Introduction
N-(2-Cyanophenyl)chloromethanimidoyl chloride can be used as a suitable starting compound for the synthesis of fused heterocyclic compounds and their precursors. Dyson and Harrington [
1] have shown that the reaction of phenylisothiocyanate with chlorine in an inert solvent provides an unstable chloro-addition intermediate that is converted into the N-phenylchloromethanimidoyl chloride on further action of chlorine. Another method for the synthesis of chloromethanimidoyl chlorides is chlorination of isocyanides. Nef [
2] described this addition reaction for the first time in 1892. Bly, Perkins and Lewis [
3] have observed that formanilide chlorinated in the presence of thionyl chloride gives a series of products identified as 2,4-dichloroformanilide, N-phenylchloromethanimidoyl chloride, N-(4-chlorophenyl)chloromethanimidoyl chloride and N-(2,4-dichlorophenyl)chloromethanimidoyl chloride, respectively. Yet another method of chloromethanimidoyl chloride preparation is the reaction of isocyanates with phosphorus pentachloride [
4]. Good yields of chloromethanimidoyl chloride are reported for the chlorination of aliphatic isocyanates [
5].
Results and Discussion
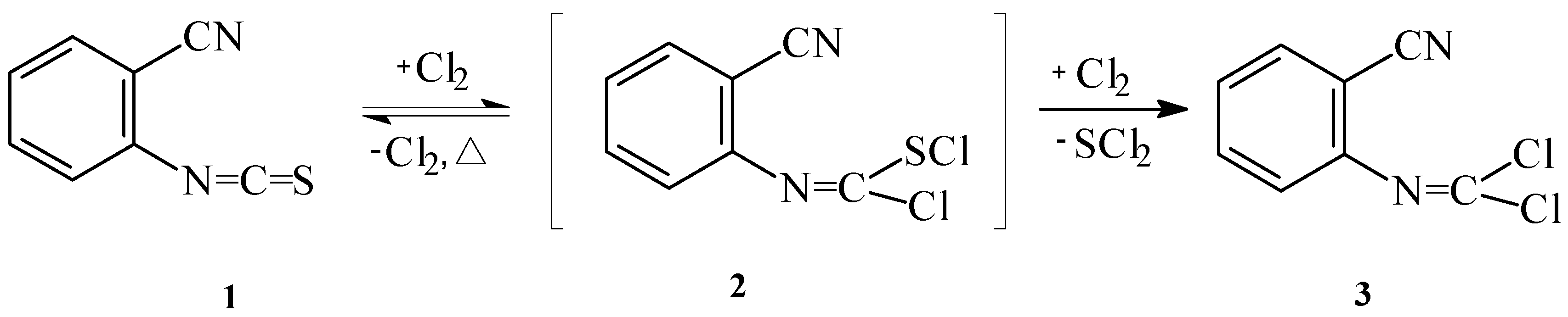
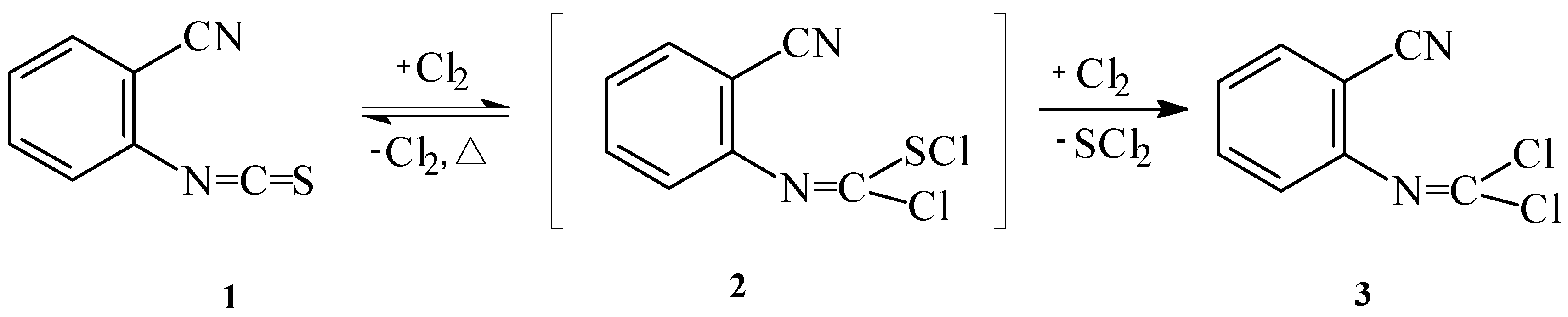
We have attempted to prepare N-(2-cyanophenyl)chloromethanimidoyl chloride (
3) from 2-cyanophenylisothiocyanate [
6] (
1) and N-(2-cyanophenyl)formamide (
4). First, we performed chlorination of 2-cyanophenylisothiocyanate in chloroform with an excess of sulfuryl chloride. The reaction proceeded for 48h under reflux and the title compound was isolated in 73% yield. When gaseous chlorine was used as chlorinating agent and the reaction was carried out at room temperature, the reaction time was reduced to 3 hours (
Scheme 1). In this case, the yield of chloromethanimidoyl chloride
3 was 75%.
