
2-MCPD-Induced Effects in the Heart: Toxicological and Mechanistic Implications from Comparative Proteomic Analyses in Rats

 , ,
, ,
Abstract
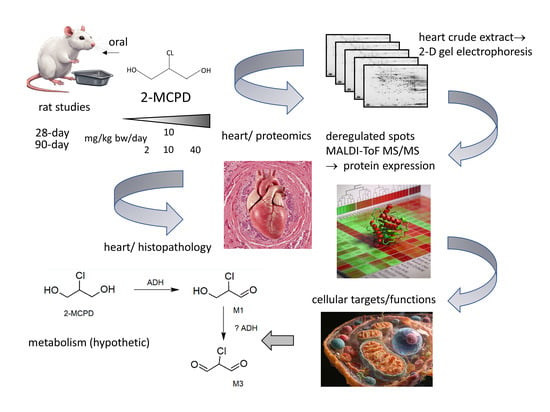
1. Introduction
2. Results
2.1. Animal Studies
2.2. Proteomic Analysis of Differentially Expressed Proteins
2.3. Ingenuity Pathway Analysis (IPA)
2.3.1. Toxicity Functions and Categories
2.3.2. Related Pathways
2.3.3. Network Analysis
3. Discussion
3.1. Considerations on Possible Modes of Action of 2-MCPD Regarding Cardiac Stress Response and Maintenance of Cardiac Structural Integrity and Function
3.2. Mechanistic Implications Regarding 2-MCPD Biotransformation, Mitochondrial Metabolism and Function in the Heart
3.3. Enzymes Utilizing or Producing Acetyl-Coenzyme A Necessary for Pyruvate Metabolism and Fatty Acid Degradation
3.4. Hypothesis of Reactive Intermediates Formed from 2-MCPD in Rat Heart
3.5. Summary
4. Materials and Methods
4.1. Data Sources/Animal Studies
4.2. Proteomic Analysis
4.3. Comparative Analyses of the 28- and 90-Day Studies
4.3.1. Evaluation and Characterization of Deregulated Protein Spots
4.3.2. Gel Spot Identification
4.3.3. Data Interpretation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamikata, K.; Vicente, E.; Arisseto-Bragotto, A.P.; Miguel, A.M.R.d.O.; Milani, R.F.; Tfouni, S.A.V. Occurrence of 3-MCPD, 2-MCPD and glycidyl esters in extra virgin olive oils, olive oils and oil blends and correlation with identity and quality parameters. Food Control 2019, 95, 135–141. [Google Scholar] [CrossRef]
- Matthäus, B.; Pudel, F.; Fehling, P.; Vosmann, K.; Freudenstein, A. Strategies for the reduction of 3-MCPD esters and related compounds in vegetable oils. Eur. J. Lipid Sci. Technol. 2011, 113, 380–386. [Google Scholar] [CrossRef]
- EFSA. Risks for human health related to the presence of 3- and 2-monochloropropanediol (MCPD), and their fatty acid esters, and glycidyl fatty acid esters in food. EFSA J. 2016, 14, 4426–4584. [Google Scholar] [CrossRef]
- Eisenreich, A.; Monien, B.H.; Götz, M.E.; Buhrke, T.; Oberemm, A.; Schultrich, K.; Abraham, K.; Braeuning, A.; Schafer, B. 3-MCPD as contaminant in processed foods: State of knowledge and remaining challenges. Food Chem. 2023, 403, 134332. [Google Scholar] [CrossRef]
- Kaze, N.; Watanabe, Y.; Sato, H.; Murota, K.; Kotaniguchi, M.; Yamamoto, H.; Inui, H.; Kitamura, S. Estimation of the Intestinal Absorption and Metabolism Behaviors of 2-and 3-Monochloropropanediol Esters. Lipids 2016, 51, 913–922. [Google Scholar] [CrossRef]
- Barocelli, E.; Corradi, A.; Mutti, A.; Petronini, P.G. Comparison between 3-MCPD and its palmitic esters in a 90-day toxicological study. EFSA Support. Publ. 2011, 8, 187E. [Google Scholar] [CrossRef]
- Cho, W.-S.; Han, B.S.; Lee, H.; Kim, C.; Nam, K.T.; Park, K.; Choi, M.; Kim, S.J.; Kim, S.H.; Jeong, J.; et al. Subchronic toxicity study of 3-monochloropropane-1,2-diol administered by drinking water to B6C3F1 mice. Food Chem. Toxicol. 2008, 46, 1666–1673. [Google Scholar] [CrossRef]
- Sunahara, G.; Perrin, I.; Marchesini, M. Carcinogenicity Study on 3-Monochloropropane-1, 2-Diol (3-MCPD) Administered in Drinking Water to Fischer 344 Rats; Report No. RE-SR93003; Nestec Ltd. Research & Development: Vevey, Switzerland, 1993. [Google Scholar]
- EFSA. Update of the risk assessment on 3-monochloropropane diol and its fatty acid esters. EFSA J. 2018, 16, e05083. [Google Scholar] [CrossRef]
