Introduction
Trans-stilbene is an important organic compound carrying a wide variety of useful applications in or- ganic synthesis [
1]. These applications include, but are not limited to, asymmetric dihydroxylation [
1a], photocyclization [
1b], photoisomerization [
1c], and synthesis of diphenyl acetylenes [
1d], bromohydrins [
1e], and benzils [
1f]. Heterocyclic analogues of
trans-stilbene are of interest not only for exploring these various applications but also for their inherent potential to serve as key precursors to the synthesis of a host of novel as well as known heterocycles. We present here the synthesis of
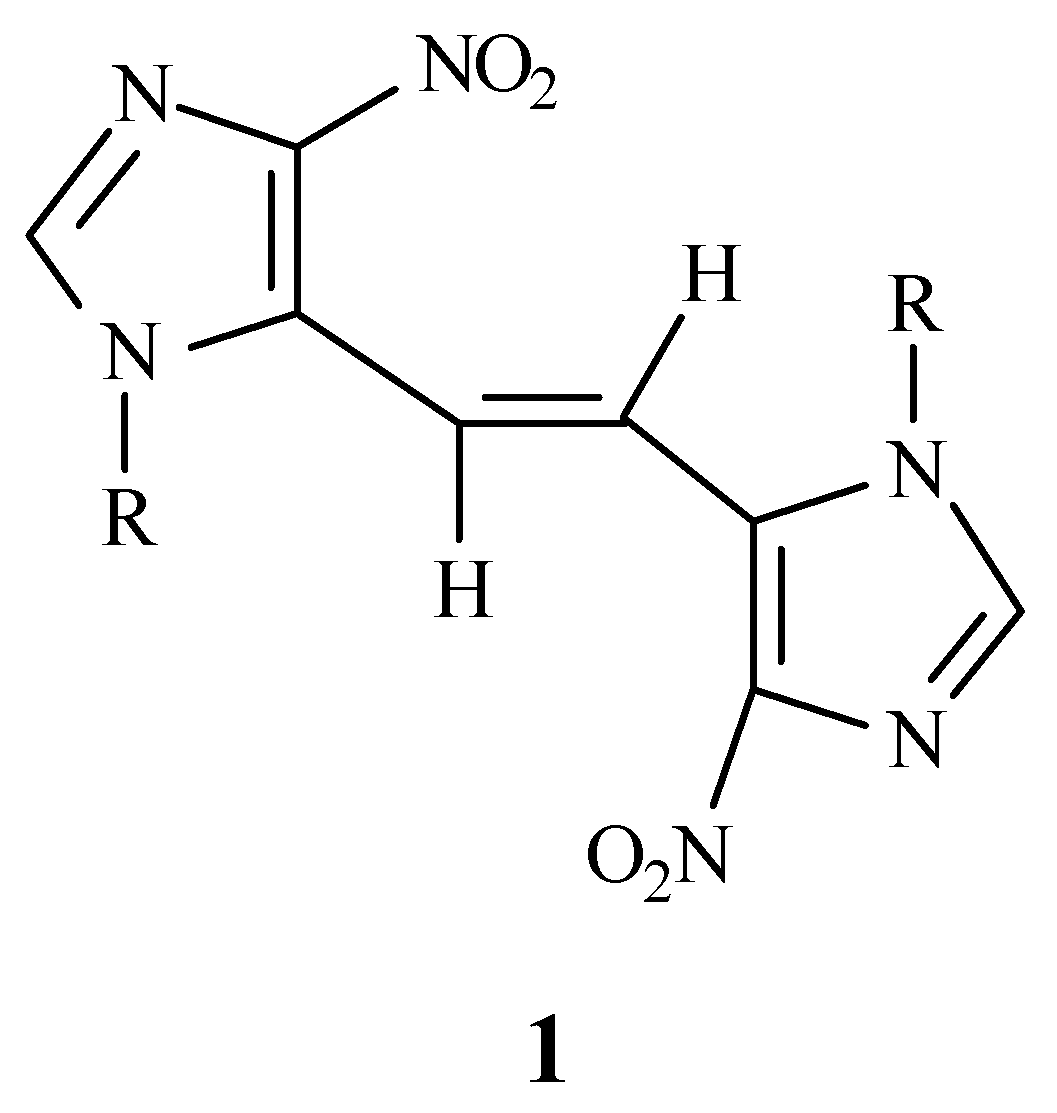
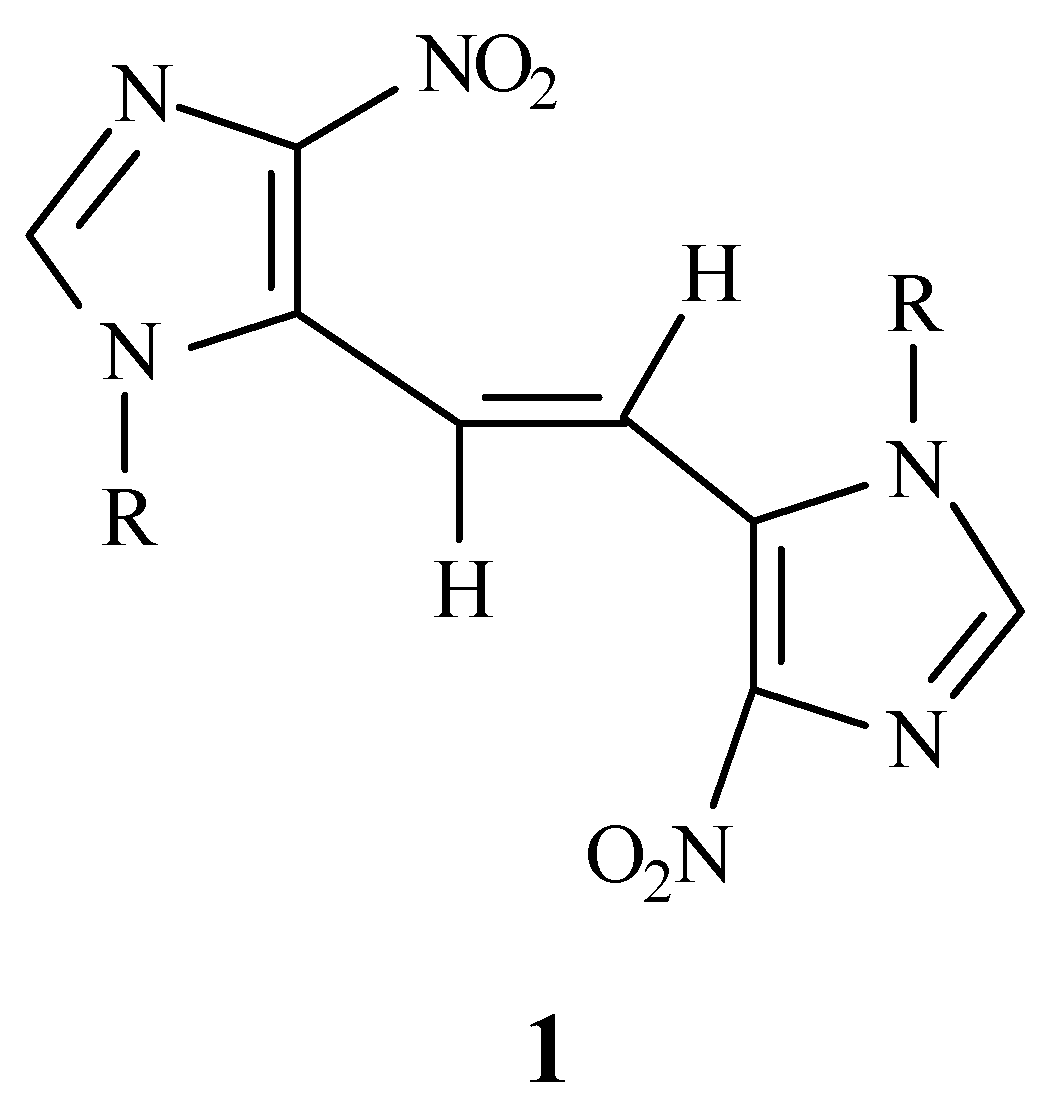
trans-1,2-bis(4-nitro- 1-
p-methoxybenzylimidazol-5-yl)ethene (
1) as an analogue of
trans-stilbene containing an imidazole ring. In order to confirm the
trans geometry of
1 using ultraviolet spectroscopy, a mixture of both
trans (
1) and
cis isomer (
3) was also prepared. The comparison of the UV spectrum of
1 with that of the mixture corroborated the
trans geometry of
1.
