Results and Discussion
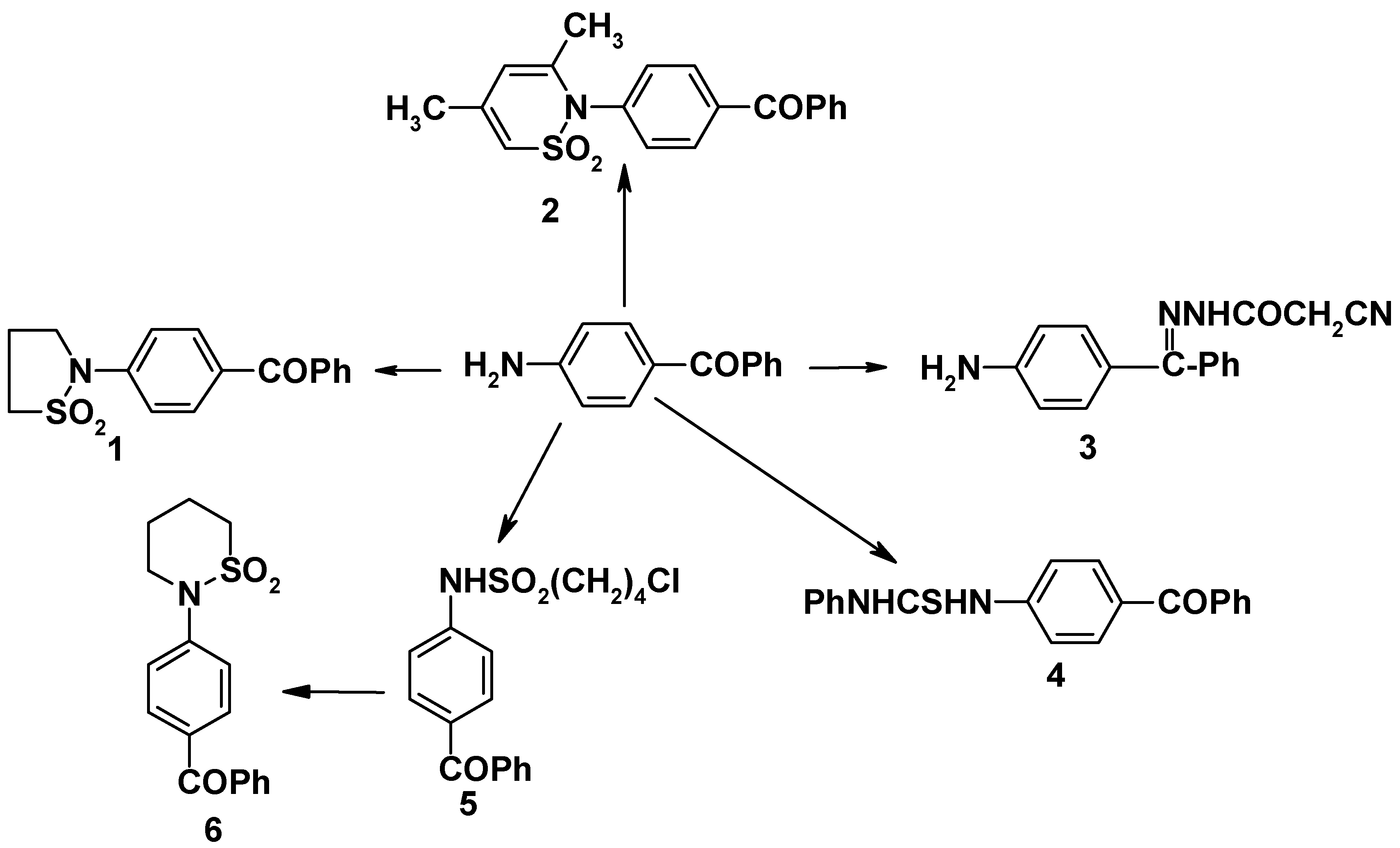
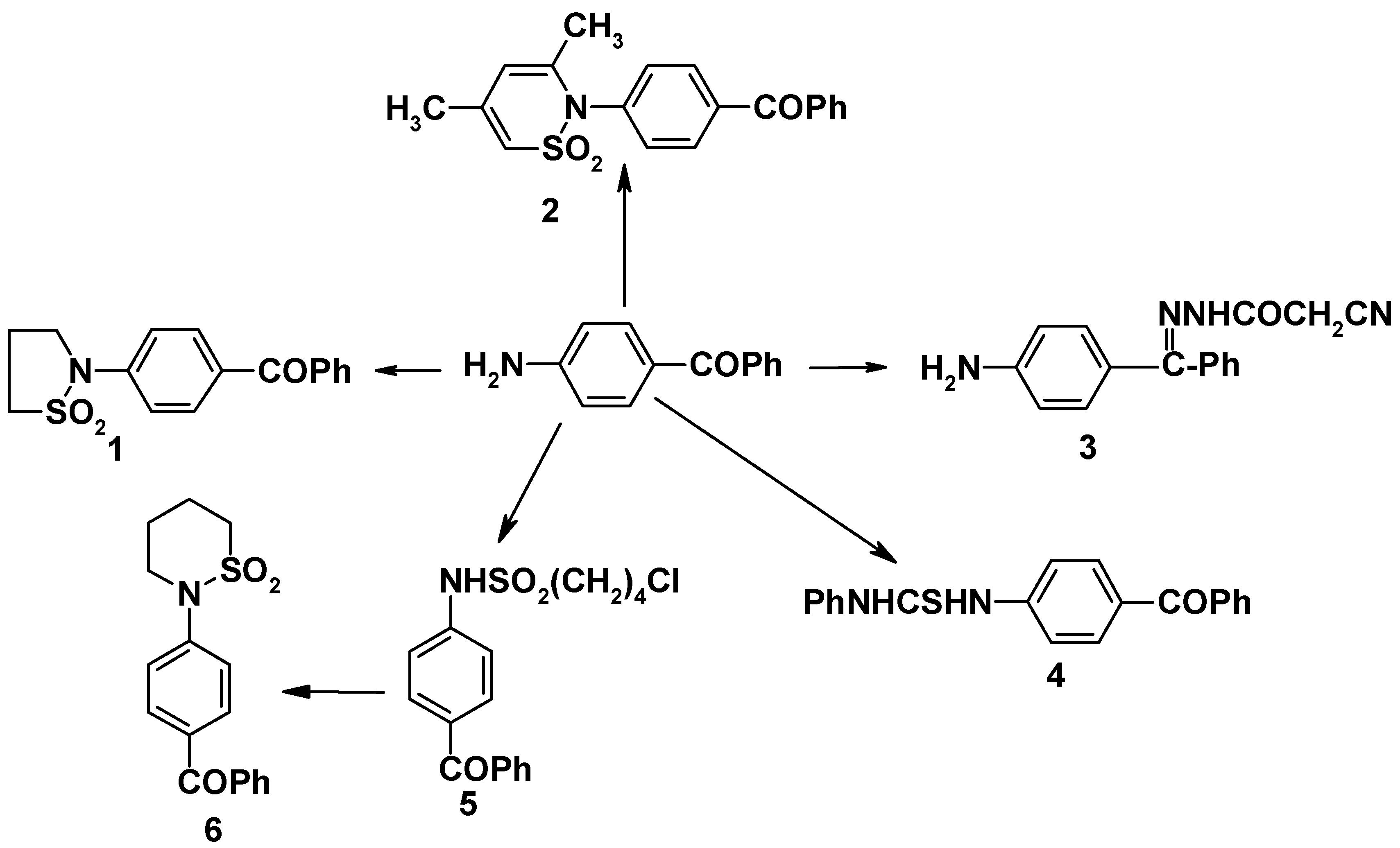
4-Aminobenzophenone [
3] was condensed with oxathiolan-2,2-dioxide, 4,6-dimethyl-1,2-oxathiin-2,2-dioxide, cyanoacetohydrazide and phenyl isothiocyanate to give the corresponding derivatives
1-4, respectively. 4-(tetrahydro-2H-1,2-thiazin-1,1-dioxide-2-yl) benzophenone (
6) was also prepared (
cf.
Scheme 1). The UV spectra of the sultamyl derivatives exhibited maximum absorption band at λ
max 235-254 nm (EtOH) assigned to the vibrational structure of the sultamyl group (n-π
*) transition [
4]. Additional bands at λ
