Synthesis and X-Ray Crystal Structure of the First Pure and Air-Stable Salt of Peroxymonosulphuric Acid: (Ph)4PHSO5

Abstract
:Introduction
| H2SO5 | 2(KHSO5)(KHSO4)(K2SO4) | (Bu)4NHSO5 | Ph4PHSO5 | |||
| 1 | 2 | 3 | 4 |
Results and Discussion
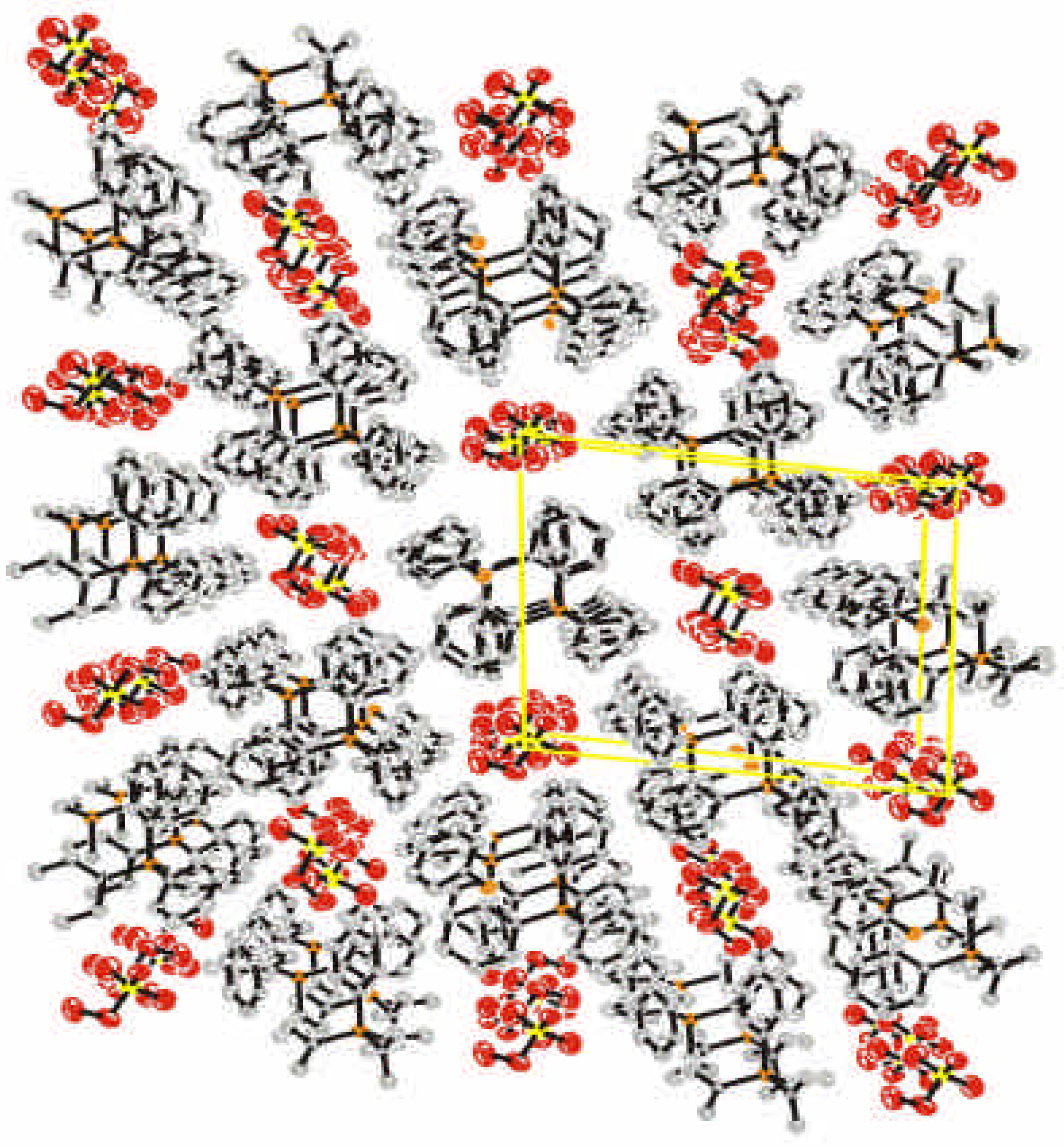
Crystal Data and Structure
Infrared Absorption
Conclusions
Experimental
Synthesis and purification of Ph4PHSO5
Acknowledgements
References and Notes
- Caro, H.Z. Angew. Chem. 1898, 11, 845.