Results and Discussion
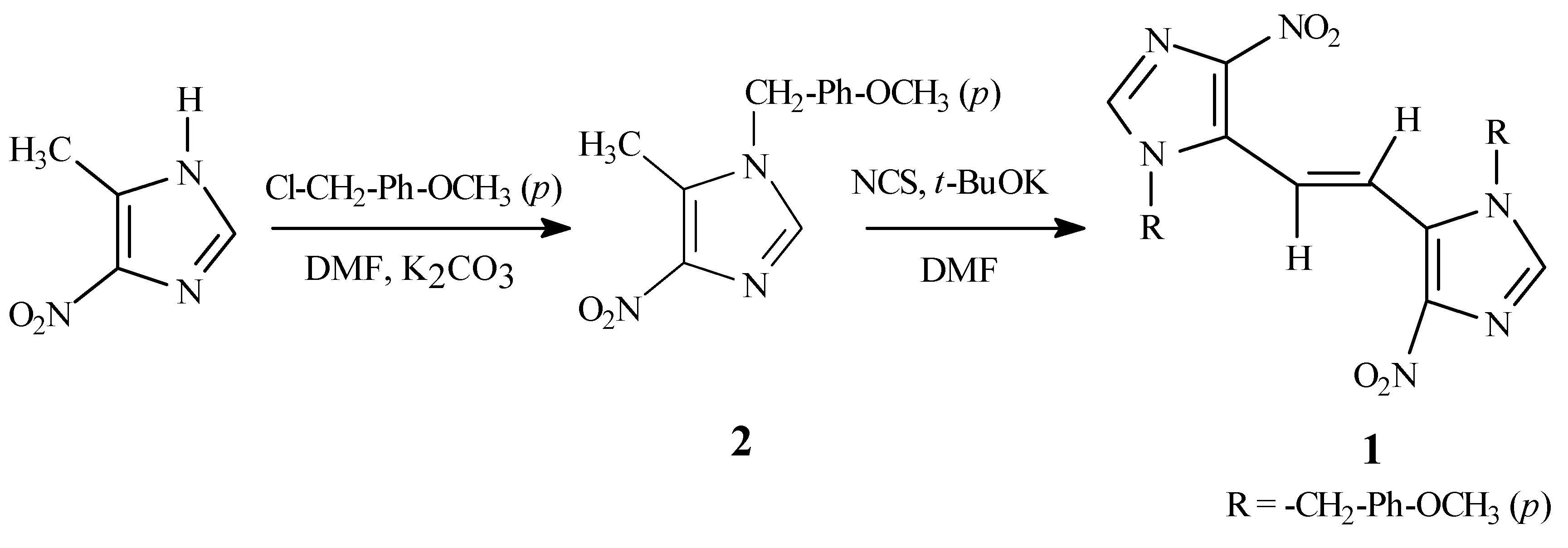
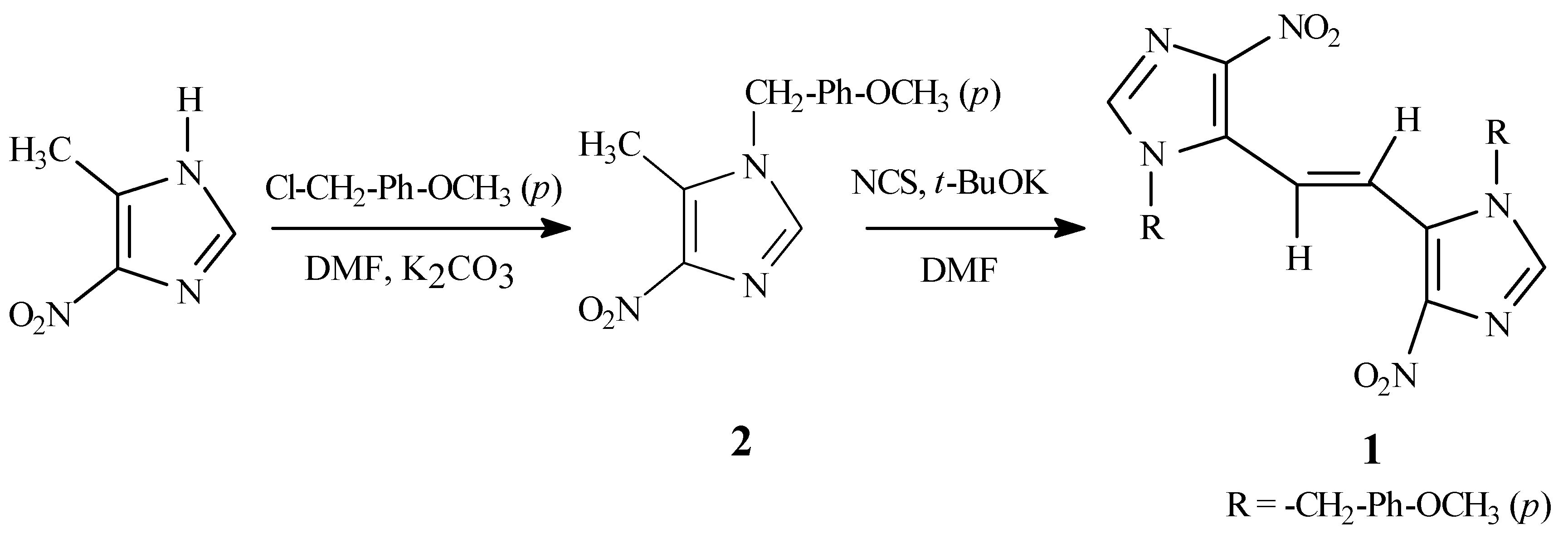
The required synthetic precursor, 4-nitro-1-
p-methoxybenzyl-5-methyl-1
H-imidazole (
2), was pre- pared by alkylation of 5-methyl-4-nitro-1
H-imidazole [
2] with
p-methoxybenzyl chloride in the presence of potassium carbonate in dimethylformamide (DMF) (
Scheme 1). Compound
2 was obtained as a sin- gle regioisomer as determined from NMR data. The regioisomeric assignment of
2 was based upon (a) comparison of the benzyl absorptions of
2 in its
1H NMR spectrum with those of several other benzyl- substituted nitroimidazoles previously synthesized in this laboratory [
3,
4,
5], and (b) the established fact that the alkylation of nitroimidazoles predominantly yield substitutions at the nitrogen atom farther away from the nitro group [
6].