max 305-335 nm (EtOH) were present, also characteristic of the sultamyl group, but which were absent from the spectrum of the thiourea derivative.
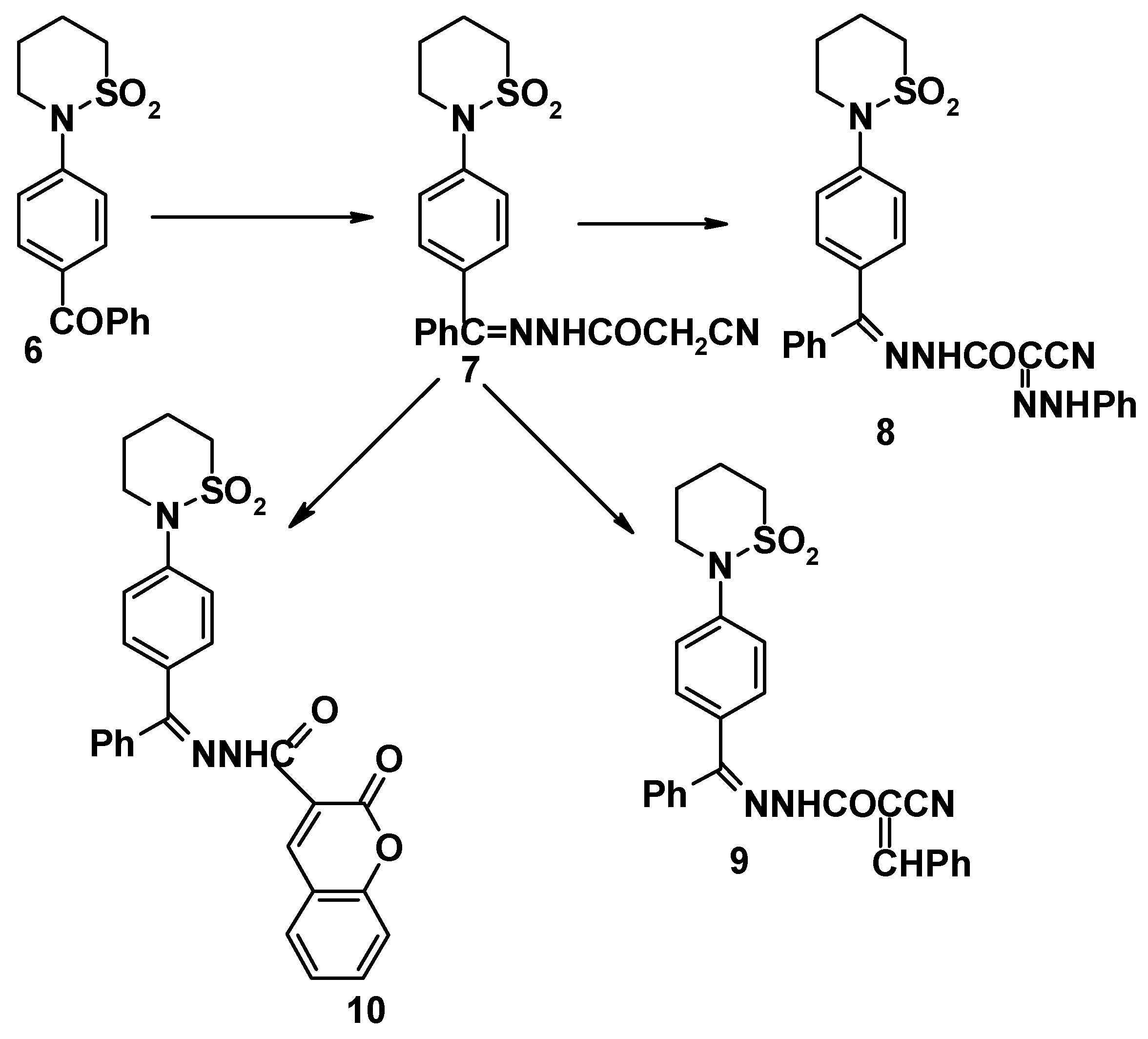
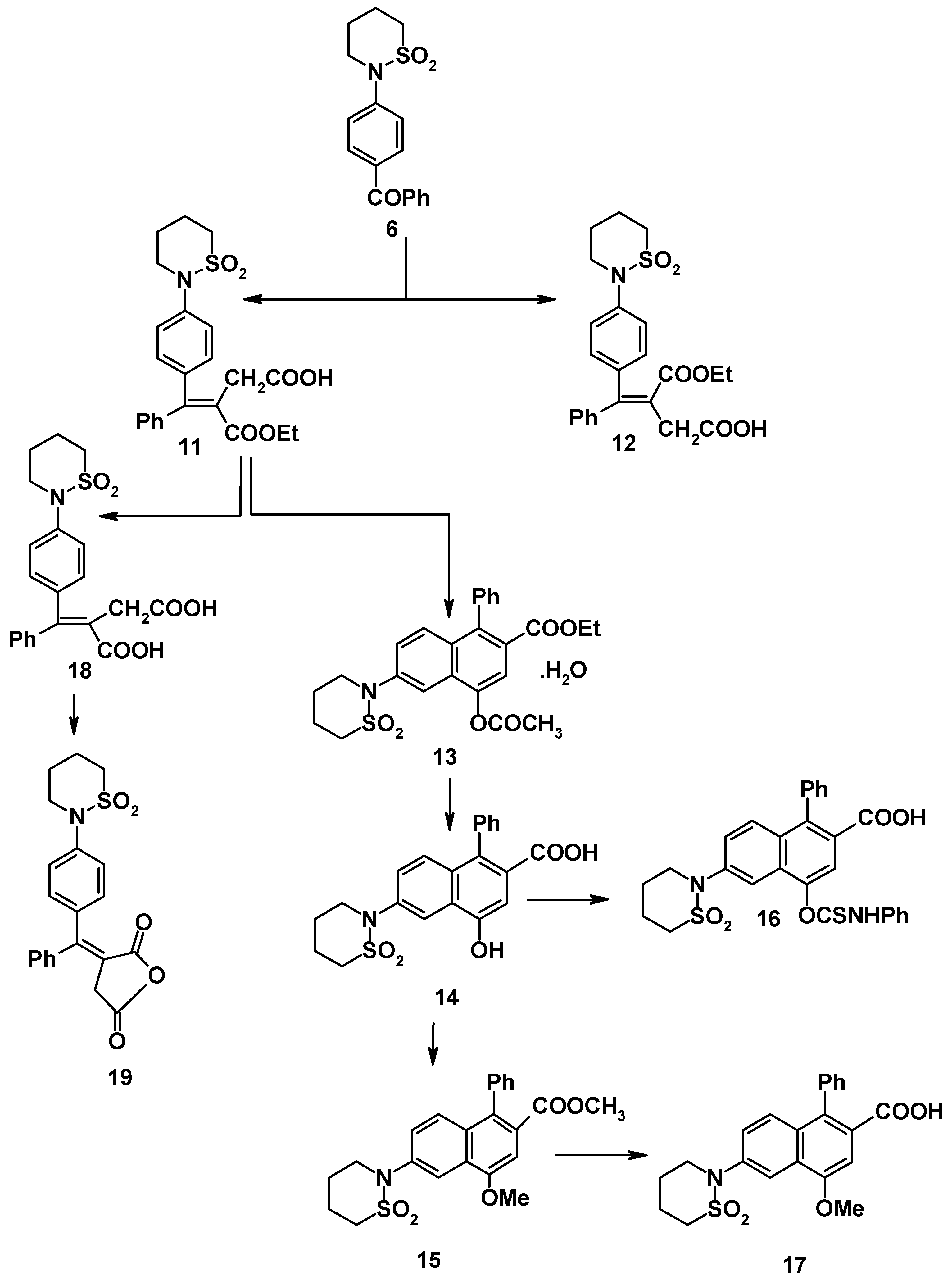
Compound
6 reacted with cyanoacetohydrazide to give 4-(tetrahydro-2H-1,2-thiazine-1,1-dioxide-2-yl)benzophenonecyanoacetohydrazone (
7). The structure of the latter product was elucidated on the basis of its elemental analysis and spectral data (
cf. Experimental). Sultam derivatives
8-
10 were obtained by the reaction of compound
7 with benzenediazonium chloride, benzaldehyde and salicylaldehyde, respectively (
cf.
Scheme 2). Structures
8-
10 were confirmed on the basis of elemental analyses and spectral data (
cf. Experimental).
Ketone
6 was condensed with diethyl succinate to give an oily half-ester (82%). The (
E-) and (
Z-) 3-ethoxycarbonyl-4-phenyl-4-(4-tetrahydro-1,2-thiazine-1,1-dioxide-2-yl)-3-butenoic acids were separated by fractional crystallization; the (
E-) isomer was predominant. The relative ratio was estimated by the application of the spectrophotometric method [
5], the ratio was almost the same as the ratio obtained by fractional crystallization (6:1).