- Balej, J. J. Electroanal. Chim. 1986, 214, 481.
- Kennedy, R.J.; Stock, A.M. J. Am. Chem. Soc. 1960, 25, 1901.
- Arnau, J.L.; Giguère, P.A. Can. J. Chem. 1970, 48, 3903.
- Frank, W.; Bertsch-Frank, B. Angew. Chem. Int. Ed. Engl. 1992, 31, 436.
- Ball, D.L.; Edwards, J.O. J. Am. Chem. Soc. 1956, 78, 1125.
- Flanagan, J.; Griffith, W.P.; Skapski, A.C. J. Chem. Soc. Chem. Commun. 1984, 1574.
- Schlemper, E.O.; Thompson, R.C.; Kay Fair, C.; Ross, F.K.; Appelman, E.H.; Basile, L.J. Acta Cryst. 1984, 1781.
- Block, R.; Abecassis, J.; Hassan, D. J. Org. Chem. 1985, 50, 1544.
- de Poorter, B.; Meunier, B. J. Chem. Soc. Perkin Trans. 2 1985, 1735.
- Mello, R.; Fiorentino, M.; Fusco, C.; Curci, R. J. Am. Chem. Soc. 1989, 111, 6749.
- Trost, B.M.; Braslau, R. J. Org. Chem. 1988, 53, 532.
- Campestrini, S.; Di Furia, F.; Labat, G.; Novello, F. J. Chem. Soc. Perkin Trans. 2 1994, 2175.
- Sheldrick, G.M. SHELXS 86. Program for the solution of crystal structures. University of Göttingen: Germany, 1985. [Google Scholar]
- Sheldrick, G.M. SHELXL 93. Program for refining crystal structures. University of Göttingen: Germany, 1993. [Google Scholar]
- Curci, R.; Edwards, J.O. Organic Peroxides; Swern, D., Ed.; Wiley-Interscience: New York, 1970; Vol. 1, pp. 199–264. [Google Scholar]