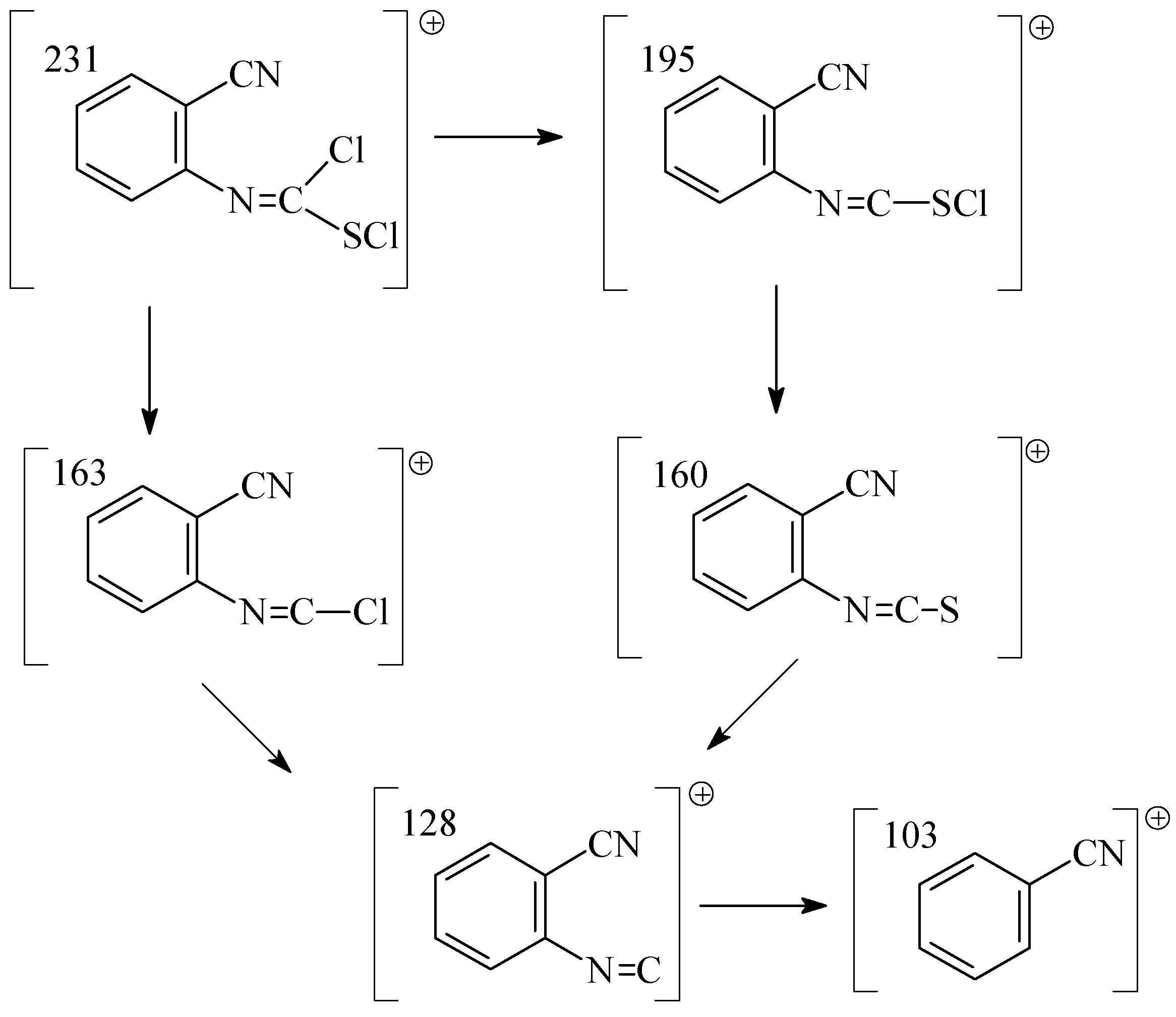
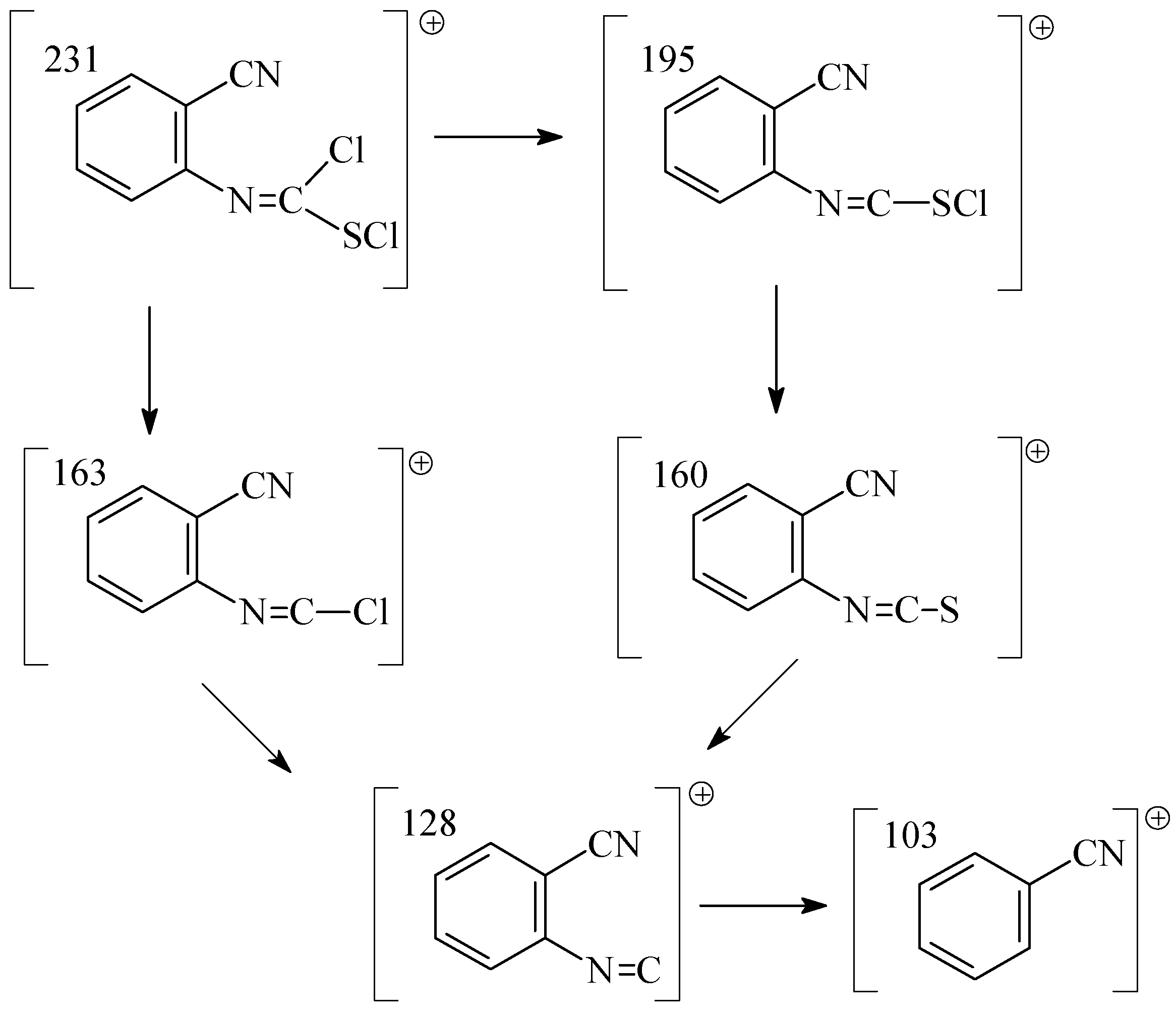
A GC study combined with mass spectroscopy has shown that this reaction proceeded in two steps. In the first step, the addition of chlorine to the C=S bond of the isothiocyanate substrate yields the dichloro adduct,
i. e. N-(2-cyanophenyl)chloromethanimidothioic chloride (
2). We found the following fragments in the mass spectrum of
2: m/z (I
r/%) (C
8H
4N
2SCl
2, M
r= 231,09): 232 (8), 231 (12), 163 (43), 160 (100), 103 (52) (
Scheme 2).