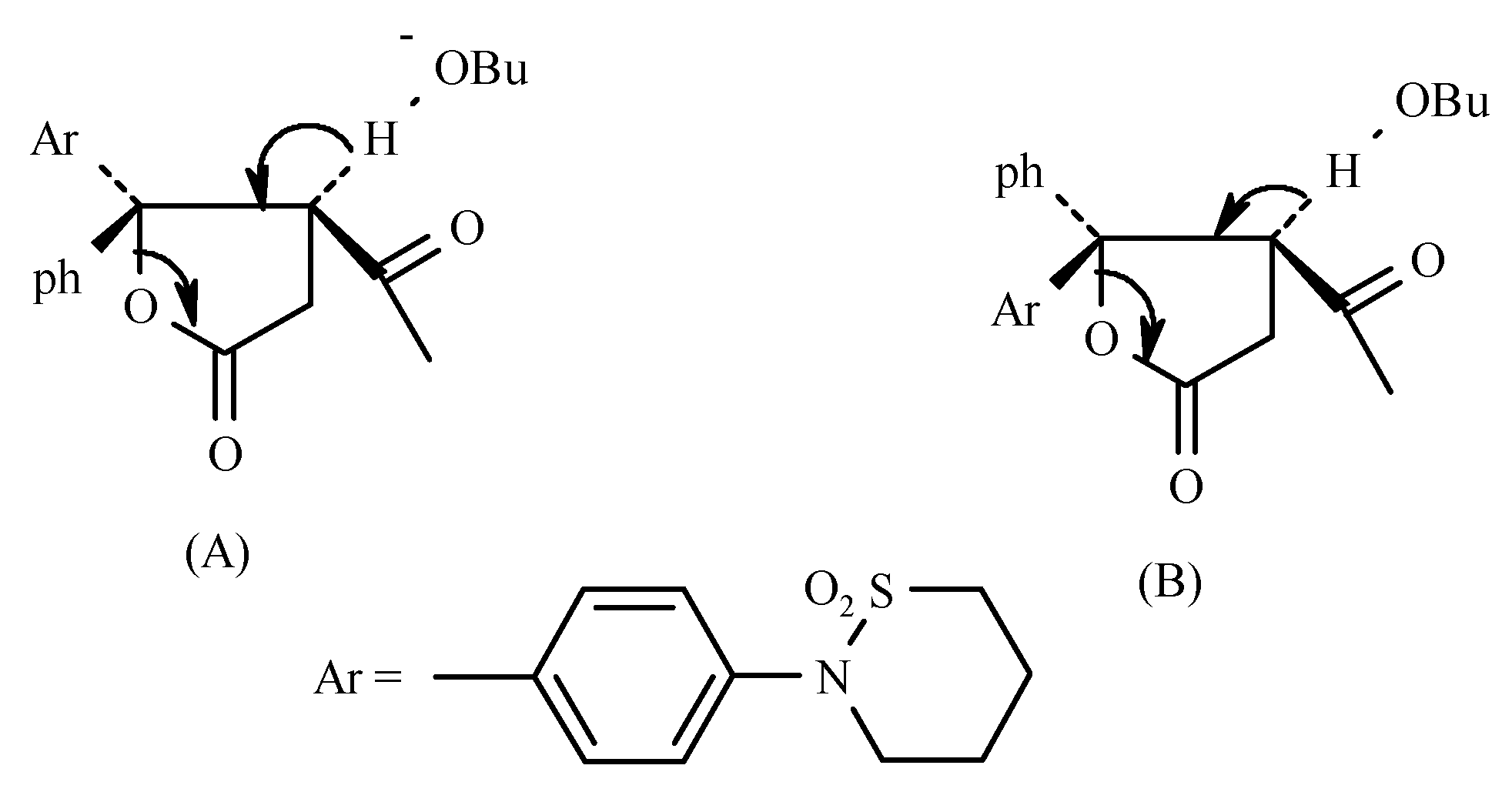
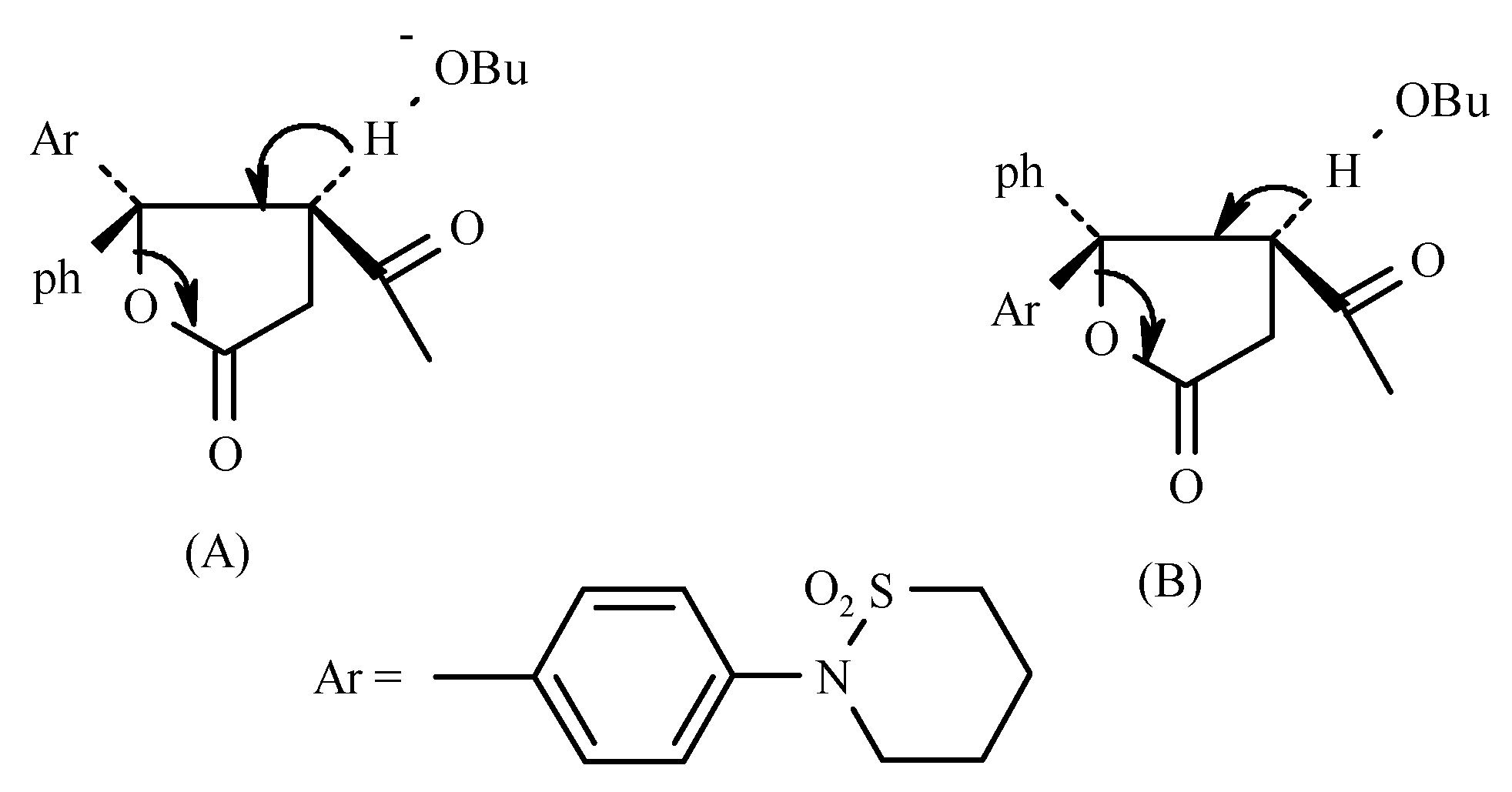
The relative proportion of the two hemiesters obtained is determined by polar non-bonded interactions existing between the groups attached the carbonyl of the ketone and the carbanion. The predominance of the (
E-) isomer is most likely probably due to the polar factor since repulsion between Ph and -COOR is expected to be less significant than that between Ar and –COOR. The polar factor i.e., repulsion between Ph and COOEt and between Ar and COOEt probably plays the more important role (See
Figure 1).