- Steiner, T.; Saenger, W. J. Am. Chem. Soc. 1993, 115, 4540.
- Sample Availability: Available from the authors.
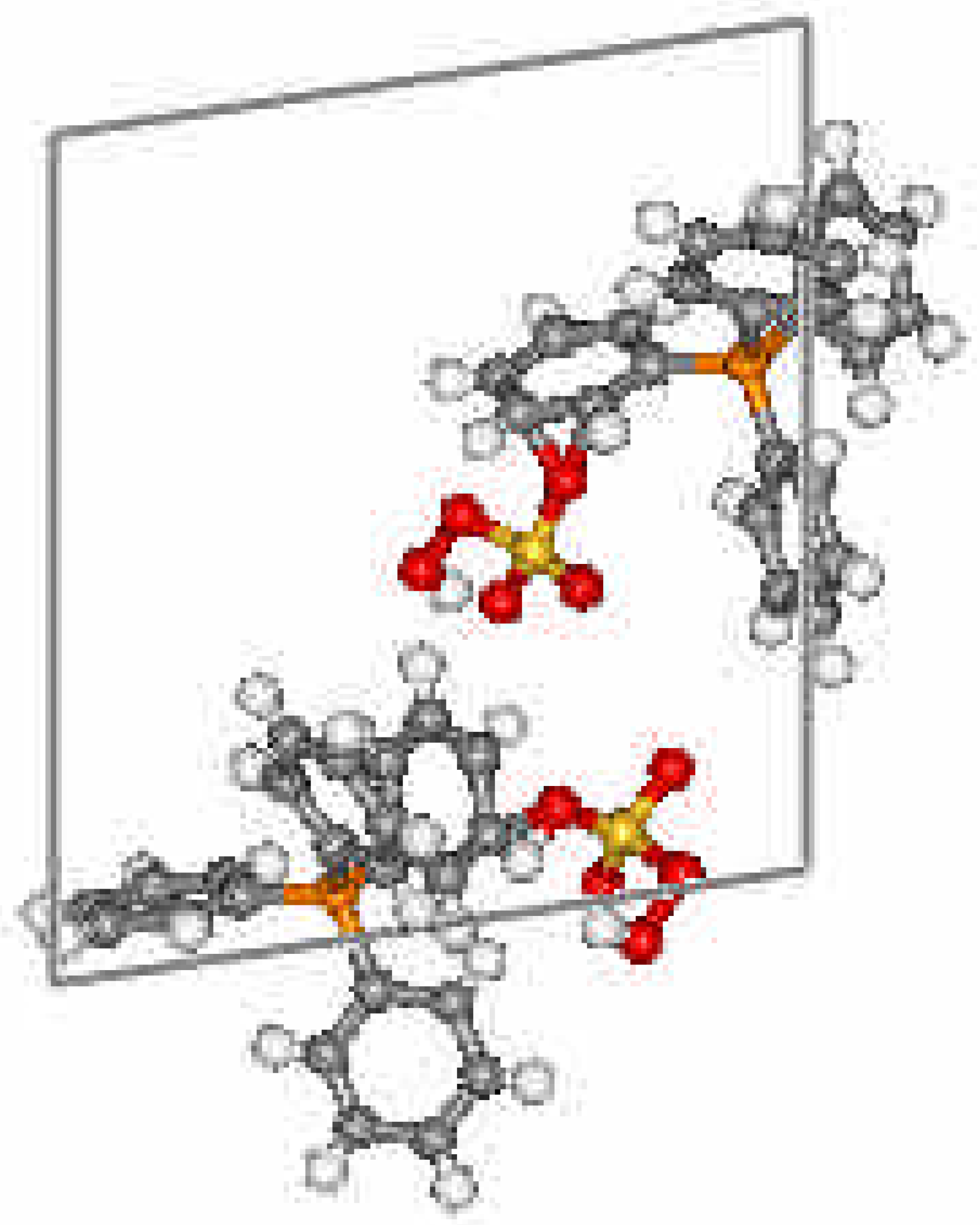

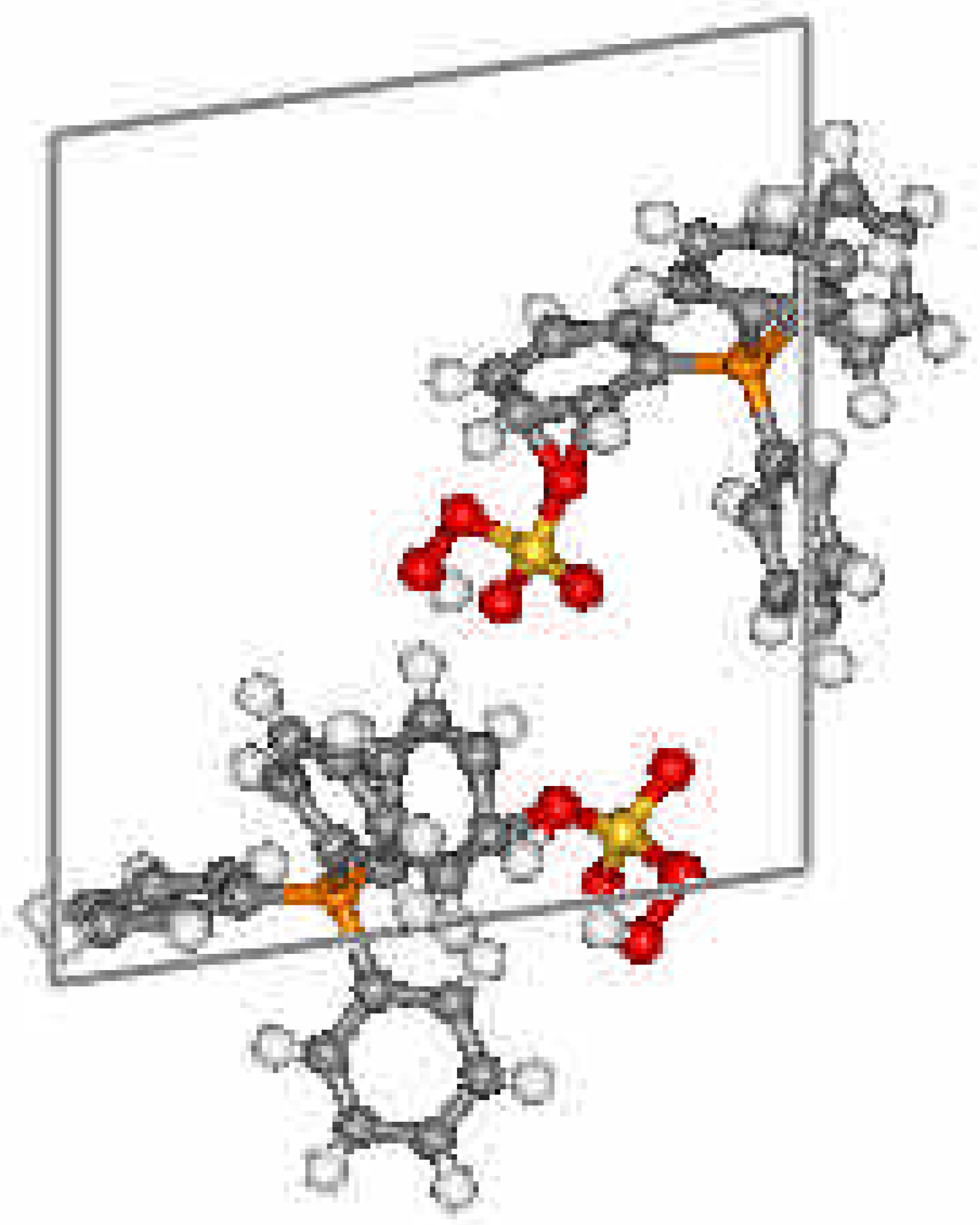{kind=link}
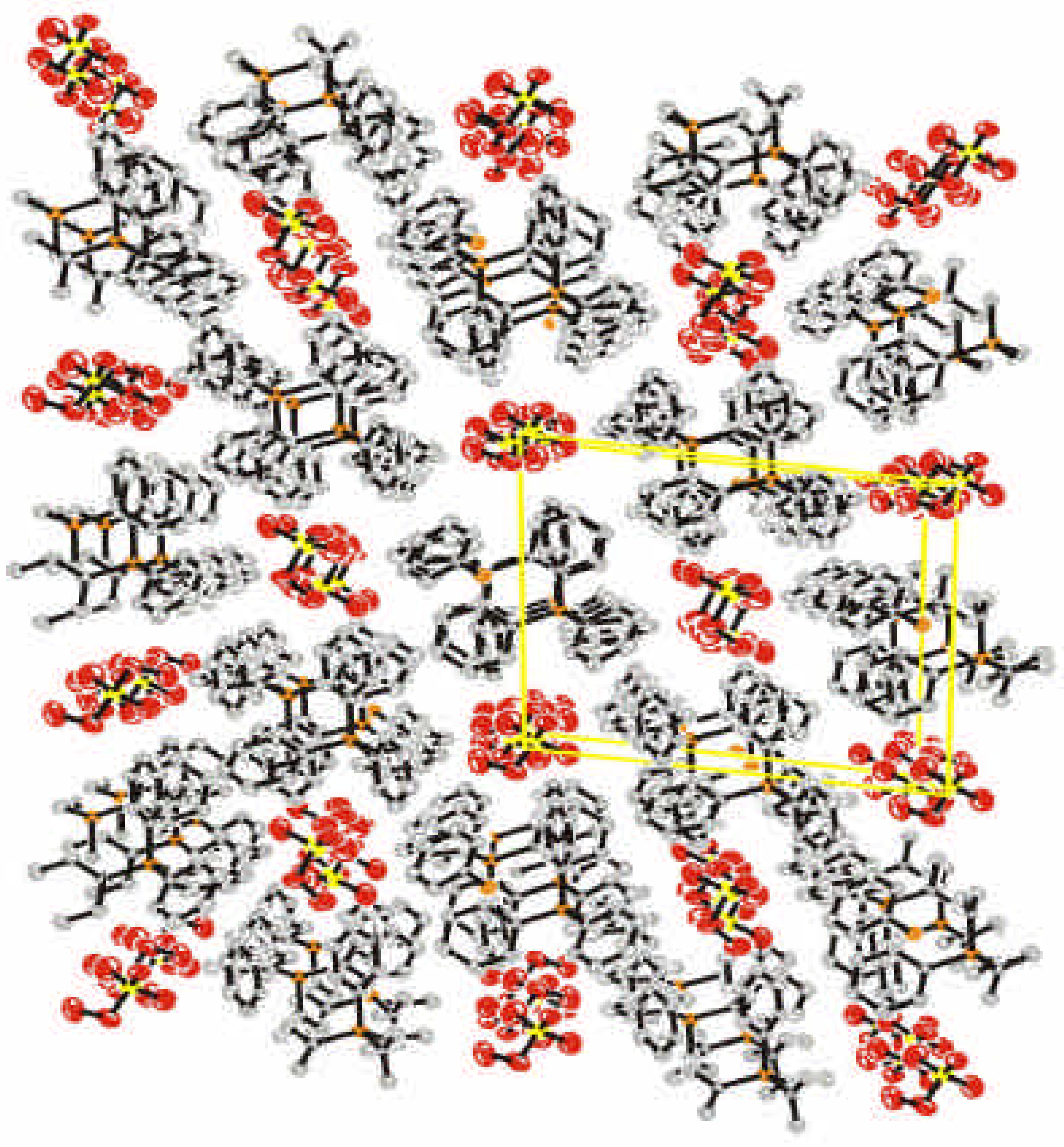{kind=link}
| Atom | x | y | z | Ueq | Atom | x | y | z | Ueq | |
| S1 | 5778(2) | 5092(2) | 2327(2) | 97(1) | S2 | -193(2) | 259(1) | 7759(2) | 81(1) | |
| O1 | 6179(4) | 4688(4) | 1408(5) | 107(2) | O6 | -827(3) | 531(3) | 8504(5) | 95(2) | |
| O2 | 6181(5) | 5936(4) | 2808(6) | 147(3) | O7 | -612(6) | -351(4) | 6803(6) | 155(3) | |
| O3 | 5728(6) | 4478(5) | 3219(7) | 185(3) | O8 | 183(4) | 1026(4) | 7369(6) | 131(2) | |
| O4 | 4626(4) | 5543(4) | 1607(6) | 123(2) | O9 | 858(4) | -262(4) | 8683(5) | 113(2) | |
| O5 | 4046(4) | 4902(4) | 943(6) | 131(2) | O10 | 637(5) | -1073(4) | 9178(6) | 134(2) | |
| P1 | 5463(1) | 891(1) | 7658(2) | 55(1) | P2 | 487(1) | 5857(1) | 7797(2) | 56(1) | |
| C1 | 5761(5) | 1437(4) | 9144(6) | 53(2) | C25 | 869(5) | 6441(4) | 9240(6) | 55(2) | |
| C2 | 6675(5) | 1160(4) | 10077(6) | 69(2) | C26 | 1884(5) | 6555(4) | 9732(6) | 63(2) | |
| C3 | 6933(6) | 1644(5) | 11167(7) | 80(2) | C27 | 2251(6) | 6982(4) | 10853(7) | 75(2) | |