- Buhrke, T.; Schultrich, K.; Braeuning, A.; Lampen, A. Comparative analysis of transcriptomic responses to repeated-dose exposure to 2-MCPD and 3-MCPD in rat kidney, liver and testis. Food Chem. Toxicol. 2017, 106, 36–46. [Google Scholar] [CrossRef]
- Schultrich, K.; Henderson, C.J.; Buhrke, T.; Braeuning, A. Effects of 2-MCPD on oxidative stress in different organs of male mice. Food Chem. Toxicol. 2020, 142, 111459. [Google Scholar] [CrossRef]
- Jones, A.R. Antifertility actions of alpha-chlorohydrin in the male. Aust. J. Biol. Sci. 1983, 36, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Kwack, S.J.; Kim, S.S.; Choi, Y.W.; Rhee, G.S.; Da Lee, R.; Seok, J.H.; Chae, S.Y.; Won, Y.H.; Lim, K.J.; Choi, K.S.; et al. Mechanism of antifertility in male rats treated with 3-monochloro-1,2-propanediol (3-MCPD). J. Toxicol. Environ. Health A 2004, 67, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Porter, L.M. Inhibition of glycolysis in boar spermatozoa by alpha-chlorohydrin phosphate appears to be mediated by phosphatase activity. Reprod. Fertil. Dev. 1995, 7, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Lynch, B.S.; Bryant, D.W.; Hook, G.J.; Nestmann, E.R.; Munro, I.C. Carcinogenicity of monochloro-1,2-propanediol (α-chlorohydrin, 3-MCPD). Int. J. Toxicol. 1998, 17, 47–76. [Google Scholar] [CrossRef]
- Mohri, H.; Suter, D.A.; Brown-Woodman, P.D.; White, I.G.; Ridley, D.D. Identification of the biochemical lesion produced by alpha-chlorohydrin in spermatozoa. Nature 1975, 255, 75–77. [Google Scholar] [CrossRef]
- Jones, A.R.; Fakhouri, G. Epoxides as obligatory intermediates in the metabolism of alpha-halohydrins. Xenobiotica 1979, 9, 595–599. [Google Scholar] [CrossRef]
- Jones, A.R.; Gadiel, P.; Stevenson, D. The fate of oxalic acid in the Wistar rat. Xenobiotica 1981, 11, 385–390. [Google Scholar] [CrossRef]
- Schultrich, K.; Frenzel, F.; Oberemm, A.; Buhrke, T.; Braeuning, A.; Lampen, A. Comparative proteomic analysis of 2-MCPD- and 3-MCPD-induced heart toxicity in the rat. Arch. Toxicol. 2017, 91, 3145–3155. [Google Scholar] [CrossRef]
- Cayer, L.G.J.; Buhrke, T.; Roberts, J.; Nunnikhoven, A.; Sommerkorn, K.; Reinhold, A.; Braeuning, A.; Raju, J.; Aukema, H.M.; Karakach, T. An integrated multi-omics analysis of the effects of the food processing-induced contaminant 2-monochloropropane-1,3-diol (2-MCPD) in rat heart. Arch. Toxicol. 2024, 98, 4033–4045. [Google Scholar] [CrossRef]
- BfR. 28-Day Oral Toxicity Study with 2-MCPD Diester, 2-MCPD and 3-MCPD Using Proteomics Analysis; BfR: Berlin, Germany, 2016. [Google Scholar]
- Roberts, J.; Caldwell, D.; Zhao, T.; Aziz, S.A.; Raju, J. Toxicological responses in F344 rats following repeated dose sub-acute and sub-chronic dietary exposures to the food processing-induced contaminant 2-monochloro-1,3-propanediol (2-MCPD). Food Chem. Toxicol. 2025, 206, 115730. [Google Scholar] [CrossRef]