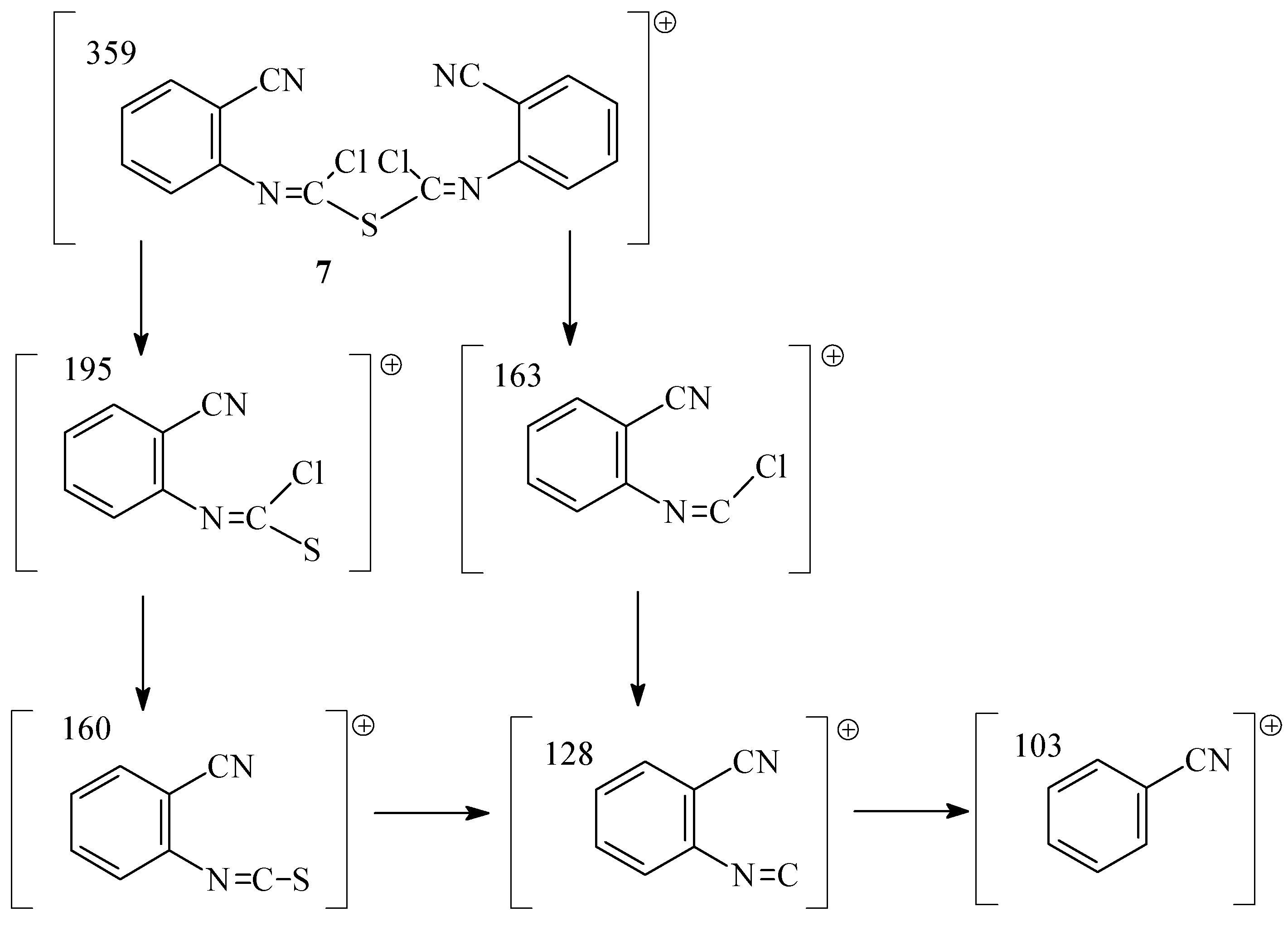
The first step of the reaction was found to be reversible, in contrast to analogous reaction described in literature [
5]. These findings may be explained by the instability of the S-Cl bond due to the presence of cyano group in the molecules. Furthermore, the activation energy of the retro–formation of isothiocyanate
1 is probably lower than that of
3. The product