| C4 | 6277(6) | 2397(5) | 11309(7) | 79(2) | C28 | 1582(7) | 7305(5) | 11458(7) | 83(2) | |
| C5 | 5356(6) | 2674(5) | 10391(7) | 81(2) | C29 | 564(6) | 7212(4) | 10987(7) | 80(2) | |
| C6 | 5078(5) | 2189(4) | 9288(7) | 72(2) | C30 | 183(5) | 6757(4) | 9851(7) | 72(2) | |
| C7 | 6143(4) | -258(4) | 7813(5) | 55(2) | C31 | 854(4) | 6462(4) | 6704(6) | 59(2) | |
| C8 | 7212(5) | -407(5) | 8097(6) | 71(2) | C32 | 567(5) | 7413(5) | 6582(6) | 68(2) | |
| C9 | 7738(5) | -1280(5) | 8194(6) | 79(2) | C33 | 782(5) | 7901(5) | 5703(7) | 78(2) | |
| C10 | 7213(6) | -2030(5) | 7989(7) | 85(2) | C34 | 1279(6) | 7457(6) | 4961(7) | 85(2) | |
| C11 | 6159(5) | -1888(4) | 7699(7) | 83(2) | C35 | 1599(6) | 6515(6) | 5106(7) | 88(2) | |
| C12 | 5628(5) | -1016(4) | 7617(6) | 69(2) | C36 | 1395(5) | 6014(5) | 5991(7) | 68(2) | |
| C13 | 4107(5) | 877(4) | 7025(6) | 53(2) | C37 | -866(5) | 5835(4) | 7203(6) | 59(2) | |
| C14 | 3594(5) | 593(4) | 7780(6) | 68(2) | C38 | -1489(5) | 6287(4) | 6088(6) | 69(2) | |
| C15 | 2558(5) | 530(4) | 7307(8) | 73(2) | C39 | -2528(6) | 6212(5) | 5638(7) | 86(2) | |
| C16 | 2038(5) | 744(4) | 6075(8) | 76(2) | C40 | -2958(6) | 5714(5) | 6287(8) | 88(2) | |
| C17 | 2516(5) | 1030(4) | 5307(7) | 77(2) | C41 | -2344(6) | 5264(5) | 7399(8) | 83(2) | |