- Marunouchi, T.; Inomata, S.; Sanbe, A.; Takagi, N.; Tanonaka, K. Protective effect of geranylgeranylacetone via enhanced induction of HSPB1 and HSPB8 in mitochondria of the failing heart following myocardial infarction in rats. Eur. J. Pharmacol. 2014, 730, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, M.; Xu, J.; Song, E.; Lv, Y.; Tang, S.; Zhang, X.; Kemper, N.; Hartung, J.; Bao, E. In vitro evaluation of aspirin-induced HspB1 against heat stress damage in chicken myocardial cells. Cell Stress Chaperones 2016, 21, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bian, X.; Li, L.; Liu, L.; Feng, C.; Wang, Y.; Ni, J.; Li, S.; Lu, D.; Li, Y.; et al. SENP1-Mediated HSP90ab1 DeSUMOylation in Cardiomyocytes Prevents Myocardial Fibrosis by Paracrine Signaling. Adv. Sci. 2024, 11, e2400741. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Orekhov, N.A.; Grechko, A.V.; Orekhov, A.N. Heat Shock Protein 90 as Therapeutic Target for CVDs and Heart Ageing. Int. J. Mol. Sci. 2022, 23, 649. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Ma, J.; Meng, X.W.; Liu, H.; Wang, H.; Song, S.Y.; Chen, Q.C.; Liu, H.Y.; Zhang, J.; Peng, K.; et al. Heat Shock Protein 70 Protects the Heart from Ischemia/Reperfusion Injury through Inhibition of p38 MAPK Signaling. Oxid. Med. Cell. Longev. 2020, 2020, 3908641. [Google Scholar] [CrossRef]
- Mohamed, B.A.; Barakat, A.Z.; Zimmermann, W.H.; Bittner, R.E.; Mühlfeld, C.; Hünlich, M.; Engel, W.; Maier, L.S.; Adham, I.M. Targeted disruption of Hspa4 gene leads to cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol. 2012, 53, 459–468. [Google Scholar] [CrossRef]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.; May, H.I.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef]
- Gorey, M.A.; Mericskay, M.; Li, Z.; Decaux, J.F. Interrelation between α-Cardiac Actin Treadmilling and Myocardin-Related Transcription Factor-A Nuclear Shuttling in Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 7394. [Google Scholar] [CrossRef]
- Angelini, A.; Gorey, M.-A.; Dumont, F.; Mougenot, N.; Chatzifrangkeskou, M.; Muchir, A.; Li, Z.; Mericskay, M.; Decaux, J.-F. Cardioprotective effects of α-cardiac actin on oxidative stress in a dilated cardiomyopathy mouse model. FASEB J. 2020, 34, 2987–3005. [Google Scholar] [CrossRef]
- Parlakian, A.; Charvet, C.; Escoubet, B.; Mericskay, M.; Molkentin, J.D.; Gary-Bobo, G.; De Windt, L.J.; Ludosky, M.-A.; Paulin, D.; Daegelen, D.; et al. Temporally Controlled Onset of Dilated Cardiomyopathy Through Disruption of the SRF Gene in Adult Heart. Circulation 2005, 112, 2930–2939. [Google Scholar] [CrossRef]
- Rottbauer, W.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Hassel, D.; Burns, C.G.; Katus, H.A.; Fishman, M.C. Cardiac Myosin Light Chain-2. Circ. Res. 2006, 99, 323–331. [Google Scholar] [CrossRef]
- Sheikh, F.; Lyon, R.C.; Chen, J. Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene 2015, 569, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Markandeya, Y.S.; Gregorich, Z.R.; Feng, L.; Ramchandran, V.; O’Hara, T.; Vaidyanathan, R.; Mansfield, C.; Keefe, A.M.; Beglinger, C.J.; Best, J.M.; et al. Caveolin-3 and Caveolae regulate ventricular repolarization. J. Mol. Cell. Cardiol. 2023, 177, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, K.H.; Schilling, J.M.; Leem, J.; Dhanani, M.; Head, B.P.; Roth, D.M.; Zemljic-Harpf, A.E.; Patel, H.H. Protective role of cardiac-specific overexpression of caveolin-3 in cirrhotic cardiomyopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G531–G541. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.T.; Panneerselvam, M.; Kawaraguchi, Y.; Tsutsumi, Y.M.; Ali, S.S.; Balijepalli, R.C.; Murray, F.; Head, B.P.; Niesman, I.R.; Rieg, T.; et al. Cardiac-specific overexpression of caveolin-3 attenuates cardiac hypertrophy and increases natriuretic peptide expression and signaling. J. Am. Coll. Cardiol. 2011, 57, 2273–2283. [Google Scholar] [CrossRef]
