Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide

Abstract
:Introduction
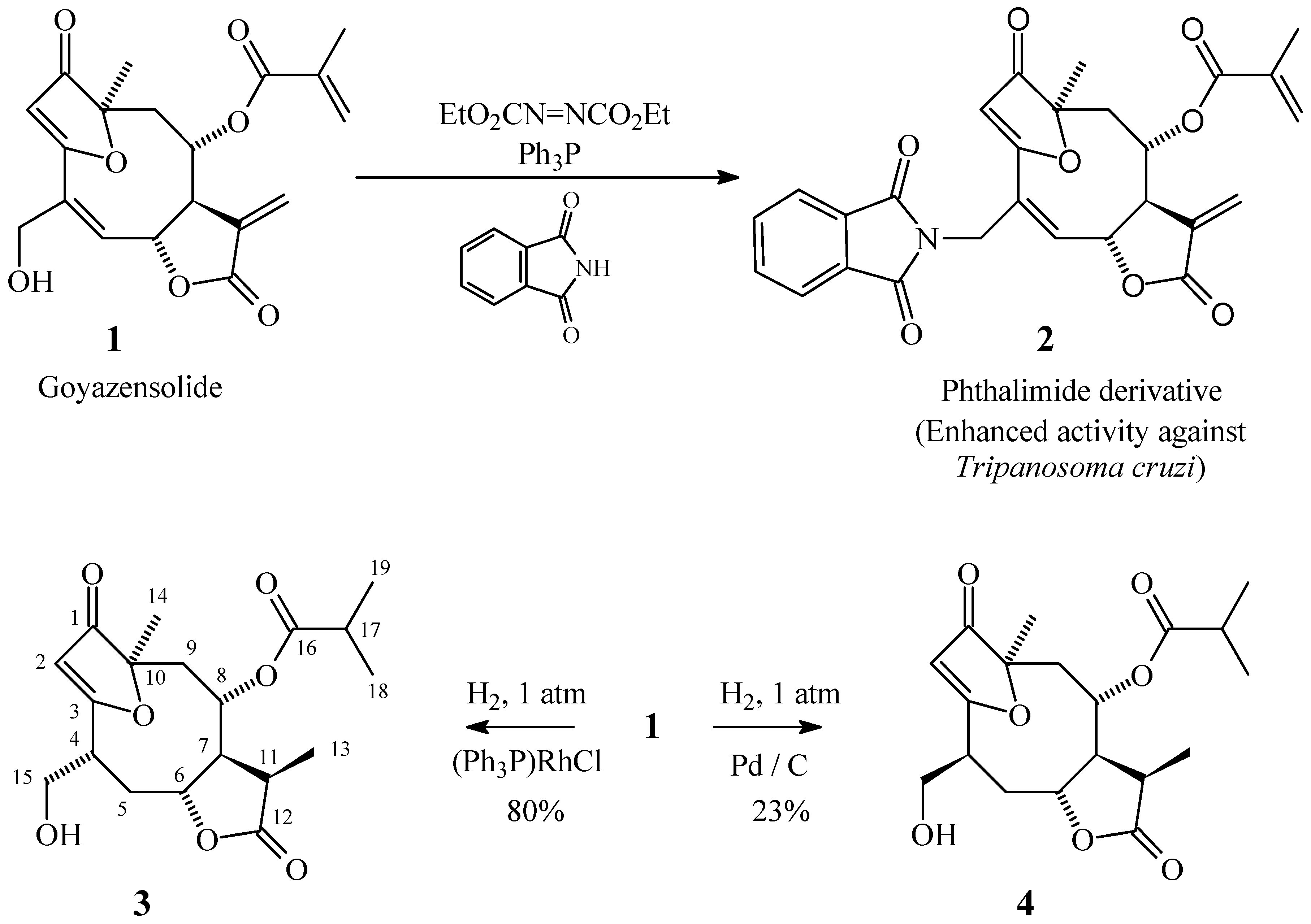
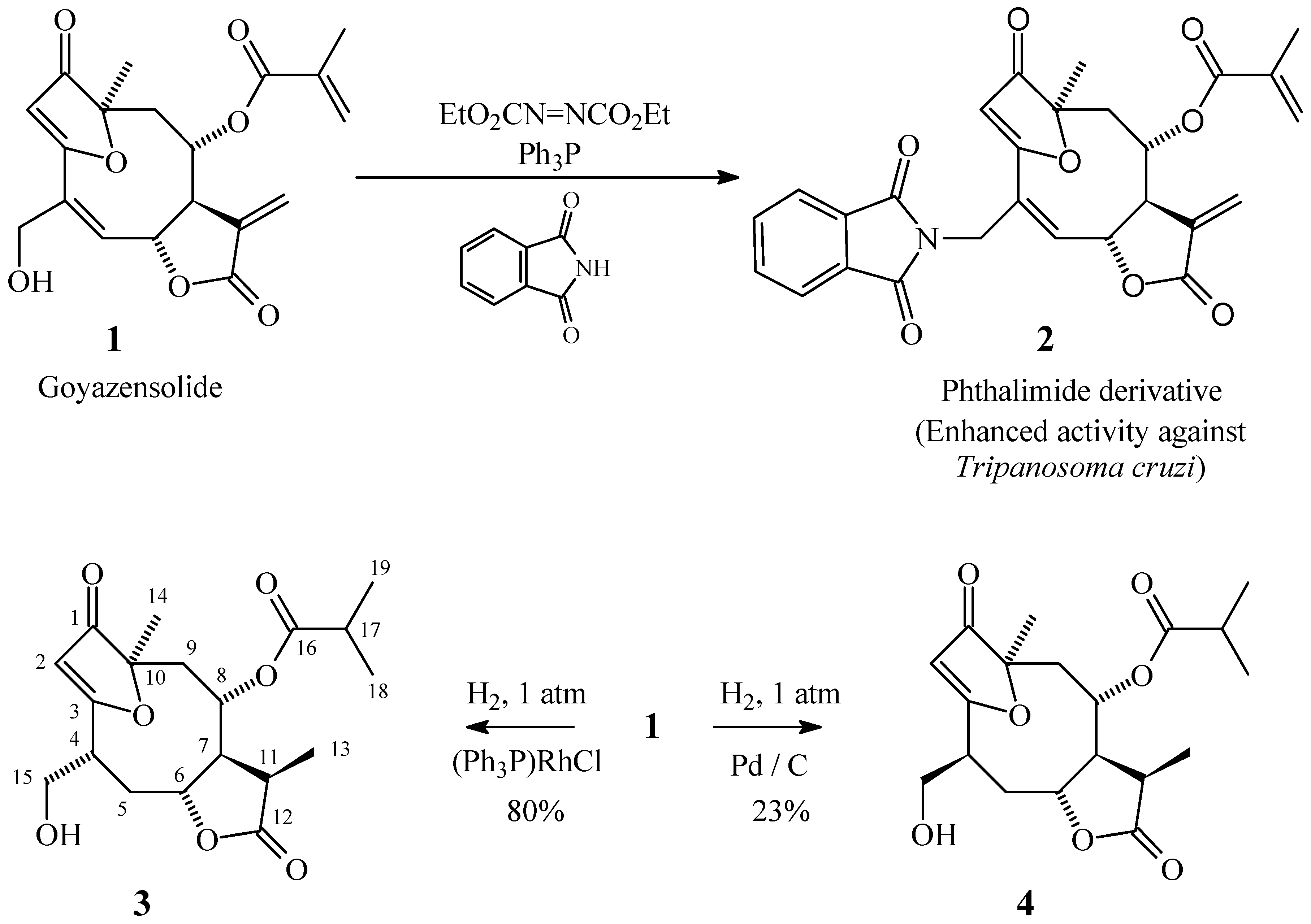
Results and Discussion

{kind=link}
| H | 3 | 4 | δ(4) - δ(3) |
|---|---|---|---|
| 5α | 2.14 | 2.44 | 0.30 |
| 4 | 3.33 | 3.10 | -0.23 |
| 5β | 2.35 | 2.14 | -0.21 |
| 7 | 3.06 | 3.26 | 0.20 |
| 2 | 5.70 | 5.89 | 0.19 |
| 15a | 3.85 | 3.99 | 0.14 |
| 15b | 3.80 | 3.93 | 0.13 |
| 8 | 4.93 | 5.01 | 0.08 |
| 9β | 2.07 | 2.10 | 0.03 |
| 6 | 4.23 | 4.26 | 0.03 |
| 9α | 2.20 | 2.18 | -0.02 |
| 17 | 2.53 | 2.55 | 0.02 |
| 18 | 1.18 | 1.20 | 0.02 |
| 19 | 1.17 | 1.19 | 0.02 |
| 11 | 2.88 | 2.89 | 0.01 |
| 13 | 1.11 | 1.12 | 0.01 |
| 14 | 1.48 | 1.49 | 0.01 |
| J | 3 | 4 | J(4) – J(3) |
|---|---|---|---|
| 4,5β | 7.0 | 3.4 | -3.6 |
| 8,9α | 6.8 | 5.3 | -1.5 |
| 6,7 | 5.5 | 4.1 | -1.4 |
| 4,5α | 10.0 | 8.7 | -1.3 |
| 4,15a | 7.9 | 6.7 | -1.2 |
| 2,4 | 0.0 | 1.1 | 1.1 |