The target
1 was prepared by the slow addition of a solution of
2 and potassium
t-butoxide in DMF to a solution of
N-chlorosuccinimide (NCS) [
7] in DMF.
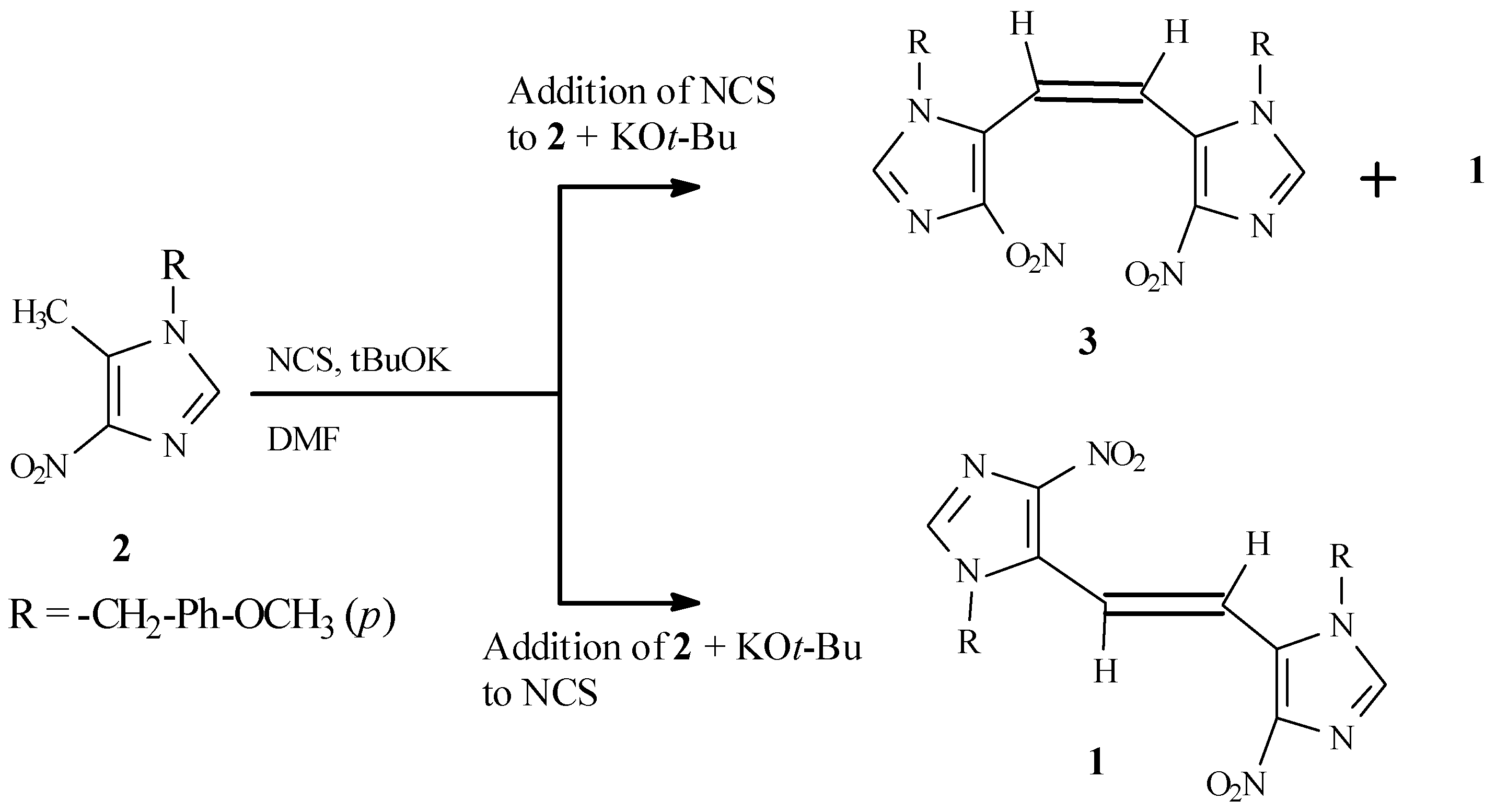
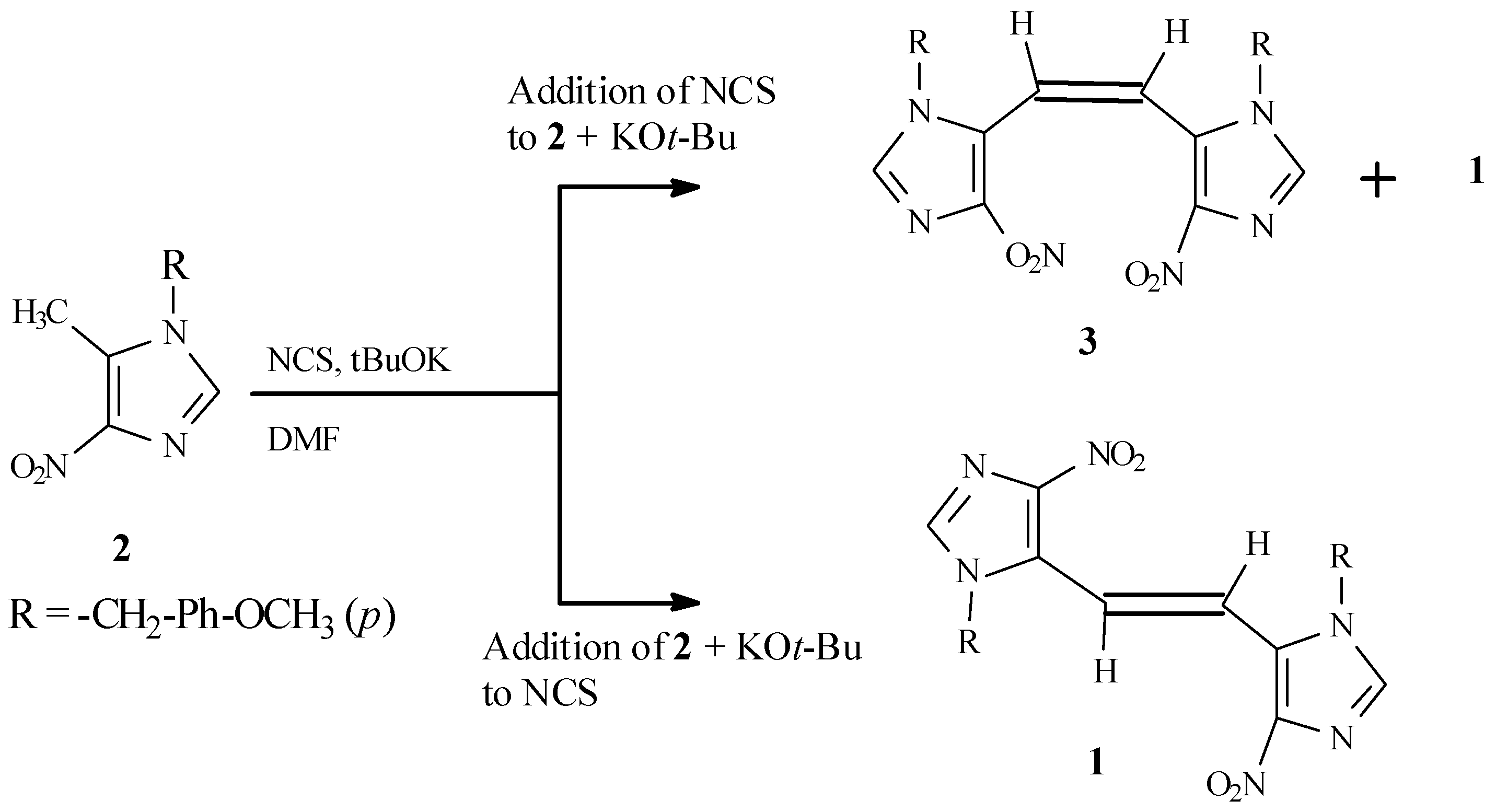
The energetically less-favored
cis isomer (
3), required for structural assignment by UV comparison, was prepared in low yield as a mixture of
