2 Hz); 5.12 (d, 1H, J=6.3 Hz); 5.55 (t, 1H, J=6.3 Hz); 5.6 (d, 1H, J=6.6 Hz); 5.9 (d, 1H, J=6.2 Hz) |
| 3ca | 0.8 (d, 3H, J=7.2 Hz); 2.4 (dq, 1H, J=1.7 and 7.2 Hz); 3.8 (s, 3H); 4.95 (d, 1H, J=1.7 Hz); 5.25 (t, 1H, J=6.3 Hz); 5.6 (d, 1H, J=6.8 Hz); 5.82 (t, 1H, J=6.8 Hz); 6.0 (d, 1H, J=6.3 Hz) | 4fc | 1.1 (s, 9H); 3.5 (d, 1H, J=4.0 Hz); 4.0 (s, 3H); 4.88 (t, 1H, J=6.2 Hz); 5.02 (d, 1H, J=6.8 Hz); 5.18 (d, 1H, J=4.0 Hz); 5.6 (t, 1H, J=6.8 Hz); 6.71 (d, 1H, J=6.2 Hz) |
| 3da | 1.15 (d, 3H, J=7.1 Hz); 2.25 (dq, 1H, J=3.5 and 7.1 Hz); 3.8 (s, 3H); 4.4 (d, 1H, J=3.5 Hz); 5.25 (t, 1H, J=6.3 Hz); 5.58 (d, 1H, J=6.8 Hz); 5.8 (t, 1H, J=6.8 Hz); 6.98 (d, 1H, J=6.3 Hz) | 5ac | 0.88 (s, 3H); 1.6 (s, 3H); 3.8 (s, 3H); 5.45 (s, 1H); 6.8-7.4 (m, 4H) |
| 3ea | 1.12 (s, 9H); 2.62 (d, 1H, J=7.7 Hz); 3.72 (s, 3H); 4.88 (t, 1H, J=6.2 Hz); 4.96 (d, 1H, J=6.5 Hz); 5.14 (d, 1H, J=7.7 Hz); 5.56 (t, 1H, J=6.5 Hz); 5.92 (d, 1H, J=6.2 Hz) | 5bc | 0.9 (s, 3H); 1.65 (s, 3H); 5.48 (s, 1H); 7.1-7.5 (m, 4H) |
| 3fa | 1.17 (s, 9H); 2.59 (d, 1H, J=2 Hz); 3.79 (s, 3H); 4.88 (t, 1H, J=6.2 Hz); 5.19 (br s, 1H)b; 5.2 (d, 1H, J=6.5 Hz); 5.49 (t, 1H, J=6.5 Hz); 5.76 (d, 1H, J=6.2 Hz) | 5cc | 0.9 (d, 3H, J=7.6 Hz); 3.8 (s, 3H); 4.0 (dq, 1H, J=7.6 and 6.5 Hz); 5.8 (d, 1H, J=6.5 Hz); 6.95-7.4 (m, 4H) |
| 4ac | 1.05 (s, 3H); 1.5 (s, 3H); 3.77 (s, 3H); 4.95 (t, 1H, J=6.3 Hz); 5.07 (d, 1H, J=6.6 Hz); 5.4 (s, 1H); 5.5 (t, 1H, J=6.6 Hz); 5.77 (d, 1H, J=6.3 Hz) | 5ec | 0.81 (s, 9H); 3.5 (d, 1H, J=6.7 Hz); 3.82 (s, 3H); 5.82 (d, 1H, J=6.7 Hz); 6.8-7.5 (m, 4H) |
| 4bc | 1.08 (s, 3H); 1.6 (s, 3H); 5.15 (t, 1H, J=6.2 Hz); 5.31 (s, 1H); 5.41 (t, 1H, J=6.3 Hz); 5.49 (d, 1H, J=6.3 Hz); 5.68 (d, 1H, J=6.2 Hz) | 5fc | 1.1 (s, 9H); 3.4 (d, 1H, J=4.1 Hz); 3.8 (s, 3H); 5.55 (d, 1H, J=4.1 Hz); 6.8-7.3 (m, 4H) |
Acknowledgments
References
- Pommier, A.; Pons, J. M. The synthesis of natural 2-oxetanones. Synthesis 1995, 729–744. [Google Scholar] [CrossRef]
- Pommier, A.; Pons, J.M. Recent advances in β-lactone chemistry. Synthesis 1993, 441–459. [Google Scholar] [CrossRef]
- Abe, H.; Matsubara, I.; Doi, Y. Physical properties and enzymatic degradability of polymer blends of bacterial poly[(R)-3-hydroxybutyrate] and poly[(R,S)-3-hydroxybutyrate] stereoisomers. Macromolecules 1995, 28, 844–853. [Google Scholar] [CrossRef]
- Baldoli, C.; Del Buttero, P.; Licandro, E.; Maiorana, S.; Papagni, A. Tricarbonyl(η6-arene)chromium(0) complexes as chiral auxiliaries. Homochiral β-lactams synthesis “via” [2+2] cycloaddition reaction. Tetrahedron: Asymmetry 1994, 5, 809–812. [Google Scholar] [CrossRef]
- Baldoli, C.; Del Buttero, P.; Licandro, E.; Papagni, A.; Pilati, T. Tricarbonyl(η6-arene)chromium(0) complexes as chiral auxiliaries. Asymmetric Synthesis of β-aminoesters and β- lactams by Reformatsky condensation. Tetrahedron 1996, 52, 4849–4856. [Google Scholar] [CrossRef]
- Baldoli, C.; Del Buttero, P.; Perdicchia, D.; Pilati, T. Stereoselective synthesis of β-sultams using chiral tricarbonyl(η6-arene)chromium(0) complexes. Tetrahedron 1999, 55, 14089–14096. [Google Scholar] [CrossRef]
- Del Buttero, P.; Baldoli, C.; Molteni, G.; Pilati, T. Stereoselective synthesis of a new enantiopure tricyclic β-lactam <<via>> tricarbonyl(η6-arene)chromium(0) complex. Tetrahedron: Asymmetry 2000, 11, 1927–1941. [Google Scholar] [CrossRef]
- Yang, H. W.; Romo, D. Methods for the synthesis of optically active β-lactones (2-oxetanones). Tetrahedron 1999, 55, 6403–6434. [Google Scholar] [CrossRef]