3 is formed by the reaction of chlorine with dichloro adducts
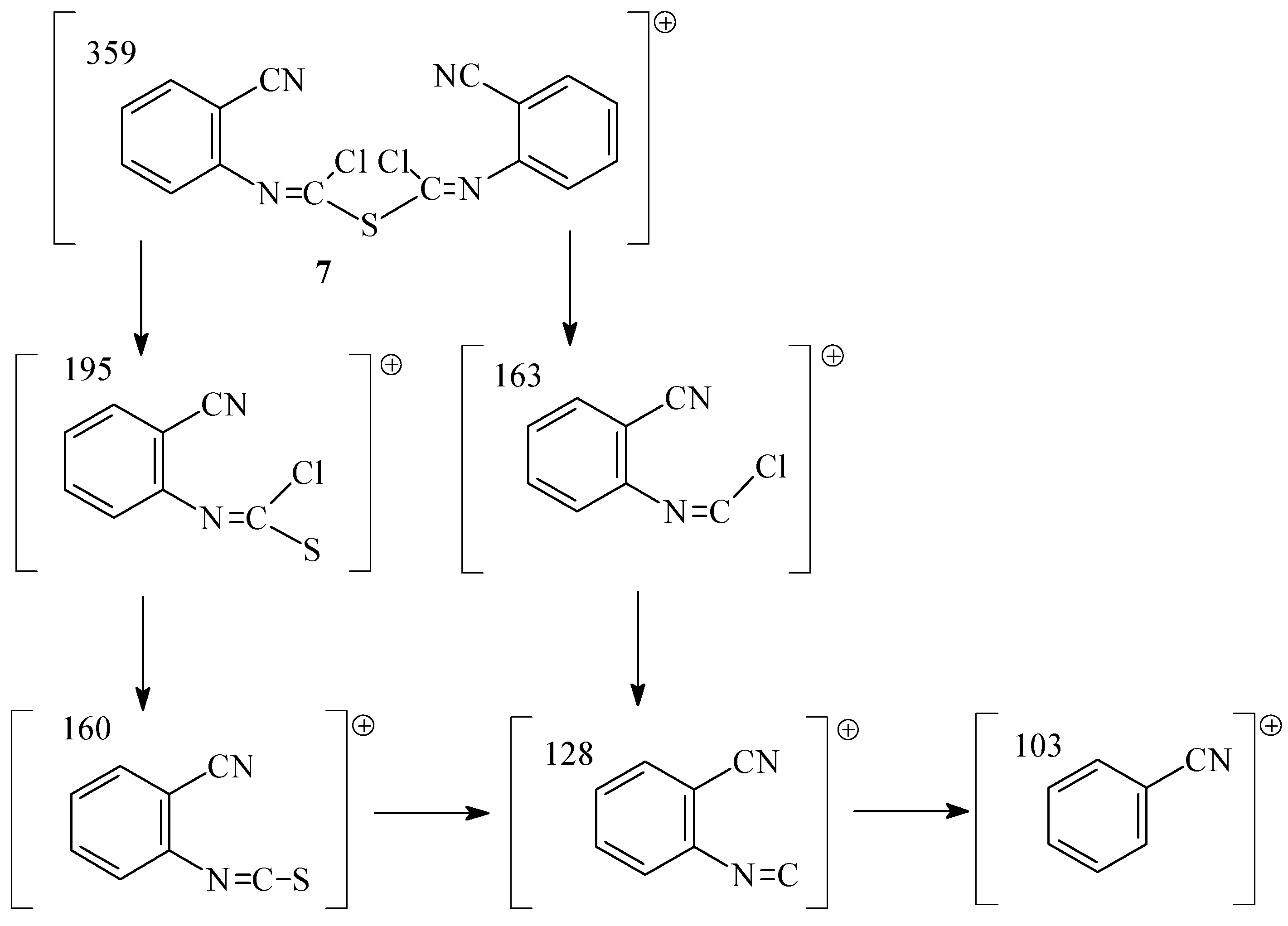
2 with simultaneous elimination of SCl
2 in the following step. The GC of samples from the reaction mixture showed the presence of a by product which was identified as bis[N-(cyanophenyl)chloromethanimidoyl] sulfide (
7) by MS (fragments m/z (I
r/%) (C
16H
8N
4SCl
2, M
r=359,14): 359 (4), 195 (4), 163 (33), 160 (100), 128 (15), 103 (78)). (
Scheme 3).