cis and
trans isomers by reversal of the above addition proce- dure, consisting of a slow addition of a solution of NCS in DMF to a mixture of
2 and potassium
t- butoxide in DMF (
Scheme 2). The
1H NMR of the product mixture thus obtained exhibited two sets of peaks, while the product obtained by the first method showed only a single set of peaks.
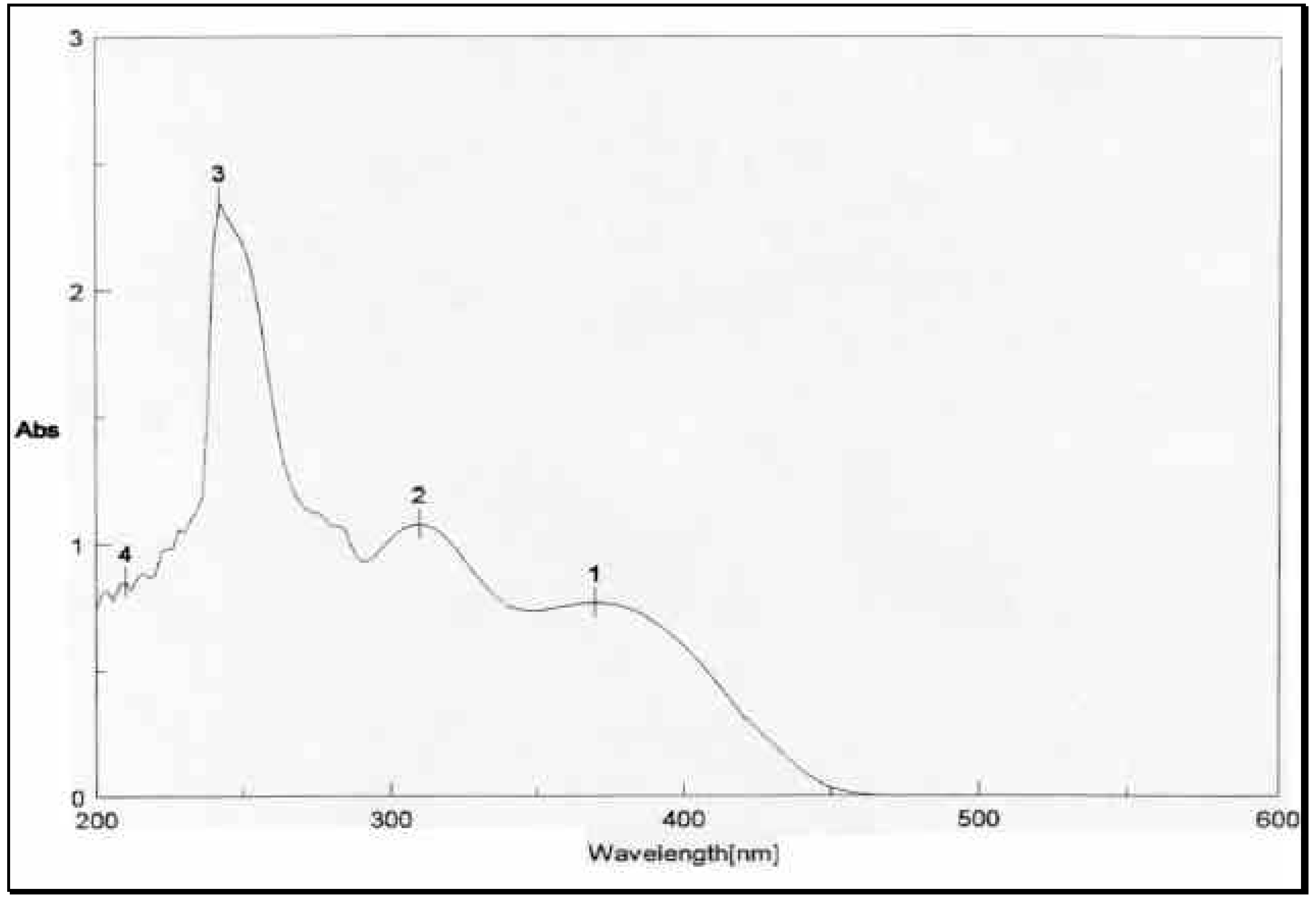