The conjugation of the lone pair of N in the sultam ring will increase electron density in the Ar group. Thus the (E-) Ar/-COOR configuration is expected to be predominant, as found experimentally. Evidence for the (E-) configuration of the hemiester was provided by its cyclization to the corresponding 1-phenylnaphthalene derivative 13 with the sultam ring in position 6. The structure of 13 was proven by:
(a) analytical data;
(b) IR,
1H-NMR and mass spectra (
cf. Experimental and
Table 1);
(c) No fragment of m/e 210 was obtained (
![Molecules 05 00816 i001]()
, C
10H
12O
2NS); which is a reliable proof that the sultam ring is in position 6 of the 1-phenylnaphthalene and not in position 4'. Furthermore, the presence of five and not four adjacent hydrogen atoms [
6] revealed the presence of an unsubstituted phenyl group (
cf. mechanism [
2b]).
Cyclization of the predominant (
E-) hemiester (
11) using sodium acetate and acetic anhydride [
7] gave 4-acetoxy-2-ethoxycarbonyl-1-phenyl-6-(tetrahydro-1,2-thiazin-1,1-dioxide-2-yl)-naphthalene (
13). Compound
13 was hydrolyzed with aqueous sodium hydroxide (8%) to give the phenolic acid
14. The latter acid was either treated with phenyl isothiocyanate in benzene to give
16 or methylated to give methoxy ester
15. The latter was hydrolyzed with sodium hydroxide (2N) to give 2-carboxy-4-methoxy-1-phenyl-6-(tetrahydro-1,2-thiazine-1,1-dioxide-2-yl)naphthalene (
17) (cf.
Scheme 3).
Saponification of the (
E-) hemiester
11 with 2N sodium hydroxide (at 50-60
oC) gave the dibasic acid
18 in 87% yield. Use more of concentrated alkali or alcoholic alkali was not successful. This may be due to the destruction of sultam ring. The dibasic acid was converted by refluxing with acetic anhydride to the corresponding cyclic anhydride which exhibited the expected carbonyl coupling bands (1830 and 1780 cm
-1) [
4,
6].
Biological testing of all reported compounds, in particular for antitumor and antiepileptic activity, is currently under way and results will be published elsewhere.
Experimental
General
All melting points were determined on a Electrothermal melting point apparatus and are uncorrected. IR spectra (KBr disks) were recorded on a Beckman IR 4220 spectrophotometer. 1H-NMR spectra were recorded on a Varian EM 390-90 MHz. spectrometer and chemical shifts are expressed in δ (ppm) units using TMS as internal reference. Electronic absorption spectra were recorded in ethanol solutions a Shimadzu graphic printer Pr-1 spectrophotometer. Mass spectra were recorded using a GCMS-QP 1000 EX Shimadzu spectrometer. The Microanalytical units of Cairo University and of the National Research Center performed elemental analyses.
4-(Trihydro-2H-1,2-thiazole-1,1-dioxide-2-yl)benzophenone (1)
4-Aminobenzophenone (1.97g, 0.01 mol) was fused with oxathiolan-2,2-dioxide (1.2 g, 0.01 mol) at 90-100
oC in oil bath for one hour. The reaction mixture changed to yellow, then brownish oil, which solidified. This solid was washed with hydrochloric acid and then with water. Yield 69%. Compound
1 had mp. 230-32
oC (methanol), Found for C
16H
15NO
3S (301.36): C, 63.51; H, 4.80; N, 4.80 and S, 10.10. Calcd.: C, 63.77; H, 5.01; N, 4.65; S, 10.63.
1H NMR: 1.7 (m, 2H, CH
2CH
2SO
2), 2.6 (t, 2H, NCH
2), 3.2 (t, 2H, CH
2SO
2) and 7.4-7.7 (m, 9H, ArH’s)[
8]. UV: λ
max 218 (log ε = 4.47), 285 (log ε = 4.32) and 332 (log ε = 4.90) nm ; IR (cm
-1) 1648 (CO), 1157 (SO
2), 1317 (SO
2 asym.), 1283 (sultam band) and 738-705 (5 phenylic adjacent H atoms).
4-(3,5-Dimethyl-1',2'-thiazine-1',1'-dioxide-2-yl)benzophenone (2)
4-Aminobenzophenone (1.97 g, 0.01 mol) was fused with 4,6-dimethyl-1,2-oxathiin-2,2-dioxide (1.6g, 0.01 mol) at 110oC for one hour in an oil bath. The solid formed was washed with dil. hydrochloric acid and then with water. Yield: 35%. Compound 2 had mp. 185-86oC (methanol), Found for C19H17NO3S (339.41): C, 66.80; H, 5.00; N, 3.90; S, 9.50. Calcd.: C, 67.23; H, 5.40; N, 4.12; S, 9.44. UV λmax 245 (log ε = 4.00) and 334 (log ε = 4.03); IR (cm-1) 1658 (CO), 1581 (conjugated double bond of the sultam ring), 1280 (sultam ring).
4-Aminobenzophenone cyanoacetohydrazone (3)
To a mixture of 4-aminobenzophenone (1.97g, 0.01 mol) and cyanoacetohydrazide [
8] (0.99g, 0.01 mol) in ethanol (50 mL), 3 drops of hydrochloric acid are added. The mixture was left at room temperature for 72 hrs. Yield: 99%. Compound
3 had mp., 145-46
oC. Found for C
16H
14N
4O (278.31): C, 69.10; H, 5.10; N, 20.04. Calcd.: C, 69.05, H, 5.07; N, 20.13.
1H NMR: 3.8 (s, 2H, NH
2), 4.2 (d, 2H, CH
2CN), 6.5-7.6 (m, 9H, ArH’s) and 9.6 (s, 1H, NH). IR (cm
-1), 3460, 3375 (NH
2), 2260 (CN), 1687 (CONH), 1140 (SO
2), 1322 (SO
2 asym).