| C18 | 3561(5) | 1099(4) | 5778(6) | 64(2) | C42 | -1309(5) | 5327(4) | 7831(6) | 73(2) | |
| C19 | 5800(4) | 1552(4) | 6602(6) | 56(2) | C43 | 1095(4) | 4682(4) | 8005(6) | 58(2) | |
| C20 | 6276(5) | 1153(5) | 5805(7) | 74(2) | C44 | 1885(5) | 4368(4) | 9078(6) | 71(2) | |
| C21 | 6461(6) | 1661(6) | 4926(7) | 89(2) | C45 | 2342(5) | 3452(5) | 9226(7) | 86(2) | |
| C22 | 6167(6) | 2610(6) | 4844(7) | 83(2) | C46 | 1974(6) | 2844(5) | 8296(9) | 97(3) | |
| C23 | 5705(5) | 3024(5) | 5644(7) | 76(2) | C47 | 1187(6) | 3156(5) | 7241(8) | 96(3) | |
| C24 | 5516(5) | 2509(4) | 6524(6) | 64(2) | C48 | 745(5) | 4050(4) | 7092(7) | 79(2) |
| S1-O3 | 1.396(6) | C13-C14 | 1.394(8) | C26-C27 | 1.381(8) |
| S1-O1 | 1.426(5) | C13-C18 | 1.387(8) | C27-C28 | 1.365(9) |
| S1-O2 | 1.458(6) | C14-C15 | 1.382(8) | C28-C29 | 1.368(9) |
| S1-O4 | 1.588(6) | C15-C16 | 1.364(9) | C29-C30 | 1.419(9) |
| O4-O5 | 1.378(7) | C16-C17 | 1.369(9) | C31-C36 | 1.380(8) |
| P1-C7 | 1.790(6) | C17-C18 | 1.396(8) | C31-C32 | 1.390(8) |
| P1-C19 | 1.779(6) | C19-C20 | 1.371(8) | C32-C33 | 1.378(8) |
| P1-C1 | 1.803(6) | C19-C24 | 1.397(8) | C33-C34 | 1.363(9) |
| P1-C13 | 1.790(6) | C20-C21 | 1.372(9) | C34-C35 | 1.384(10) |
| C1-C2 | 1.370(8) | C21-C22 | 1.388(9) | C47-C48 | 1.353(9) |
| C1-C6 | 1.395(8) | C22-C23 | 1.369(9) | C35-C36 | 1.386(9) |