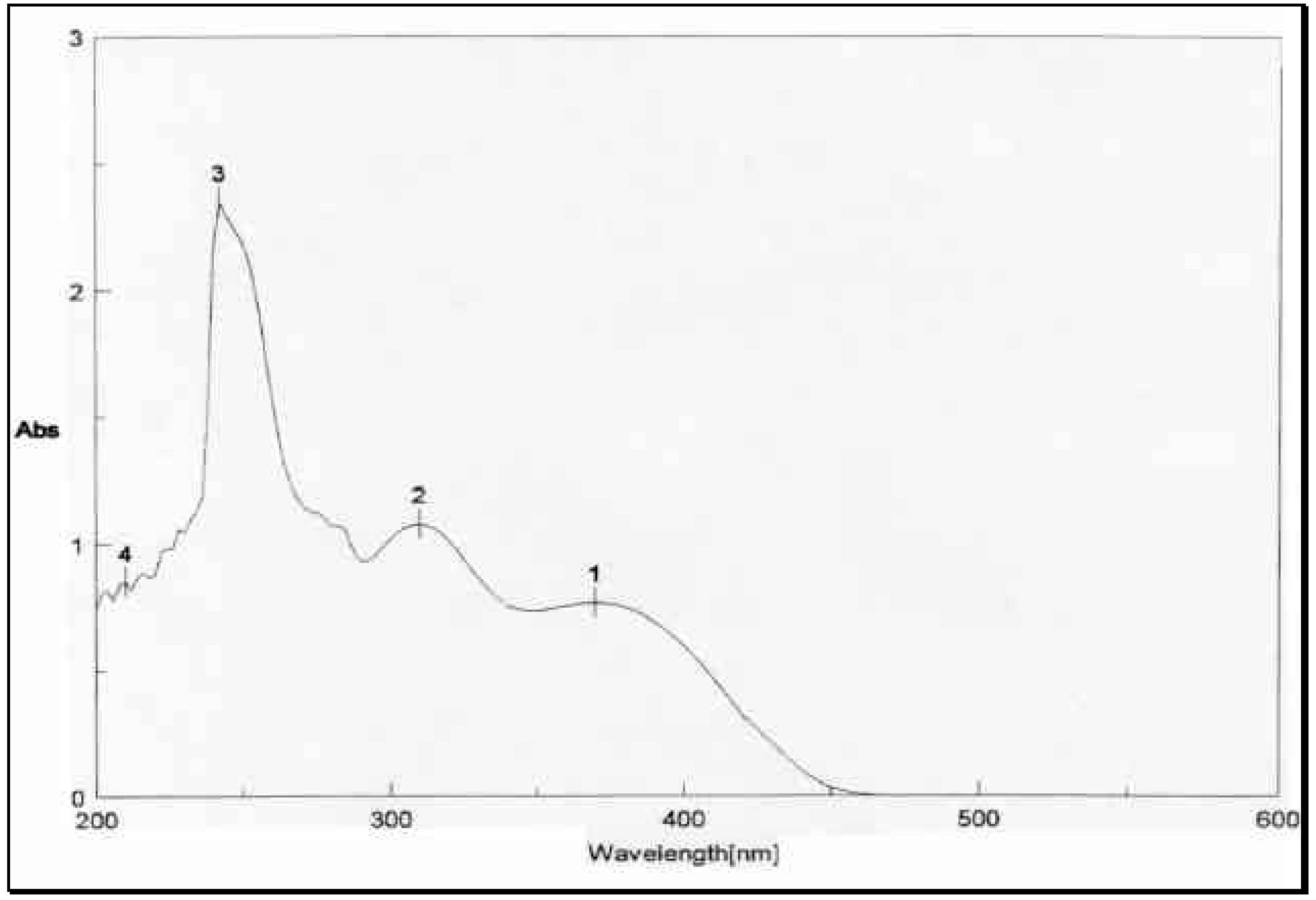
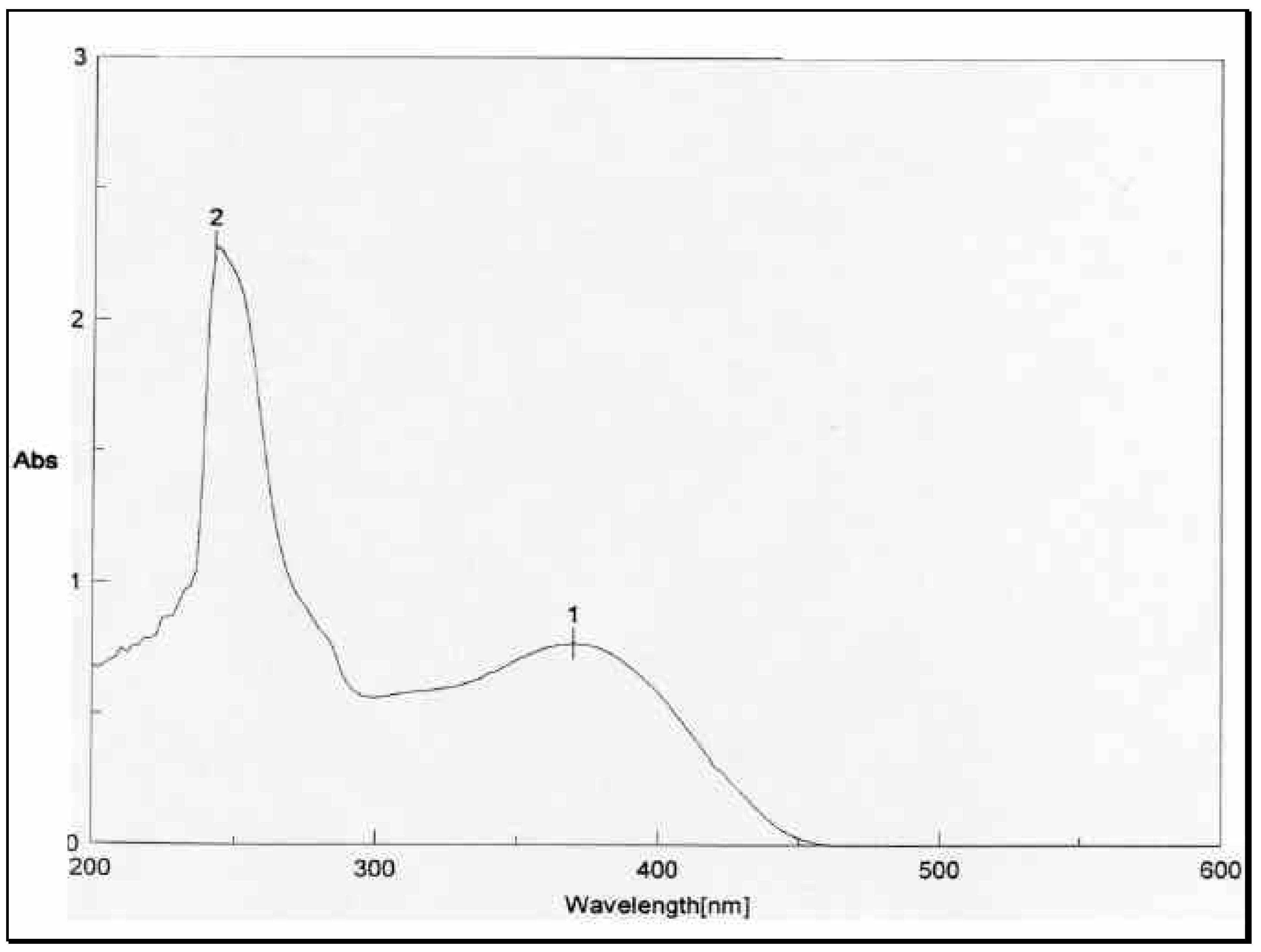
The structural distinction between the two geometric isomers was achieved by comparing the UV absorbance data of the mixture
1 and
3 with that of
1 alone. The UV absorbance spectra of a mixture of
1 and
3, depicted in
Figure 2, showed two peaks above 300 nm, one at 8max 310 nm and 370 nm.