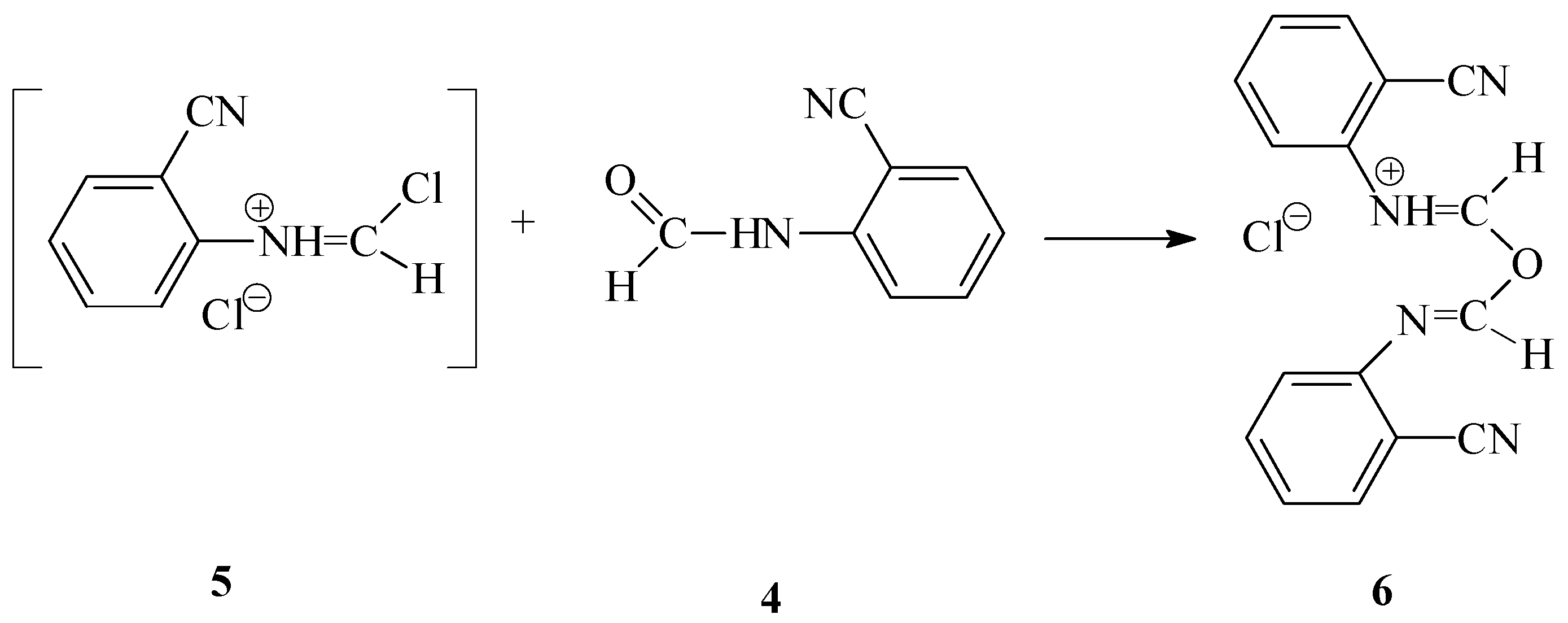
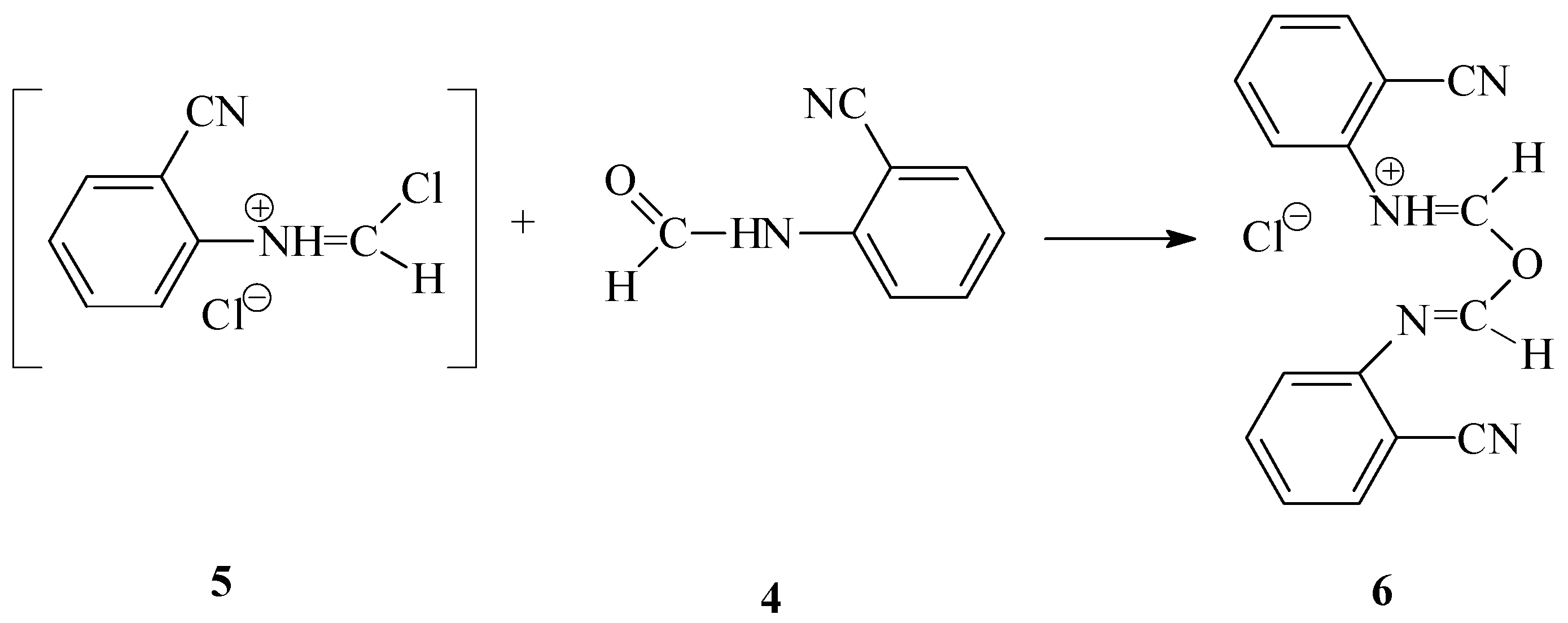
The preparation of N-(2-cyanophenyl)chloromethanimidoyl chloride (
3) from N-(2-cyanophenyl)formamide (
4), thionyl chloride and sulfuryl chloride [
7]
via a three-component
one-pot reaction appeared to be difficult. A solid product formed during the reaction that did not dissolve in the reaction medium and could not be reacted with sulfuryl chloride, even by extending the reaction time. We identified this solid product as (2-cyanophenyl)(1-{[(2-cyanophenyl)imino]methoxy}methylidene)ammonium chloride (
6). We assume that compound
6 was generated as the product of the competitive reaction by the interaction of oxygen atom of N-(2-cyanophenyl)formamide
4 with the carbon atom of N-(2-cyanophenyl)chloroformamidinium chloride (
5) (formed
in situ from the formamide by the action of thionyl chloride [
7]) (
Scheme 4).
1H and
13C NMR experiments supported our findings. We observed a proton signal associated with the C=N group (chemical shift δ 7.82 ppm).
When we carried out the reaction of N-formanilide 4 with thionyl chloride at a temperature of 0–5°C, we prevented from side reaction. Chlorination by sulfuryl chloride proceeded under reflux under elimination of hydrogen chloride and sulfur(IV)oxide. We