| C2-C3 | 1.383(8) | C23-C24 | 1.383(8) | C37-C38 | 1.390(8) |
| C3-C4 | 1.372(9) | S2-O7 | 1.421(6) | C37-C42 | 1.380(8) |
| C4-C5 | 1.369(9) | S2-O6 | 1.432(5) | C38-C39 | 1.389(9) |
| C5-C6 | 1.397(8) | S2-O8 | 1.445(5) | C39-C40 | 1.374(9) |
| C7-C8 | 1.402(8) | S2-O9 | 1.584(6) | C40-C41 | 1.383(9) |
| C7-C12 | 1.387(7) | O9-O10 | 1.442(7) | C41-C42 | 1.381(9) |
| C8-C9 | 1.367(8) | P2-C25 | 1.792(6) | C43-C44 | 1.375(8) |
| C9-C10 | 1.385(8) | P2-C31 | 1.791(6) | C43-C48 | 1.392(8) |
| C10-C11 | 1.381(9) | P2-C43 | 1.788(6) | C44-C45 | 1.388(8) |
| C11-C12 | 1.369(8) | C25-C26 | 1.374(8) | C45-C46 | 1.384(9) |
| C25-C30 | 1.387(8) | C46-C47 | 1.359(10) |
| O3-S1-O1 | 115.6(4) | C14-C13-P1 | 119.7(5) | C30-C25-P2 | 122.1(5) |
| O3-S1-O2 | 115.6(5) | C18-C13-P1 | 121.3(5) | C25-C26-C27 | 121.0(7) |
| O1-S1-O2 | 113.0(4) | C13-C14-C15 | 121.2(7) | C28-C27-C26 | 118.7(7) |
| O3-S1-O4 | 105.9(4) | C16-C15-C14 | 118.8(7) | C27-C28-C29 | 121.8(7) |
| O1-S1-O4 | 106.7(3) | C15-C16-C17 | 121.6(7) | C30-C29-C28 | 120.0(7) |
| O2-S1-O4 | 97.8(4) | C18-C17-C16 | 120.0(7) | C29-C30-C25 | 117.6(7) |
| O5-O4-S1 | 111.7(5) | C17-C18-C13 | 119.4(6) | C36-C31-C32 | 120.2(6) |
| C7-P1-C19 | 109.6(3) | C20-C19-C24 | 118.3(6) | C36-C31-P2 | 122.2(5) |