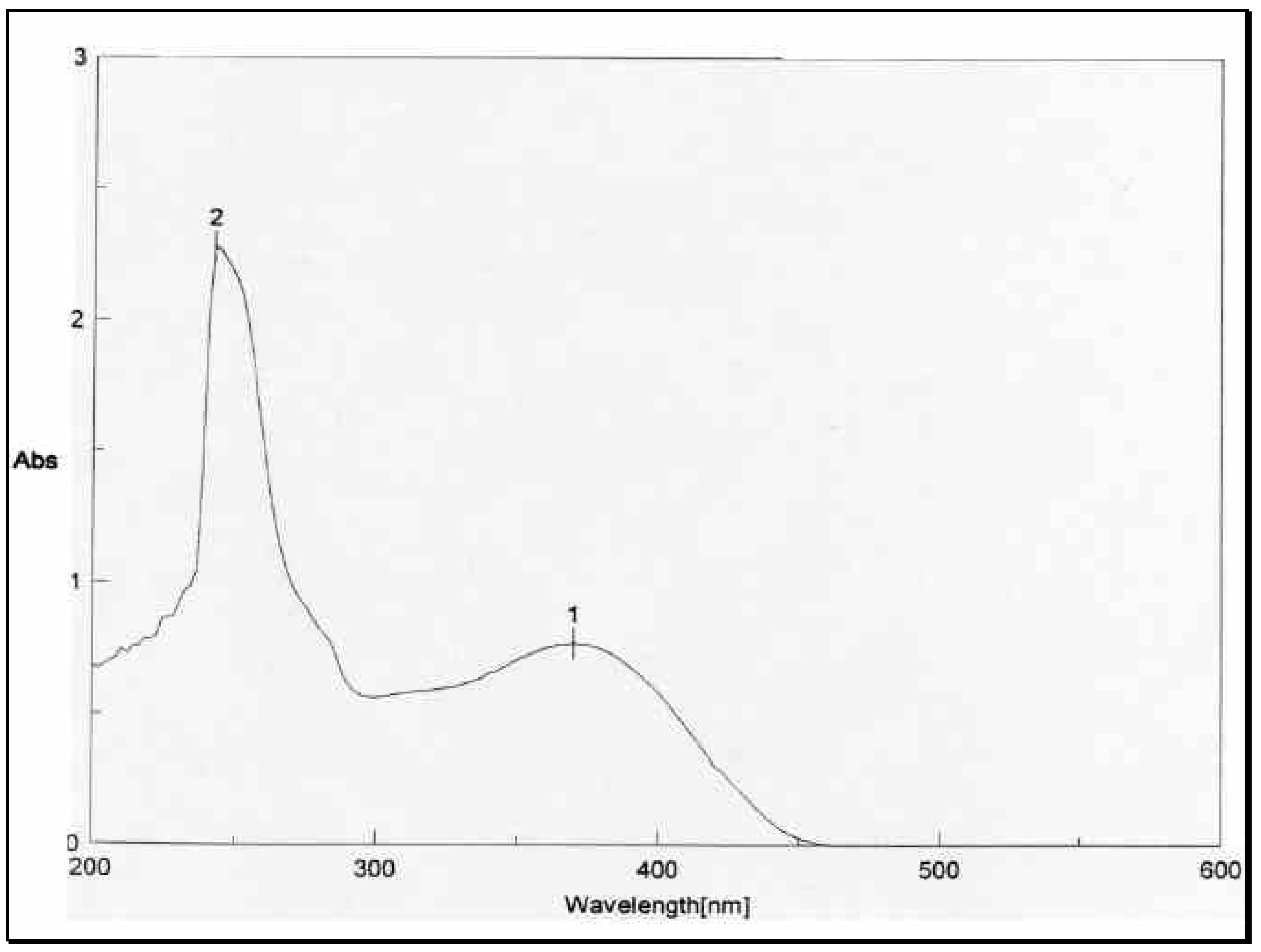
The UV absorbance spectrum of
1 in
Figure 3, on the other hand, showed only one peak at 8
max 370 nm. The energetically less-favored
cis isomer
3, with its two-charged nitro groups in close spatial proximity, is anticipated to absorb at a lower wave number (higher energy) than the
trans isomer
1. Thus
3 and
1 were assigned the
cis and
trans configurations, respectively. The mass spectrometric and microanalytical data were consistent with the dimeric structures of
1 and
3.