| 5α,6 | 8.6 | 9.7 | 1.1 |
| 7,11 | 8.3 | 9.3 | 1.0 |
| 5β,6 | 0.8 | 1.4 | 0.6 |
| 9α,9β | 15.0 | 15.5 | 0.5 |
| 8,9β | 2.5 | 3.0 | 0.5 |
| 7,8 | 9.3 | 9.8 | 0.5 |
| 17,18 | 7.2 | 6.8 | -0.4 |
| 5α,5β | 14.0 | 14.3 | 0.3 |
| 15a,15b | 10.7 | 10.5 | -0.2 |
| 4,15b | 5.9 | 5.8 | -0.1 |
| 11,13 | 7.4 | 7.5 | 0.1 |
| 3 | 4 | |
|---|---|---|
| C-1 | 205.5 | 205.9 |
| C-2 | 105.9 | 102.8 |
| C-3 | 189.0 | 189.0 |
| C-4 | 42.5 | 39.6 |
| C-5 | 35.2 | 34.5 |
| C-6 | 79.5 | 78.1 |
| C-7 | 51.9 | 49.8 |
| C-8 | 70.4 | 71.1 |
| C-9 | 40.4 | 37.4 |
| C-10 | 89.3 | 88.2 |
| C-11 | 38.4 | 37.3 |
| C-12 | 177.7 | 177.8 |
| C-13 | 10.8 | 10.3 |
| C-14 | 19.9 | 19.9 |
| C-15 | 63.4 | 62.0 |
| C-16 | 175.3 | 175.4 |
| C-17 | 34.2 | 34.2 |
| C-18 | 18.6 | 18.7 |
| C-19 | 18.8 | 18.7 |
| Irradiated Hydrogen | Enhanced Signals | |
|---|---|---|
| Compound 3 | Compound 4 | |
| 5α | 5β, 7, 15, 6 | 5β, 4, 7, 6 (small) |