- Wang, X.; Pierre, V.; Liu, C.; Senapati, S.; Park, P.S.; Senyo, S.E. Exogenous extracellular matrix proteins decrease cardiac fibroblast activation in stiffening microenvironment through CAPG. J. Mol. Cell. Cardiol. 2021, 159, 105–119. [Google Scholar] [CrossRef]
- McLendon, P.M.; Robbins, J. Desmin-related cardiomyopathy: An unfolding story. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1220–H1228. [Google Scholar] [CrossRef]
- Heckmann, M.B.; Bauer, R.; Jungmann, A.; Winter, L.; Rapti, K.; Strucksberg, K.H.; Clemen, C.S.; Li, Z.; Schröder, R.; Katus, H.A.; et al. AAV9-mediated gene transfer of desmin ameliorates cardiomyopathy in desmin-deficient mice. Gene Ther. 2016, 23, 673–679. [Google Scholar] [CrossRef]
- Yuan, B.; Wan, P.; Chu, D.; Nie, J.; Cao, Y.; Luo, W.; Lu, S.; Chen, J.; Yang, Z. A Cardiomyocyte-Specific Wdr1 Knockout Demonstrates Essential Functional Roles for Actin Disassembly during Myocardial Growth and Maintenance in Mice. Am. J. Pathol. 2014, 184, 1967–1980. [Google Scholar] [CrossRef]
- Huang, X.; Li, Z.; Hu, J.; Yang, Z.; Liu, Z.; Zhang, T.; Zhang, C.; Yuan, B. Knockout of Wdr1 results in cardiac hypertrophy and impaired cardiac function in adult mouse heart. Gene 2019, 697, 40–47. [Google Scholar] [CrossRef]
- Lambert, M.R.; Gussoni, E. Tropomyosin 3 (TPM3) function in skeletal muscle and in myopathy. Skelet Muscle 2023, 13, 18. [Google Scholar] [CrossRef]
- Ma, S.; Xu, Q.; Bai, R.; Dong, T.; Peng, Z.; Liu, X. Generation of a TPM1 homozygous knockout embryonic stem cell line by CRISPR/Cas9 editing. Stem. Cell Res. 2021, 55, 102470. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Wang, L.; Kawai, M. A study of tropomyosin’s role in cardiac function and disease using thin-filament reconstituted myocardium. J. Muscle Res. Cell Motil. 2013, 34, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Yang, W.; Shi, M.; Wang, S.; Li, Y.; Xu, Z. The Role of TPM3 in Protecting Cardiomyocyte from Hypoxia-Induced Injury via Cytoskeleton Stabilization. Int. J. Mol. Sci. 2024, 25, 6797. [Google Scholar] [CrossRef] [PubMed]
- Gagat, M.; Grzanka, D.; Izdebska, M.; Grzanka, A. Effect of L-homocysteine on endothelial cell-cell junctions following F-actin stabilization through tropomyosin-1 overexpression. Int. J. Mol. Med. 2013, 32, 115–129. [Google Scholar] [CrossRef]
- Caporizzo, M.A.; Prosser, B.L. The microtubule cytoskeleton in cardiac mechanics and heart failure. Nat. Rev. Cardiol. 2022, 19, 364–378. [Google Scholar] [CrossRef]
- Chaurembo, A.I.; Xing, N.; Chanda, F.; Li, Y.; Zhang, H.J.; Fu, L.D.; Huang, J.Y.; Xu, Y.J.; Deng, W.H.; Cui, H.D.; et al. Mitofilin in cardiovascular diseases: Insights into the pathogenesis and potential pharmacological interventions. Pharmacol. Res. 2024, 203, 107164. [Google Scholar] [CrossRef]
- Gudmundsson, H.; Hund, T.J.; Wright, P.J.; Kline, C.F.; Snyder, J.S.; Qian, L.; Koval, O.M.; Cunha, S.R.; George, M.; Rainey, M.A.; et al. EH domain proteins regulate cardiac membrane protein targeting. Circ. Res. 2010, 107, 84–95. [Google Scholar] [CrossRef]
- Yang, F.; Aiello, D.L.; Pyle, W.G. Cardiac myofilament regulation by protein phosphatase type 1alpha and CapZ. Biochem. Cell Biol. 2008, 86, 70–78. [Google Scholar] [CrossRef]