isolated N-(2-cyanophenyl)chloromethanimidoyl chloride (3) as a yellow liquid, which afforded the product as yellow crystals when crystallized from cyclohexane. FTIR data 1650 cm-1 (C=N), 890 cm-1 (C-Cl), 2220 cm-1 (C≡ N) also confirmed these facts.
Experimental
General
Melting points of prepared compounds were measured on a Boetius Rapido PHMK 79/2106 (Wägetechnik) instrument. TLC was carried out on Silufol UV 254 plates (Kavalier, Votice) and the detection with Fluotes Universal (Quarzlampen, Hanau) and with iodine vapors. Chloroform and diethylether were used as eluents. FTIR spectra were taken on a Genesis (UNICAM) spectrometer in potassium bromide pellets. NMR spectra were measured on a Bruker Avance DRX-500 spectrometer in deuterochloroform or deuterotrifluoroacetic acid. The 1H and 13C spectra were referenced to tetramethylsilane or to the solvent signals of CDCl3 and of residual CHCl3 at 77.00 ppm (13C) and 7.27 ppm (1H), respectively. The measured 13C and 1H NMR spectra were correlated with those obtained by on-line simulation (Advanced Chemistry Development, Inc., Toronto, Canada). Gas chromatography was accomplished on a Shimadzu GC – 17A apparatus and on a TRIO 1000 GC/MS system (FISONS Instruments). The electron impact method was used for ionization (70eV).