| (+9α+9β in 3) | (8, through 9α and 9β) | |
| 5β | 5α, 4, 15, 6 | 5α, 6, 15 (small) |
| (+9α+9β in 4) | (8, through 9α and 9β) | |
| 6 | 5α, 5β, 13, 7 | 5β, 13 (small) |
| 7 | 11, 5α [8, 6 small] | 11, 5α |
| 13 | 11, 8, 6 | 8, 11, 6 (small) |
| (+18+19) | (17, through 18 and 19) | (17, through 18 and 19) |
| 11 | 7, 13 | 7, 13 |
| 4 | Not effected | 15, 5α |
Experimental
General
Preparation of Compound 2
Preparation of Compound 3
Preparation of Compound 4
| Compound | % lysis | |||
| 100 | 250 | 500 μg / mL | ||
| Negative control | 0.0 | 0.0 | 0.0 | |
| Goyazensolide (1) | 9.9 | 12.9 | 22.4 | |
| Phthalimide derivative (2) | 50.0 | 52.9 | 61.5 | |
| *positive control gentian violet 250 μg / mL | ||||
Acknowledgements
References and Notes
- Vichnewski, W.; Takahashi, A. M.; Nasi, A. M. T. T.; Gonçalves, D. C. R. G.; Dias, D. A.; Lopes, J. N. C.; Goedken, V. L.; Gutiérrez, A. B.; Herz, W. Phytochemistry 1989, 28, 1441.