- Gao, Y.; Peng, L.; Zhao, C. MYH7 in cardiomyopathy and skeletal muscle myopathy. Mol. Cell Biochem 2024, 479, 393–417. [Google Scholar] [CrossRef]
- Heling, L.; Geeves, M.A.; Kad, N.M. MyBP-C: One protein to govern them all. J Muscle Res Cell Motil. 2020, 41, 91–101. [Google Scholar] [CrossRef]
- Turner, M.; Anderson, D.E.; Bartels, P.; Nieves-Cintron, M.; Coleman, A.M.; Henderson, P.B.; Man, K.N.M.; Tseng, P.Y.; Yarov-Yarovoy, V.; Bers, D.M.; et al. α-Actinin-1 promotes activity of the L-type Ca2+ channel Cav1.2. Embo. J. 2020, 39, e102622. [Google Scholar] [CrossRef] [PubMed]
- Bergau, N.; Zhao, Z.Y.; Abraham, K.; Monien, B.H. Metabolites of 2-and 3-Monochloropropanediol (2-and 3-MCPD) in Humans: Urinary Excretion of 2-Chlorohydracrylic Acid and 3-Chlorolactic Acid after Controlled Exposure to a Single High Dose of Fatty Acid Esters of 2-and 3-MCPD. Mol. Nutr. Food Res. 2021, 65, 2000736. [Google Scholar] [CrossRef]
- Fukao, T.; Kamijo, K.; Osumi, T.; Fujiki, Y.; Yamaguchi, S.; Orii, T.; Hashimoto, T. Molecular cloning and nucleotide sequence of cDNA encoding the entire precursor of rat mitochondrial acetoacetyl-CoA thiolase. J. Biochem. 1989, 106, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Harijan, R.K.; Dalwani, S.; Kiema, T.R.; Venkatesan, R.; Wierenga, R.K. Thiolase: A Versatile Biocatalyst Employing Coenzyme A-Thioester Chemistry for Making and Breaking C-C Bonds. Annu. Rev. Biochem. 2023, 92, 351–384. [Google Scholar] [CrossRef]
- Park, K.C.; Krywawych, S.; Richard, E.; Desviat, L.R.; Swietach, P. Cardiac Complications of Propionic and Other Inherited Organic Acidemias. Front. Cardiovasc. Med. 2020, 7, 617451. [Google Scholar] [CrossRef]
- Escriva, F.; Rodriguez, C.; Pascual-Leone, A.M. Glycemia, ketonemia, and brain enzymes of ketone body utilization in suckling and adult rats undernourished from intrauterine life. J. Neurochem. 1985, 44, 1358–1362. [Google Scholar] [CrossRef]
- Uchida, Y.; Izai, K.; Orii, T.; Hashimoto, T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J. Biol. Chem. 1992, 267, 1034–1041. [Google Scholar]
- Russell, J.J.; Patel, M.S. Purification and properties of succinyl-CoA:3-oxo-acid CoA-transferase from rat brain. J. Neurochem. 1982, 38, 1446–1452. [Google Scholar] [CrossRef]
- Kimmich, G.A.; Rasmussen, H. Regulation of pyruvate carboxylase activity by calcium in intact rat liver mitochondria. J. Biol. Chem. 1969, 244, 190–199. [Google Scholar] [CrossRef]
- Patel, M.S.; Harris, R.A. Mammalian alpha-keto acid dehydrogenase complexes: Gene regulation and genetic defects. FASEB J. 1995, 9, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Hackert, M.L. Structure-function relationships in dihydrolipoamide acyltransferases. J. Biol. Chem. 1990, 265, 8971–8974. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Leighton, B.; Young, M.E.; Blomstrand, E.; Newsholme, E.A. Inactivation of aconitase and oxoglutarate dehydrogenase in skeletal muscle in vitro by superoxide anions and/or nitric oxide. Biochem. Biophys. Res. Commun. 1998, 249, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hakozaki, M.; Kimura, A.; Kochi, H. Purification, resolution, and reconstitution of rat liver branched-chain alpha-keto acid dehydrogenase complex. J. Biochem. 1987, 101, 19–27. [Google Scholar] [CrossRef]