N-(2-Cyanophenyl)chloromethanimidoyl chloride (3)
Procedure A
2-Cyanophenylisothiocyanate (1) (24.00g, 0.15 mol) was dissolved in dry chloroform (50 ml) at room temperature under stirring. Sulfuryl chloride (24 ml, 0.30 mol) was added. The reaction mixture was heated under reflux for 48 hours, excess sulfuryl chloride and solvent were removed on a vacuum evaporator and the crude product was crystallized from dry cyclohexane.
Procedure B
N-(2-Cyanophenyl)formamide (5) (2.90g, 20mmol) was mixed with thionyl chloride (20ml) at 0–5°C. After 20 hours stirring at 0–5°C sulfuryl chloride (2.6g, 20mmol) was added dropwise. The reaction mixture was refluxed for 1 hour then concentrated in vacuo. The crude product was suspended in chloroform. The suspension was filtered through silica and the resulting filtrate evaporated in vacuo to yield a product that was crystallized from dry cyclohexane.
Procedure C
A solution of 2-cyanophenylisothiocyanate (1) (30.0g, 0.19 mol) in dry chloroform (50 ml) was saturated with gaseous chlorine (28.0g, 0.40 mol) with stirring at room temperature for 3 hours. Chloroform and sulfur dichloride were stripped off on a vacuum evaporator and the product was crystallized from dry cyclohexane.
Yield (A) 22.0g (73%); Yield (B) 2.0g (51%); Yield (C) 28.5g (75%). M.p. 62–64°C; For C8H4Cl2N2 (199.04) calculated: 48.28% C, 2.03% H, 14.07% N, 35.62% Cl; found: 48.02% C, 1.92% H, 14.14% N, 35.30% Cl; FTIR, /cm-1: 2200 (CN), 1650 (C=N), 1480 (C=C), 890 (C-Cl); 1H NMR (CDCl3) δ: 7.69 – 7.68 (1H, m, ArH), 7.63 – 7.59 (1H, m, ArH), 7.32 – 7.29 (1H, m, ArH), 7.13 – 7.12 (1H, m, ArH); 13H NMR (CDCl3) δ: 147.58 (Cq), 141.54 (Cq), 133.54 (CHAr), 133.26 (CHAr), 126.02 (CHAr), 121.20 (CHAr), 116.09 (CN), 105.92 (Cq). Mass spectrum, m/z (Ir/%): 200 (16), 198 (25), 165 (37), 163 (100), 102 (25).
(2-Cyanophenyl)(1-[(2-cyanophenyl)imino]methoxymethylidene)ammonium chloride (6)
This by-product was separated by suction from suspension formed in procedure B. Yield 1.5g (25%); M.p. 298–300°C. For C16H11N4OCl (310.74) calculated: 61.84% C, 3.57% H, 18,03% N; found: 61.39% C, 3.21% H, 17.59% N; FTIR, /cm-1: 2212 (CN), 2220 (CN), 1653 (C=N+), 1633 (C=N), 1085 (COC); 1H NMR (CF3COOD) δ: 9.83 (1H, bs, HC=N+), 8.46 – 8.29 (4H, m, ArH), 8.17 – 8.14 (1H, m, ArH), 8.04 – 7.96 (1H, m, ArH), 7.85 – 7.79 (2H, m, 1ArH+1HC=), 7.53 – 7.23 (2H, m, ArH). 13C NMR (CF3COOD) δ: 168.13 (NH+=CH), 162.49 (N=CH), 138.48 (Cq), 136.14 (Cq), 134.20 (CHAr), 134.01(CHAr), 132.91 (CHAr), 132.74 (CHAr), 132.37 (CHAr), 131.19 (Cq), 127.49 (CHAr), 124.24 (CHAr), 120.05 (CHAr), 119.55 (Cq), 119.00 (Cq), 113.49 (Cq).

{kind=link}
{kind=link}
{kind=link}
{kind=link}