- Herz, W.; Goedken, V. L. J. Org. Chem. 1982, 47, 2798. [CrossRef]
- Cf. compounds 2a and 9a of reference 1. To make some data match better we had to assume that hydrogens 5α and 5β for compound 9a are inverted in reference [1].
- Jackman, L. M.; Sternhell, S. Applications of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry, 2nd Edition; Pergamon Press: Oxford, 1978; p. 316. [Google Scholar]
- Albuquerque, S.; Nasi, A. M. T. T.; Ribeiro, R. D.; Lopes, R. A.; Carraro, A. A.; Rodrigues, E. R. Mem. Inst. Oswaldo Cruz 1991, 86 Suppl.I, 234.
- Mitsunobu, O. Synthesis 1981, 1.
- Mitsunobu, O.; Wada, M.; Sano, T. J. Amer. Chem. Soc. 1972, 94, 679. [CrossRef]
- The results of biological activity measurements are summarized in the table below:
- Rabjohn, N. Organic Syntheses; Horning, E. C., Ed.; John Wiley & Sons: New York, 1963; Coll. Vol. III, p. 375. [Google Scholar]
- Kauer, J. C. Organic Syntheses; Rabjohn, N., Ed.; John Wiley & Sons: New York, 1967; Coll. Vol. IV, p. 411. [Google Scholar]
- Harwood, L. M.; Moody, C. J. Experimental Organic Chemistry: Principles and Practice; Blackwell Scientific Publications: Oxford, 1989; pp. 511–513. [Google Scholar]
- March, J. Advanced Organic Chemistry, 4th Edition; John Wiley & Sons: New York, 1992; p. 771. [Google Scholar]
- Ireland, R. E.; Bey, P. Organic Syntheses; Noland, W. E., Ed.; John Wiley & Sons: New York, 1988; Coll. Vol. VI, p. 459. [Google Scholar]
- Sample Availability: Not available.
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Da Silva, G.V.J.; Heleno, V.C.G.; Constantino, M.G. Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide. Molecules 2000, 5, 908-915. https://doi.org/10.3390/50600908
Da Silva GVJ, Heleno VCG, Constantino MG. Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide. Molecules. 2000; 5(6):908-915. https://doi.org/10.3390/50600908
Chicago/Turabian StyleDa Silva, Gil Valdo José, Vladimir Constantino Gomes Heleno, and Mauricio Gomes Constantino. 2000. "Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide" Molecules 5, no. 6: 908-915. https://doi.org/10.3390/50600908
APA StyleDa Silva, G. V. J., Heleno, V. C. G., & Constantino, M. G. (2000). Reduction and Preparation of a Phthalimide Derivative from a Furo-heliangolide. Molecules, 5(6), 908-915. https://doi.org/10.3390/50600908