- Zhang, B.; Kuntz, M.J.; Goodwin, G.W.; Harris, R.A.; Crabb, D.W. Molecular cloning of a cDNA for the E1 alpha subunit of rat liver branched chain alpha-ketoacid dehydrogenase. J. Biol. Chem. 1987, 262, 15220–15224. [Google Scholar] [CrossRef]
- Zhang, X.; Vincent, A.S.; Halliwell, B.; Wong, K.P. A mechanism of sulfite neurotoxicity: Direct inhibition of glutamate dehydrogenase. J. Biol. Chem. 2004, 279, 43035–43045. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Rougraff, P.M.; Davis, E.J.; Harris, R.A. Purification and characterization of methylmalonate-semialdehyde dehydrogenase from rat liver. Identity to malonate-semialdehyde dehydrogenase. J. Biol. Chem. 1989, 264, 14965–14971. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Williams, N.; Hullihen, J. Mitochondrial ATP synthase: Dramatic Mg2+-induced alterations in the structure and function of the F1-ATPase moiety. Biochemistry 1987, 26, 8631–8637. [Google Scholar] [CrossRef]
- Xian, C.; Luo, Q.; Li, W.; Zou, L.; Liu, J. ATP5F1A deficiency causes developmental delay and motor dysfunction in humans and zebrafish. J. Transl. Med. 2025, 23, 1054. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, H.; Chang, Y.; Hua, X.; Chen, X.; Sheng, Y.; Shan, D.; Bao, M.; Hu, S.; Song, J. Overexpression of ATP5F1A in Cardiomyocytes Promotes Cardiac Reverse Remodeling. Circ. Heart Fail. 2024, 17, e011504. [Google Scholar] [CrossRef]
- Yu, T.; Li, X.; Wang, C.; Yang, Y.; Fu, X.; Li, T.; Wang, W.; Liu, X.; Jiang, X.; Wei, D.; et al. Lactylation of Mitochondrial Adenosine Triphosphate Synthase Subunit Alpha Regulates Vascular Remodeling and Progression of Aortic Dissection. Research 2025, 8, 0799. [Google Scholar] [CrossRef]
- Muller, M.; Moser, R.; Cheneval, D.; Carafoli, E. Cardiolipin is the membrane receptor for mitochondrial creatine phosphokinase. J. Biol. Chem. 1985, 260, 3839–3843. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.M.; Strauss, A.W. Developmental expression of sarcomeric and ubiquitous mitochondrial creatine kinase is tissue-specific. Biochim. Biophys. Acta 1994, 1219, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.Z.; Shoulkamy, M.I.; Salem, A.M.H.; Oba, S.; Goda, M.; Nakano, T.; Ide, H. Aldehydes with high and low toxicities inactivate cells by damaging distinct cellular targets. Mutat. Res.-Fund. Mol. M. 2016, 786, 41–51. [Google Scholar] [CrossRef]
- Bach, R.D.; Canepa, C. Electronic Factors Influencing the Decarboxylation of beta-Keto Acids. A Model Enzyme Study. J. Org. Chem. 1996, 61, 6346–6353. [Google Scholar] [CrossRef]
- Siu, G.M.; Draper, H.H. Metabolism of malonaldehyde in vivo and in vitro. Lipids 1982, 17, 349–355. [Google Scholar] [CrossRef]
- Matalon, R.; Michaels, K.; Kaul, R.; Whitman, V.; Rodriguez-Novo, J.; Goodman, S.; Thorburn, D. Malonic aciduria and cardiomyopathy. J. Inherit. Metab. Dis. 1993, 16, 571–573. [Google Scholar] [CrossRef]
- Huth, W.; Worm-Breitgoff, C.; Möller, U.; Wunderlich, I. Evidence for an in vivo modification of mitochondrial proteins by coenzyme A. Biochim. Biophys. Acta 1991, 1077, 1–10. [Google Scholar] [CrossRef]
- Luo, M.J.; Smeland, T.E.; Shoukry, K.; Schulz, H. Delta 3,5, delta 2,4-dienoyl-CoA isomerase from rat liver mitochondria. Purification and characterization of a new enzyme involved in the beta-oxidation of unsaturated fatty acids. J. Biol. Chem. 1994, 269, 2384–2388. [Google Scholar]