4-(Phenyl thioureido)benzophenone (4)
A mixture of 4-aminobenzophenone (1.97g, 0.01 mol), phenyl isothiocyanate (1.35g, 0.01 mol) and triethylamine (3 drops) in benzene (50 mL) was refluxed for 5 hrs. The solvent was evaporated under vacuum to give an oily residue. It was triturated with light petroleum (60-80oC) to afford a solid. Yield: 45%. Compound 4 had mp. 156-57oC (benzene-acetone). Found for C20H16N2OS (332.426): C, 72.50; H, 4.50; N, 8.00; S, 9.20. Calcd.: 72.26; H, 4.85; N, 8.42; S, 9.64. (UV λmax 217, 274 nm (log ε= 4.16 and 4.06, respectively). IR (cm-1), 3322-3029 (NH), 1690 (CO), 1253-1023 (CS).
4-(Tetrahydro-2H-1,2-thiazine-1,1-dioxide-2-yl)benzophenone (6)
4-chloro-1-butanesulfonyl chloride (0.01 mol) was added dropwise at room temperature over 0.5 hr to a stirred solution of 4-aminobenzophenone (1.97g, 0.01 mol) in benzene (20 mL) and pyridine (3 mL) to give 4-benzoyl-4-chlorobutanesulfonanilide (5). Compound 5 had mp., 105-106oC (benzene-petroleum ether). Found for C17H18ClNO3 (351.85): C, 58.30; H, 5.20; N, 3.92; S, 9.00. Calcd.: C, 58.03; H, 5.12; N; 3.98; S, 9.11. Its 2,4-DNP derivative, mp. 233-34oC (acetic acid, 60%). Found for C23H22ClN5O6S (531.98): C, 52.00; H, 4.10; N, 13.20; S, 6.00. Calcd.: C, 51.93; H, 4.16; N, 13.16; S, 6.03.
A solution of
5 was warmed at 40-50
oC for 3 hrs with sodium hydroxide (2N) to give
6 [
4]. Yield: 63%, mp. 136-37
oC (benzene). Found for C
17H
17NO
3S (315.39): C, 64.70; H, 5.30; N, 4.40; S, 9.90. Calcd., C, 64.74; H, 5.43; N, 4.44; S, 10.15.
1H NMR: 1.8 (m, 2H,
CH2CH
2SO
2), 2.3 (m, 2H, SO
2CH
2CH
2CH2), 3.2 (t, 2H, SO
2CH2), 3.8 (t, 2H, NCH
2) and 7.2-7.7 (m, 9H, ArH’s. IR (cm
-1), 1690 (CO), 1325 (SO
2N), 1280 (sultam) and 770-700 (five adjacent hydrogen atoms). UV λ
max 235, 305nm (log ε = 4.68 and 4.89, respectively). Its DNP had mp. 275-76
oC (acetic acid). Found for C
23H
21N
5O
6S (495.52); C, 56.10; H, 4.40; N, 13.60; S, 6.20. Calcd.: C, 55.75; H, 4.26; N, 14.19; S, 6.67.
4-(Tetrahydro-2H-1,2-thiazine-1,1-dioxide-2-yl)benzophenonecyanoacetohydrazone (7)
Prepared from 6 in a similar fashion as above, yield 75%. Mp. 122-23oC (ethanol). Found for C20H20N4O3S (396.47): C, 60.50; H, 5.10; N, 14.00; S, 8.20. Calcd.: C, 60.59; H, 5.08; N, 14.13, S, 8.08. 1H NMR: 1.8 (m, 2H, SO2CH2CH2), 2.3 (m, 2H, SO2CH2CH2CH2), 3.2 (t, SO2CH2), 3.8 (t, NCH2), 7.4-7.7 (m, 9H, ArH’s), 9.3 (s, 1H, NHCO).
(a) The hydrazone derivative 7 (0.01 mol) was coupled with benzenediazonium chloride (0.01 mol) to give 8, yield at room temperature 64%, mp. 130-31oC (acetic acid). Found for C26H24N6O3S (500.58): C, 61.40; H, 4.80; N, 16.80; S, 6.40. Calcd.: C, 61.38; H, 4.83; N, 16.78; S, 6.40. IR (cm-1), 2152 (CN), 1648 (CONH) and 1280 (sultam).
(b) To hydrazone derivative 7 (0.01 mol), benzaldehyde (0.01 mol) in ethanol (20 mL) and 2 drops of piperidine were added. The mixture was refluxed for 30 minutes on a water bath. After cooling, the solid was collected and crystallized to give 9 (73.5%), mp. 230-31oC (ethanol). Found for C26H24N4O3S (472.57): C, 66.30; H, 5.10; N, 11.70; S, 6.40. Calcd.: C, 66.08; H, 5.12; N, 11.85; S, 6.76. IR (cm-1), 2152 (CN), 1648 (CONH) and 1280 (sultam).
(c) To hydrazone derivative 7 (0.01 mol), salicylaldehyde (0.01 mol) in ethanolic sodium ethoxide solution [prepared from sodium metal (1.1 g-atom) in ethanol (20 mL)] was added. The mixture was refluxed for 2 hrs., then cooled. The reaction mixture was acidified with hydrochloric acid to give 10, yield 55%, mp. 243-44oC (ethanol). Found for C27H23N3O5S (501.56): C, 64.50; H, 4.60; N, 8.80, S, 6.70. Calcd.: C, 64.65; H, 4.62; N, 8.38; S, 6.39. IR (cm-1) 3446 (NH), 1700, 1648 (two CO) and 1282 (sultam ).
3-Ethoxycarbonyl-4-phenyl-4-(4'-tetrahydro-1,2-thiazine-1,1-dioxide-2-yl)phenyl-3-butenoic acids (11 and 12)
To a cooled stirred mixture of ketone 6 (0.01 mol) and diethyl succinate (0.02 mol) in t-butanol (15 mL), potassium t-butoxide was added dropwise [prepared from 1.2 g-atom potassium and t-butanol (50 mL)]. After 72 hrs, the reaction mixture was heated at 65-70oC for one hour, then worked up as usual [2b] to give an acidic brown viscous oily product (yield ~ 7.3g, 82%). The (E-) (11) and (Z-) (12) isomeric hemiesters were separated by fractional crystallization.