- Case-Green, S. C.; Davies, S. G.; Hedgecock, C. J. R. Asymmetric Synthesis of Homochiral β- lactones via the Iron Chiral Auxiliary. Synlett 1991, 779–780. [Google Scholar] [CrossRef]
- Case-Green, S. C.; Davies, S. G.; Hedgecock, C. J. R. Asymmetric Synthesis of (-)- Tetrahydrolipstatin. Synlett 1991, 781–782. [Google Scholar] [CrossRef]
- Petragnani, N.; Yonashiro, M. The reactions of dianion of carboxylic acids and ester enolates. Synthesis 1982, 521–578. [Google Scholar] [CrossRef]
- Del Buttero, P.; Baldoli, C. Arenechromiumtricarbonyl derivatives as chiral auxiliaries: synthesis of enantiomerically pure β-lactones. First International Electronic Conference on Synthetic Organic Chemistry (ECSOC-1), 1997. [Google Scholar]
- Solladié-Cavallo, A.; Solladié, G.; Tsamo, E. Chiral (Arene)tricarbonylchromium Complexes: Resolution of Aldehydes. J. Org. Chem. 1979, 44, 4189–4191. [Google Scholar] [CrossRef]
- Top, S.; Jaouen, G.; Baldoli, C.; Del Buttero, P.; Maiorana, S. Microbial resolution of organometallic planar chirality. Enantioselective reduction of ortho and meta-substituted tricarbonylchromium benzaldhydes by bakers’ yeast. J. Organomet. Chem. 1991, 413, 125–135. [Google Scholar] [CrossRef]
- Mulzer, J.; Segner, J.; Brüntrup, G. Stereoselective synthesis of threo 3-hydroxycarboxylic acids. Stereochemistry of an aldol type addition under kinetic and thermodynamic control. Tetrahedron Lett. 1977, 52, 4651–4654. [Google Scholar] [CrossRef]
- Mulzer, J.; Zippel, M.; Brüntrup, G.; Segner, J.; Finke, J. Stereochemistry of the addition of carboxylic acid dianions to aldehydes under kinetic and thermodynamic control. Synthesis and configurational assignment of 2,3-disubstituted threo- and erythro-3-hydroxycarboxylic acids. Liebigs Ann. Chem. 1980, 1108–1134. [Google Scholar] [CrossRef]
- Canceill, J.; Basselier, J.J.; Jacques, J. Sur la stéréochimie de la réaction de Réformatsky. Spectres IR et spectres de RMN des β-hydroxyesters obtenus. Dosage de leurs mélanges. Bilan des résultats. Bull. Chim. Soc. Fr. 1967, 3, 1024–1030. [Google Scholar]
- Adam, W.; Baeza, J.; Liu, J.C. Stereospecific introduction of double bonds via thermolysis of β- lactones. J. Am. Chem. Soc. 1972, 94, 2000–2006. [Google Scholar] [CrossRef]
- Mulzer, J.; Pointner, A.; Chucholowski, A.; Brüntrup, G. Threo 3-hydroxycarboxylic acids as key intermediates in a highly stereoselective synthesis of (Z) and (E)-olefins and enol ethers. J. Chem. Soc. Chem. Commun. 1979, 52–54, and ref. therein. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A. Chiral arene-chromium-carbonyl complexes in asymmetric synthesis. Adv. Met.-Org. Chem. 1989, 1, 99–133. [Google Scholar]
- Sample Availability: Samples not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Buttero, P.D.; Montrasio, D. Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones. Molecules 2001, 6, 13-20. https://doi.org/10.3390/60100013
Buttero PD, Montrasio D. Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones. Molecules. 2001; 6(1):13-20. https://doi.org/10.3390/60100013
Chicago/Turabian StyleButtero, Paola Del, and Deborah Montrasio. 2001. "Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones" Molecules 6, no. 1: 13-20. https://doi.org/10.3390/60100013
APA StyleButtero, P. D., & Montrasio, D. (2001). Tricarbonyl(h6-Arene)Chromium(0) Complexes as Chiral Auxiliaries: Asymmetric Synthesis of b-Lactones. Molecules, 6(1), 13-20. https://doi.org/10.3390/60100013