- Thorn, M.B. Inhibition by malonate of succinic dehydrogenase in heart-muscle preparations. Biochem. J. 1953, 54, 540–547. [Google Scholar] [CrossRef]
- Saggerson, D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 2008, 28, 253–272. [Google Scholar] [CrossRef]
- Görg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophor. Int. J. 2000, 21, 1037–1053. [Google Scholar] [CrossRef]
- Sawada, S.; Oberemm, A.; Buhrke, T.; Merschenz, J.; Braeuning, A.; Lampen, A. Proteomic analysis of 3-MCPD and 3-MCPD dipalmitate-induced toxicity in rat kidney. Arch. Toxicol. 2016, 90, 1437–1448. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Names | UniProt Entry | UniProt ID | Name(s) and Synonym(s) of the Protein | 28-Day Study | 90-Day Study | ||
|---|---|---|---|---|---|---|---|
| 10 mg/kg bw | 2 mg/kg bw | 10 mg/kg bw | 40 mg/kg bw | ||||
| Acaa2 | THIM_RAT | P13437 | 3-ketoacyl-CoA thiolase, mitochondrial | (-) | (-) | −0.84 | −1.62 |
| Acat1 | THIL_RAT | P17764 | Acetyl-CoA acetyltransferase, mitochondrial | (-) | 1.09 | 1.37 | 1.23 |
| Acat1 | THIL_RAT | P17764 | Acetyl-CoA acetyltransferase, mitochondrial | −3.19 | −0.51 | −1.15 | −1.57 |
| Aco2 | ACON_RAT | Q9ER34 | Aconitate hydratase, mitochondrial | 1.05 | (-) | (-) | (-) |
| Actc1 | ACTC_RAT | P68035 | Actin, alpha cardiac muscle 1 | −0.77 | (-) | −0.75 | (-) |
| Actn1 | ACTN1_RAT | Q9Z1P2 | Alpha-actinin-1 | (-) | 1.17 | 1.59 | 1.55 |
| Aldh2 | ALDH2_RAT | P11884 | Aldehyde dehydrogenase, mitochondrial | (-) | (-) | (-) | 0.62 |
| Aldh6a1 | MMSA_RAT | Q02253 | Methylmalonate semialdehyde, mitochondrial | 0.81 | (-) | 0.63 | 0.69 |
| Aldh6a1 | MMSA_RAT | Q02253 | Methylmalonate semialdehyde, mitochondrial | (-) | (-) | (-) | −1.03 |
| Atp5f1a | ATPA_RAT | P15999 | ATP synthase subunit alpha, mitochondrial | −0.74 | (-) | −0.77 | −0.85 |
| Atp5f1a | ATPA_RAT | P15999 | ATP synthase subunit alpha, mitochondrial | (-) | (-) | (-) | 1.08 |
| Atp5f1b | ATPB_RAT | P10719 | ATP synthase subunit beta, mitochondrial | −0.67 | (-) | (-) | −0.78 |
| Atp5f1c | ATPG_RAT | P35435 | ATP synthase subunit gamma, mitochondrial | (-) | (-) | (-) | 0.67 |
| Bckdha | ODBA_RAT | P11960 | 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial | 0.77 | (-) | (-) | 0.78 |
| Capg | CAPG_RAT | Q6AYC4 | Macrophage-capping protein | 0.75 | (-) | (-) | 0.86 |
| Capzb | CAPZB_RAT | Q5XI32 | F-actin-capping protein subunit beta | (-) | (-) | (-) | 0.57 |
| Cav3 | Cav3_RAT | P51638 | Caveolin-3 | 0.84 | (-) | (-) | (-) |
| Ckmt2 | KCRS_RAT | P09605 | Creatine kinase S-type, mitochondrial | −0.56 | (-) | (-) | (-) |
| Des | DESM_RAT | P48675 | Desmin | 0.81 | (-) | (-) | 0.82 |
| Dld | DLDH_RAT | Q6P6R2 | Dihydrolipoyl dehydrogenase, mitochondrial | (-) | (-) | 1.16 | 1.61 |
| Ech1 | ECH1_RAT | Q62651 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | 0.55 | (-) | (-) | 0.57 |
| Eci1 | ECI1_RAT | P23965 | Enoyl-CoA delta isomerase 1, mitochondrial | (-) | (-) | (-) | 0.68 |
| Fh | FUMH_RAT | P14408 | Fumarate hydratase, mitochondrial | (-) | (-) | (-) | −0.60 |
| Glud1 | DHE3_RAT | P10860 | Glutamate dehydrogenase 1, mitochondrial | −0.82 | (-) | (-) | (-) |
| Hadha | ECHA_RAT | Q64428 | Trifunctional enzyme subunit alpha, mitochondrial | −1.19 | −0.88 | −0.68 | −0.73 |
| Hsp90b1 | ENPL_RAT | Q66HD0 | Endoplasmin | (-) | (-) | (-) | 0.58 |