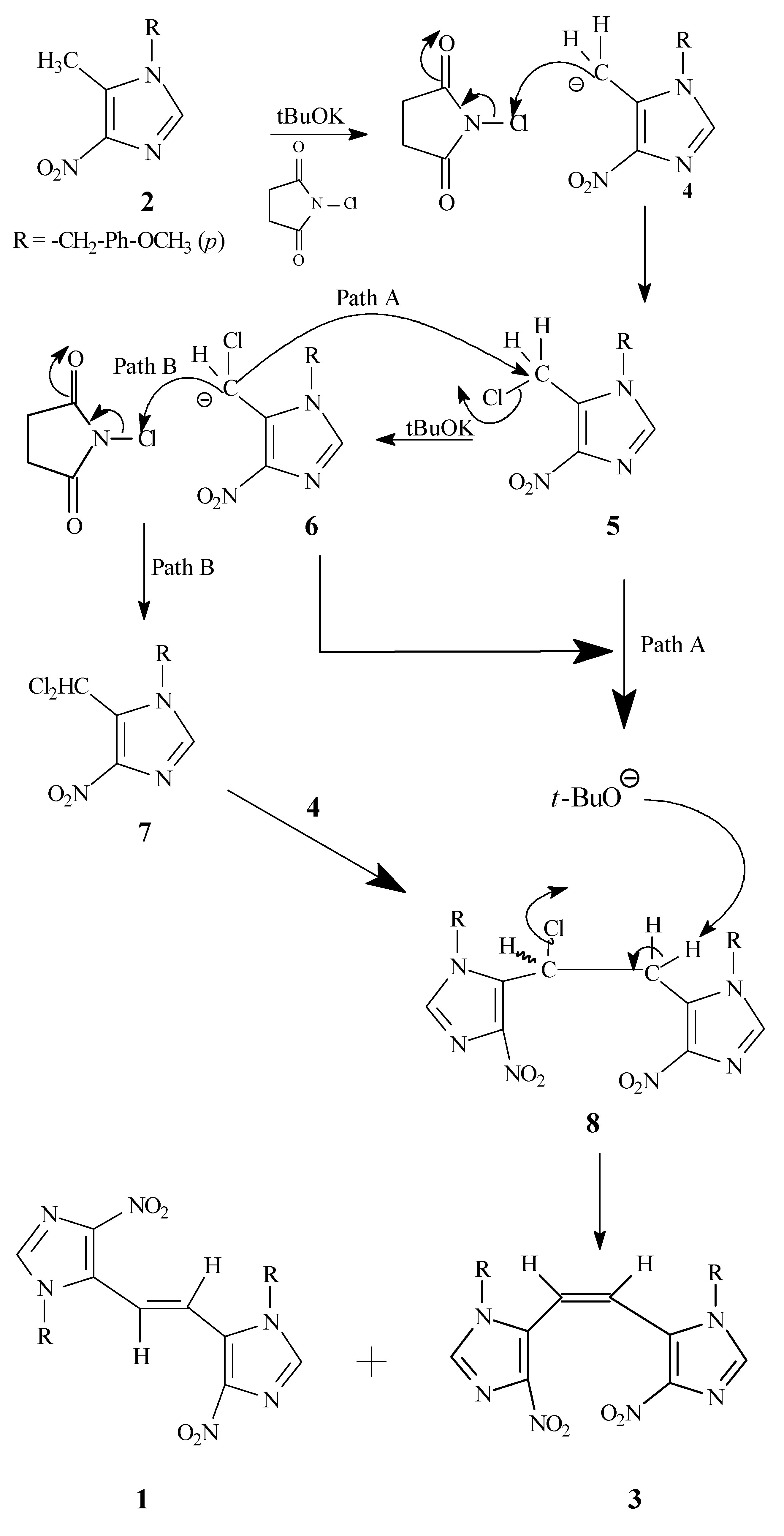
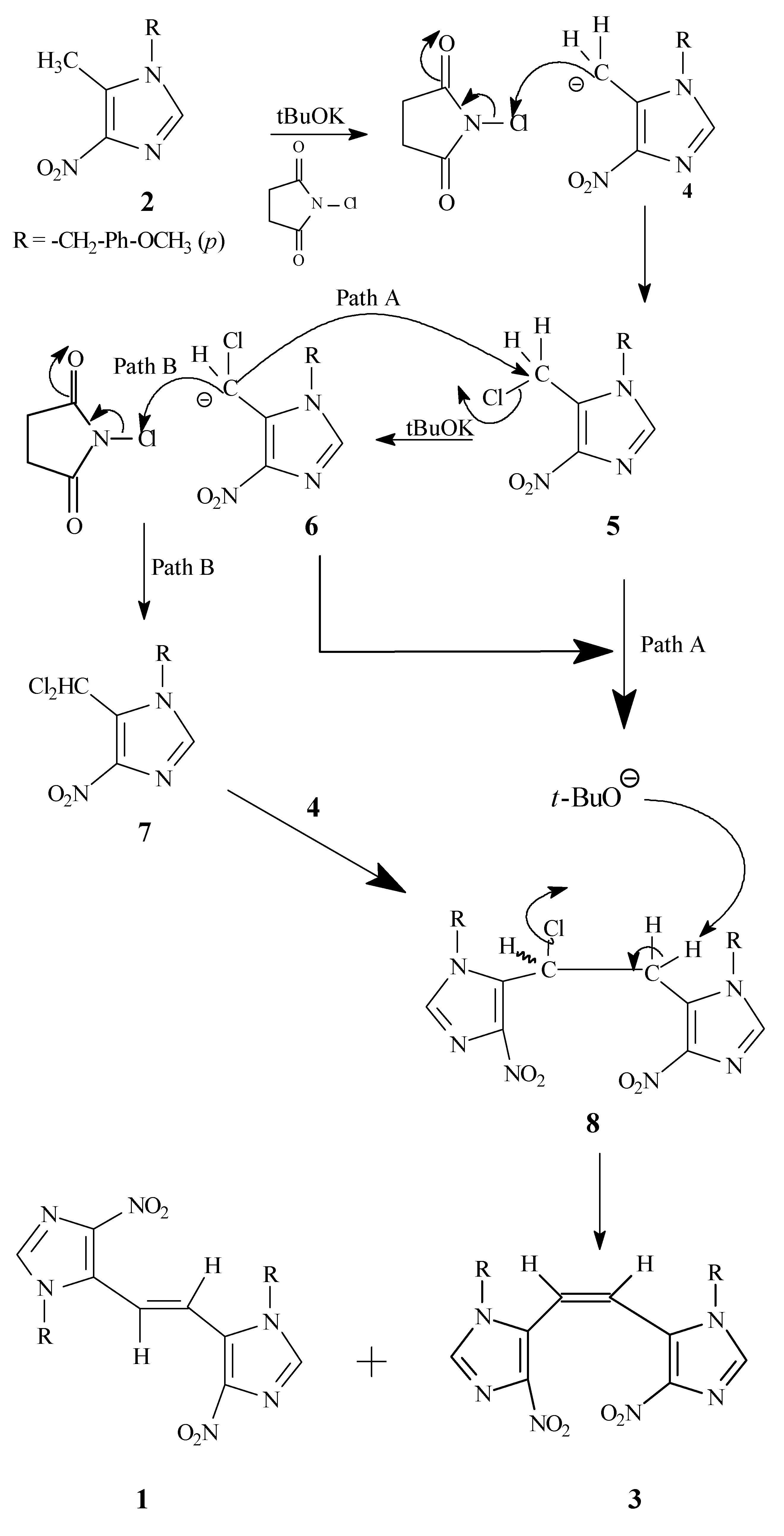
A tentative reaction pathway for the formation of
1 and
3 from
2 is outlined in
Scheme 3. It appears that the key intermediate is the monochloro dimer
8, which might be formed via a couple of different routes, including the nucleophilic displacement reaction of the carbanion
6 on the initially formed monochloro species
5 (Path A) or by the attack of carbanion
4 on the dichloro species
7 (Path B). The intermediate
8 undergoes further dehydrohalogenation in the presence of potassium
t-butoxide to yield the observed
cis and
trans dimers
3 and
1, respectively.