Compound 11 had mp. 144-45oC (benzene-acetone). Found for C23H25NO6S (443.52): C, 62.00; H, 5.50; N, 3.30; S, 7.60. Calcd.: C, 62.28; H, 5.64; N, 3.16; S, 7.23. IR (cm-1), 3300-2600 (γ OH), 1700-1620 (CO), 1270 (sultam).
Compound 12 had m.p. 108-109oC (benzene). Found for C23H25NO6S (443.52): C, 62.40; H, 5.50; N, 3.30; S, 7.50. Calcd.: C, 62.28; H, 5.64, N, 3.16; S, 7.23. IR (cm-1), 3600-2500 (OH), 1700-1640 (CO), 1280 (sultam).
Determination of the relative ratio of the two (E-) and (Z-) hemiesters by electronic absorption spectroscopy [5]
Values for Eobs/E1 were plotted vs. the values for E2s/E1 where Eobs is the observed optical density of the mixture and E1 and E2 are the optical densities of the hemiesters 11 and 12 respectively, at the same wavelength. A straight line was obtained from which the ratio of 5.9/1 was calculated. This is almost the same ratio as that obtained experimentally by fractional crystallization.
Cyclization of the (E-) hemiester
A mixture of the (E-) hemiester (1 mol) fused sodium acetate (1.2 mol) and acetic anhydride (30 mL/ 1g sodium acetate) was left overnight with occasional shaking at room temperature. The temperature was gradually raised to 70-80oC for 3hrs. The neutral cyclized product 13 was isolated and had mp. 122-23oC (ethanol). Found for C25H25NO6S.H2O (485.56): C, 62.00; H, 5.40; N, 3.00; S, 6.80. Calcd.: C, 61.84; H, 5.19; N, 2.88; S, 6.61. Mass spectrum, m/e = 485 and 1H NMR: 1.2 (t, 3H, CH3CH2O), 1.8 (m, 2H, SO2CH2CH2), 2.3 (m, 2H, CH2(CH2)2SO2, 3.2 (t, 2H, SO2CH2), 3.5 (t, 2H, NCH2), and 7.0-7.8 (m, 9H, ArH’s)9. IR (cm-1), 1760 (CO), 1290 (sultam ring), 740-700 (five adjacent hydrogen atoms).
Conversion of the acetoxy ester 13 into methoxy acid 17
The acetoxy ester was hydrolysed with 2N sodium hydroxide on water-bath (70 - 80oC) for two hrs. The hydroxy acid 14 (1 mol) was methylated by refluxing for 10 hrs with dimethyl sulphate (5 mol) and potassium carbonate (6 mol) in dry acetone. The methoxy ester was hydrolyzed to the corresponding acid by warming for 3 hrs with 8% sodium hydroxide, followed by cooling and acidification.
Compound 14 had mp. 225-26oC (acetic acid). Found for C21H19NO5S (397.45): C, 63.20; H, 5.10; N, 3.40; S, 7.70. Calcd.: C, 63.47; H, 4.81; N, 3.52; S, 8.06.
Compound 15 had mp. 167-68o (methanol). Found for C23H23NO5S (425.50): C, 64.70; H, 5.00; N, 3.60, S, 7.90. Calcd.: C, 64.92; H, 5.44; N, 3.29; S, 7.52.
Compound 17 had mp., 120-21oC (acetic acid). Found for C22H21NO5S (411.48): C, 63.90; H, 5.30; N, 3.40; S, 7.60. Calcd.: C, 64.23; H, 5.14; N, 3.40; S, 7.78. IR (cm-1), 1725 (CO), 1295 (sultam) and 740- 700 (five adjacent hydrogen atoms).
2-Carboxy-4(N-phenylthiocarbamoyl)-1-phenyl-6-(tetrahydro-2H-1,2-thiazine-1,1-dioxide-2-yl)-naphthalene (16)
A mixture of hydroxynaphthoic acid derivative 14 (0.01 mol), phenyl isothiocyanate (0.01 mol) and 3 drops of triethylamine in dry benzene (30 mL) was refluxed for 5 hrs. The benzene was evaporated under reduced pressure to give yellow solid 16, yield 71%. It had mp. 133-34oC (benzene-acetone). Found for C28H24N2O5S2 (532.64): 62.90; H, 4.90; N, 5.40; S, 11.90. Calcd.: C, 63.14; H, 4.54; N, 5.26; S, 12.01. IR (cm-1), 3247-2800 (OH), 1725(CO), 1290 (sultam) and 744-701 (five adjacent hydrogen atoms).
Saponification of the (E-) hemiester to the corresponding dibasic acid 18 and anhydride 19
The (E-) hemiester (0.1 mol) was left in NaOH (2N, 50 mL) for about 72 hrs., then kept 60-80oC for one hour. The dibasic acid 18 was refluxed with a mixture of acetic anhydride (7 mL) and acetyl chloride (5 mL) on a sand bath for 30 minutes, to give the corresponding anhydride (19).
Compound 18 had mp., 175-76oC (benzene-acetone). Found for C21H21NO6S (415.47): C, 60.20; H, 5.10; N, 3.90; S, 7.20. Calcd.: 60.72; H, 5.08; N, 3.37; S, 7.85. 1H NMR: 1.7 (m, 2H, CH2CH2SO2), 2.3 (m, 2H, CH2(CH2)2SO2), 3.2 (t, 2H, SO2CH2), 3.5 (t, 2H, NCH2), 7.1-7.4 (m, 9H, ArH’s) and 12.5 (s, 2H, (COOH)2. IR (cm-1), 3446-2500 (γ OH), 1720-1656 (CO ), 1276 (sultam), 745-707 (five adjacent hydrogen atoms).
Compound 19 had mp. 137-38oC (benzene-peteroleum ether). Found for C21H19NO5S (397.45): C, 63.20; H, 5.00; N, 3.90; S, 8.30. Calcd.: C, 63.46; H, 4.81; N, 3.52; S, 8.07. IR (cm-1), 1840,1780 (CO-O-CO), 1280 (sultam ring) and 750-700 (five adjacent hydrogen atoms).
Table 1.