| C7-P1-C1 | 111.0(3) | C20-C19-P1 | 122.4(5) | C32-C31-P2 | 117.5(5) |
| C19-P1-C1 | 109.3(3) | C24-C19-P1 | 119.1(5) | C33-C32-C31 | 119.5(7) |
| C7-P1-C13 | 110.2(3) | C19-C20-C21 | 122.1(7) | C32-C33-C34 | 120.4(7) |
| C19-P1-C13 | 107.5(3) | C20-C21-C22 | 119.3(7) | C35-C34-C33 | 120.4(8) |
| C1-P1-C13 | 109.2(3) | C23-C22-C21 | 119.6(7) | C34-C35-C36 | 119.9(7) |
| C2-C1-C6 | 120.8(6) | C24-C23-C22 | 120.9(7) | C35-C36-C31 | 119.4(7) |
| C2-C1-P1 | 120.6(5) | C23-C24-C19 | 119.8(6) | C38-C37-C42 | 118.3(6) |
| C6-C1-P1 | 118.5(5) | O7-S2-O6 | 113.5(4) | C38-C37-P2 | 121.8(5) |
| C3-C2-C1 | 119.7(6) | O7-S2-O8 | 115.3(4) | C42-C37-P2 | 119.8(5) |
| C2-C3-C4 | 120.0(7) | O6-S2-O8 | 112.9(3) | C37-C38-C39 | 119.5(7) |
| C3-C4-C5 | 120.8(7) | O7-S2-O9 | 108.2(4) | C40-C39-C38 | 121.3(7) |
| C4-C5-C6 | 119.9(7) | O6-S2-O9 | 106.0(3) | C41-C40-C39 | 119.6(7) |
| C5-C6-C1 | 118.7(7) | O8-S2-O9 | 99.2(3) | C40-C41-C42 | 118.8(7) |
| C8-C7-C12 | 118.6(6) | O10-O9-S2 | 107.6(4) | C41-C42-C37 | 122.4(7) |
| C8-C7-P1 | 120.2(5) | C25-P2-C31 | 107.1(3) | C44-C43-C48 | 118.1(6) |
| C12-C7-P1 | 121.2(5) | C25-P2-C43 | 110.5(3) | C44-C43-P2 | 121.6(5) |
| C9-C8-C7 | 120.8(6) | C31-P2-C43 | 110.8(3) | C48-C43-P2 | 120.2(5) |