| Hspa1a | HS71A_RAT | P0DMW0 | Heat shock 70 kDa protein 1A | (-) | (-) | (-) | 0.69 |
| Hspa4 | HSP74_RAT | O88600 | Heat shock 70 kDa protein 4 | (-) | (-) | 0.50 | 0.61 |
| Hspa5 | BIP_RAT | P06761 | Endoplasmic reticulum chaperone BiP | (-) | (-) | (-) | 0.87 |
| Hspb1 | HSPB1_RAT | P42930 | Heat shock protein beta-1 | 0.93 | (-) | (-) | 0.54 |
| Mybpc3 | MYPC_RAT | P56741 | Myosin-binding protein C, cardiac-type | (-) | (-) | 0.80 | 2.62 |
| Myh7 | MYH7_RAT | P02564 | Myosin-7 (Myosin heavy chain 7) | (-) | (-) | 0.74 | 0.88 |
| Myl2 | MLRV_RAT | P08733 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | (-) | (-) | (-) | −0.50 |
| Ogdh | ODO1_RAT | Q5XI78 | 2-oxoglutarate dehydrogenase complex component E1 | 0.64 | (-) | (-) | (-) |
| Oxct1 | SCOT1_RAT | B2GV06 | Succinyl-CoA:3-ketoacid coenzyme A transferase 1, mitochondrial | 0.71 | (-) | (-) | 1.12 |
| Pdha1 | ODPA_RAT | P26284 | Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial | −0.98 | −0.80 | (-) | −0.61 |
| Pdk1 | PDK1_RAT | Q63065 | [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial | −0.51 | (-) | (-) | (-) |
| Tpm1 | TPM1_RAT | P04692 | Tropomyosin alpha-1 chain | 1.04 | (-) | (-) | (-) |
| Tpm2 | TPM2_RAT | P58775 | Tropomyosin beta chain | 2.37 | (-) | (-) | (-) |
| Tpm3 | TPM3_RAT | Q63610 | Tropomyosin alpha-3 chain | 0.56 | (-) | (-) | (-) |
| Wdr1 | WDR1_RAT | Q5RKI0 | WD repeat-containing protein 1 | 1.22 | (-) | (-) | (-) |
| Experimental Details | 28-Day Study | 90-Day Study |
|---|---|---|
| according to OECD-Test No. | 407 (subacute) | 408 (subchronic) |
| species (strain) | rat (Wistar) | rat (Fischer F344/CDF) |
| sex and age | male weanlings | male and female weanlings |
| animal number | 6 | 12/12 |
| test substance supplier/Cat No. | 2-MCPD TRC Toronto | 2-MCPD TRC Toronto |
| administration | oral gavage | pellet diet |
| dose groups investigated by proteome analysis: mg/kg bw/day | 0, 10 | 0, 2, 10, 40 |
| vehicle | corn oil 7.5 mL/kg bw | corn oil, achieving 7% in the diet |
| diet | Ssniff V1534 | AIN-93G |
| necropsy/anesthesia | Whole body perfusion/pentobarbital | Whole body perfusion/isoflurane |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Oberemm, A.; Eisenreich, A.; Sommerkorn, K.; Reinhold, A.; Meckert, C.; Götz, M.E. 2-MCPD-Induced Effects in the Heart: Toxicological and Mechanistic Implications from Comparative Proteomic Analyses in Rats. Molecules 2026, 31, 692. https://doi.org/10.3390/molecules31040692
Oberemm A, Eisenreich A, Sommerkorn K, Reinhold A, Meckert C, Götz ME. 2-MCPD-Induced Effects in the Heart: Toxicological and Mechanistic Implications from Comparative Proteomic Analyses in Rats. Molecules. 2026; 31(4):692. https://doi.org/10.3390/molecules31040692
Chicago/Turabian StyleOberemm, Axel, Andreas Eisenreich, Katharina Sommerkorn, Anna Reinhold, Christine Meckert, and Mario E. Götz. 2026. "2-MCPD-Induced Effects in the Heart: Toxicological and Mechanistic Implications from Comparative Proteomic Analyses in Rats" Molecules 31, no. 4: 692. https://doi.org/10.3390/molecules31040692
APA StyleOberemm, A., Eisenreich, A., Sommerkorn, K., Reinhold, A., Meckert, C., & Götz, M. E. (2026). 2-MCPD-Induced Effects in the Heart: Toxicological and Mechanistic Implications from Comparative Proteomic Analyses in Rats. Molecules, 31(4), 692. https://doi.org/10.3390/molecules31040692