Experimental
General
1H NMR spectra were recorded on a General Electric QE-300 (300 MHz) or Varian (200 MHz) NMR instrument. The reported spectral data are in the following format: chemical shift (all relative to MeSi4 as an internal reference standard unless otherwise indicated), multiplicity (abbreviations: s = sin- glet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, b = broad and app = apparent), integration, coupling constants, exchangeability after D2O addition, and assignment of reso- nance. 13C NMR spectra were recorded on a General Electric QE-300 (300 MHz) instrument, using DMSO-d6 as the reference standard. Atlantic Microlab, Inc., Norcross Georgia, performed elemental microanalyses. The mass spectra were recorded at the Mass Spectral Facility, Department of Biochem- istry, Michigan State University. Ultraviolet spectra were recorded on a Jasco V-570 UV/VIS/NIR spectrophotometer. Thin layer chromatography was performed on Merck Kieselgel 60 GF254 plates (0.2 mm thickness). Melting points were determined on a Thomas-Hoover capillary melting point apparatus and are uncorrected. Anhydrous THF was freshly distilled from sodium. Anhydrous solvents DMF, pyridine, and chloroform were purchased from Aldrich Chemical Co. and were used without further pu- rification.
4-Nitro-1-p-methoxybenzyl-5-methylimidazole (2)
To 1.00 g of
5-methyl-4-nitro-1
H-imidazole [
2] (7.86 mmol) in a 100 mL three-neck round bottom flask equipped with a thermometer, N
2 inlet, and a magnetic stirring bar, was added potassium carbon- ate (0.868 gm, 6.29 mmol), DMF (10.0 mL) and
p-methoxybenzyl chloride (1.17 mL, 8.65 mmol) in one portion. The mixture was heated at 130°C overnight. The mixture was then cooled to room tem- perature and then further cooled in an ice bath. The resulting salts were collected under vacuum and washed with chloroform. The chloroform was then evaporated to yield a dark slurry (as a last step in evaporation process, toluene was added and the mixture evaporated again to get all the DMF out). The dark mixture was then triturated with cold ether to yield brown crystals. The crystals collected had m.p. 98-106°C, and were recrystallized in toluene to yield a light brown solid, m.p. 103-106°C, 42% yield.
1H NMR. (DMSO-d
6) δ 7.91(s, 1H, 2-H imidazole), 7.21 (d,
J=8.7Hz, 2H,
m-Ph), 6.94 (d,
J=8.4Hz, 2H,
o-Ph), 5.24 (s, 2H, CH
2Ar), 3.74 (s, 3H, OCH
3); MS (FAB)
m/z 248 (MH
+).
Anal. Calcd. for C12H13N3O3 : C, 58.29; H, 5.29; N, 16.99. Found C, 58.43; H, 5.40; N, 15.91.
Mixture of cis-1,2-bis(3-nitro-1-p-methoxybenzylimidazol-2-yl)ethene (3) and trans-1,2-bis(3-nitro- 1- p-methoxybenzyl imidazol-2-yl)ethene (1)
Compound 2 (0.200 g, 0.81 mmol) in (10.0 mL) DMF was added dropwise over 30 minutes to a stirred solution of potassium-tert butoxide (0.363 g, 3.24 mmol) in a three-neck round bottom flask, under nitrogen at 0°C. The reaction mixture was allowed to stand another 30 minutes at 0°C and then N-chlorosuccinimide (NCS) (0.216 g, 1.618 mmol) in dry THF (10.0 mL) was added to the flask. The reaction temperature was brought to room temperature, followed by refluxing for 5 hours. TLC (100:1 CHCl3 / MeOH) showed no starting material. The mixture was then rotary evaporated and neutralized to pH 7 with dil. HCl. The mixture was extracted with chloroform (3 x 50 mL). The organic layers were combined and dried over anhydrous MgSO4. The chloroform extract was mixed with silica gel and the mixture was evaporated to dryness. The residue was suspended in a minimum amount of chloroform and the resulting slurry was loaded onto a silica gel column and the column was eluted with a mixture of 100:1 (CHCl3 / MeOH). Pooling and evaporation of the appropriate eluent fractions under reduced pressure afforded an oil, which was triturated with cold ethanol to give a mixture of dimers 1 and 3, m.p. 205-210°C. 1H NMR (300 MHz, DMSO-d6) δ 8.05 & 7.87(s, 2x1, 1H, 2-H imidazole), 7.41 (s, 1H, C=C), 7.17 (d, J=8.7Hz, 2H, m-Ph), 7.04 (d, J=8.4Hz, 2H, m-Ph) 6.91 (d, J=8.7Hz, 2H, o-Ph). 6.88 (s, 1H, C=C), 6.86 (d, J=8.7Hz, 2H, o-Ph), 5.26 & 5.18 (s, 2x1, 2H, CH2Ar), 3.70 & 3.67 (s, 2x1, 3H, OCH3); MS (FAB) m/z 491 (MH+). Anal. Calcd. for C24H22N6O6·¼ H2O: C, 58.24; H, 4.58; N, 16.98. Found C, 58.22; H, 4.70; N, 16.72.
Trans-1,2-bis(3-nitro-1-p-methoxybenzylimidazol-2-yl)ethene (1)
To a vigorously stirred mixture of N-chlorosuccinimide (NCS) (0.216 g, 1.618 mmol) in DMF at room temperature was added dropwise a well-stirred (see below) mixture of potassium tert-butoxide (0.272 g, 2.427 mmol) and 2 (0.20 g, 0.809 mmol) in DMF via syringe. Before the addition, the mixture of potassium tert-butoxide and 2 was stirred for 2.0 hrs at room temperature under nitrogen. After the addition, the reaction mixture was refluxed overnight. The TLC (100:1 CHCl3 / MeOH) showed one UV-absorbing, yellow spot. The reaction mixture was evaporated and neutralized to pH 7 with 0.5 N HCl and water. The mixture was extracted with chloroform (4 x 50 mL). The organic layers were com- bined and dried over anhydrous MgSO4. The chloroform extract was mixed with silica gel and the mix- ture was evaporated to dryness. The residue was suspended in a minimum amount of chloroform and the resulting slurry was loaded onto a silica gel column and the column was eluted with a mixture of 100:1 (CHCl3 / MeOH). Pooling and evaporation of the appropriate eluent fractions under reduced pressure afforded an oil which was triturated with cold ethanol to give the dimer 1 (0.100 g, 39%), m.p. 220-223°C 1H NMR (DMSO-d6) δ 8.06 (s, 1H, 2-H imidazole), 7.41 (s, 1H, C=C), 7.04 (d, J=8.7Hz, 2H, m-Ph), 6.86 (d, J=8.7Hz, 2H, o-Ph), 5.26 (s, 2H, CH2Ar), 3.67 (s, 3H, OCH3); MS (FAB) m/z 491 (MH+). Anal. Calcd. for C24H22N6O6·1½H2O: C, 55.70; H, 4.83; N, 16.24. Found C, 55.77; H, 4.75; N, 15.44.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}