| C8-C9-C10 | 119.9(7) | C25-P2-C37 | 112.3(3) | C43-C44-C45 | 120.8(6) |
| C11-C10-C9 | 119.6(7) | C31-P2-C37 | 109.5(3) | C46-C45-C44 | 119.4(7) |
| C10-C11-C12 | 120.8(6) | C43-P2-C37 | 106.6(3) | C45-C46-C47 | 119.7(7) |
| C11-C12-C7 | 120.3(6) | C26-C25-C30 | 120.9(6) | C46-C47-C48 | 121.1(7) |
| C14-C13-C18 | 118.9(6) | C26-C25-P2 | 117.0(5) | C43-C48-C47 | 121.0(7) |
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Campestrini, S.; Crisma, M. Synthesis and X-Ray Crystal Structure of the First Pure and Air-Stable Salt of Peroxymonosulphuric Acid: (Ph)4PHSO5. Molecules 2000, 5, 886-894. https://doi.org/10.3390/50600886
Campestrini S, Crisma M. Synthesis and X-Ray Crystal Structure of the First Pure and Air-Stable Salt of Peroxymonosulphuric Acid: (Ph)4PHSO5. Molecules. 2000; 5(6):886-894. https://doi.org/10.3390/50600886
Chicago/Turabian StyleCampestrini, Sandro, and Marco Crisma. 2000. "Synthesis and X-Ray Crystal Structure of the First Pure and Air-Stable Salt of Peroxymonosulphuric Acid: (Ph)4PHSO5" Molecules 5, no. 6: 886-894. https://doi.org/10.3390/50600886
APA StyleCampestrini, S., & Crisma, M. (2000). Synthesis and X-Ray Crystal Structure of the First Pure and Air-Stable Salt of Peroxymonosulphuric Acid: (Ph)4PHSO5. Molecules, 5(6), 886-894. https://doi.org/10.3390/50600886