Mass spectra of compounds 1, 3, 6, 11, 12 and 13.
Table 1.
Mass spectra of compounds 1, 3, 6, 11, 12 and 13.
| 1 | 301 (20, C16H15NSO3+); 224 (12, C10H10NSO3+); 197 (100, C9H10NSO2+1); 180 (5, C13H9O+1); 120 (8, C3H6NSO2+); 77 (16, C6H5+). |
| 3 | 278 (100, C16H14ON4+); 238(21.69, C14H12ON3+); 210(37.08, C13H12N3+), 180 (29.44, C13H10N+); 119 (1.87, C7H5ON+); 77 (16.64, C6H5+). |
| 6 | 315 (100, C17H17NSO3+; 238 (47.13, C11H12NSO3+); 210(14.93 C10H12NSO2+); 132 (19, C9H10N+); 105 (19,C7H5O+); 77(16, C6H5+). |
| 11 | 443 (12.1, C23H25NSO6+); 339 (33.6, C22H25NSO4+1); 379 (1.8, C23H25NO4+); 365 (14.4, C22H23NO4+); 325 (100, C19H18NO4+1); 308 (7.9, C19H16O4+); 144 (1.6, C6H8O4+); 134 (0.7, C4H8NSO2+); 77 (1.5, C6H5+). |
| 12 | 443 (12.80, C23H25NSO6+); 325 (100, C19H18O4N+1); 308 (13.4, C19H16O4+); 139 (5.2, C6H2O4+1); 134 (3.4, C4H8NSO2+); 77(7.9, C6H5+). |
| 13 | 485 (100, C25H25NSO6·H2O+); 467 (5, C25H25NSO6+); 404 (5, C25H25NO4+1); 361 (5, C23H22NO+1). |

 , C10H12O2NS); which is a reliable proof that the sultam ring is in position 6 of the 1-phenylnaphthalene and not in position 4'. Furthermore, the presence of five and not four adjacent hydrogen atoms [6] revealed the presence of an unsubstituted phenyl group (cf. mechanism [2b]).
, C10H12O2NS); which is a reliable proof that the sultam ring is in position 6 of the 1-phenylnaphthalene and not in position 4'. Furthermore, the presence of five and not four adjacent hydrogen atoms [6] revealed the presence of an unsubstituted phenyl group (cf. mechanism [2b]).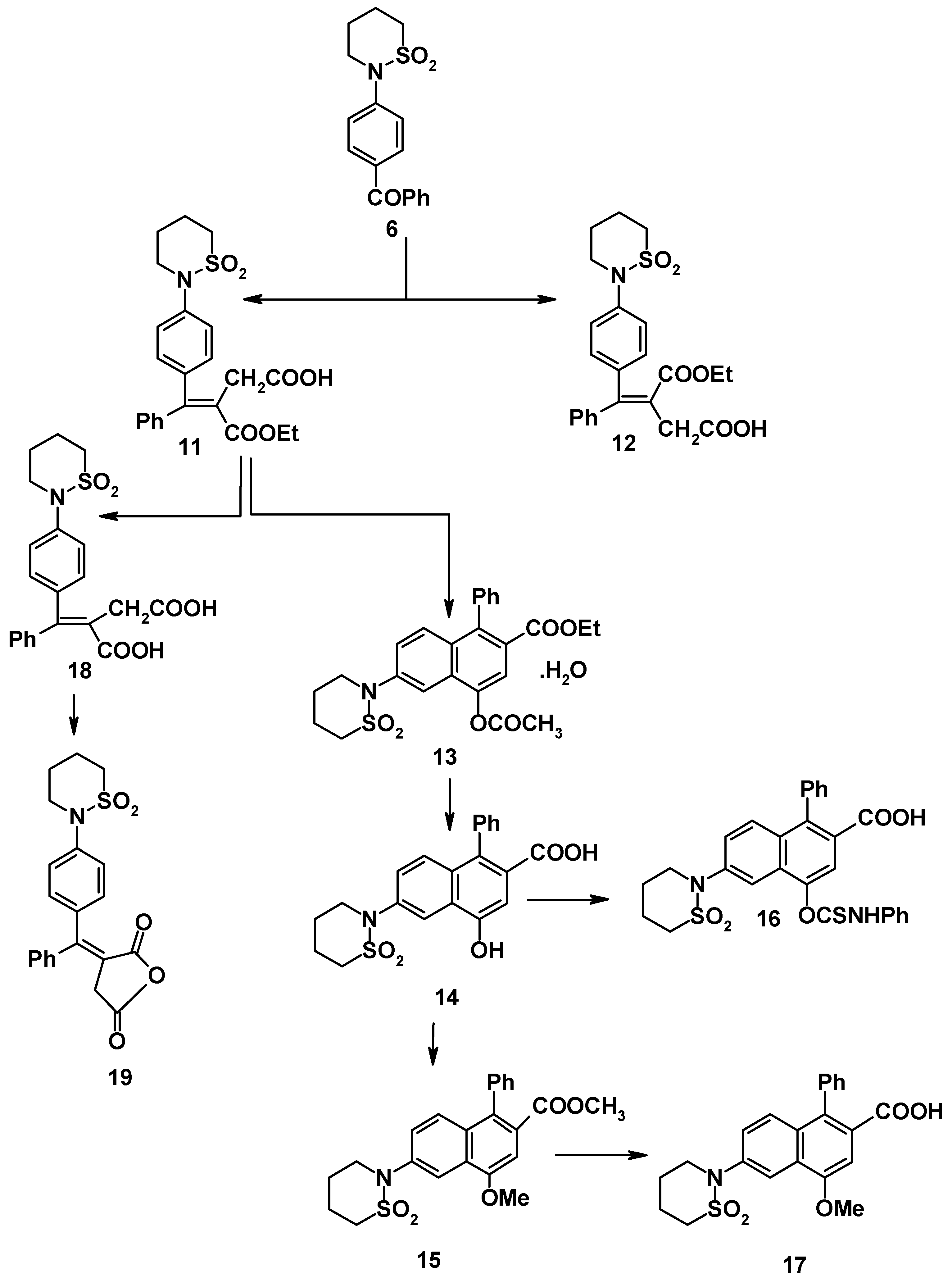

{kind=link}
{kind=link}
{kind=link}
{kind=link}