
Oxidation-Triggered Formation of Diradical Cations from Paramagnetic Molecules and Their Spin Density Evolution

 , and
, and 
Abstract

1. Introduction
2. Results
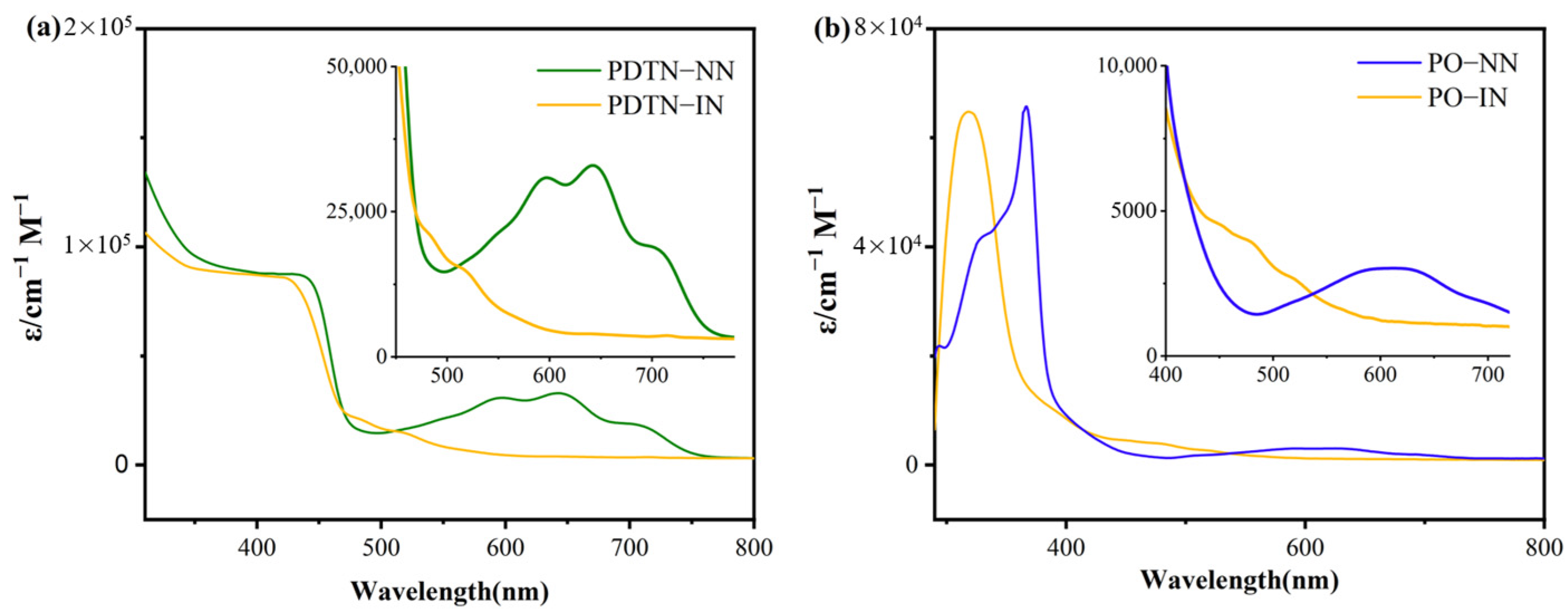
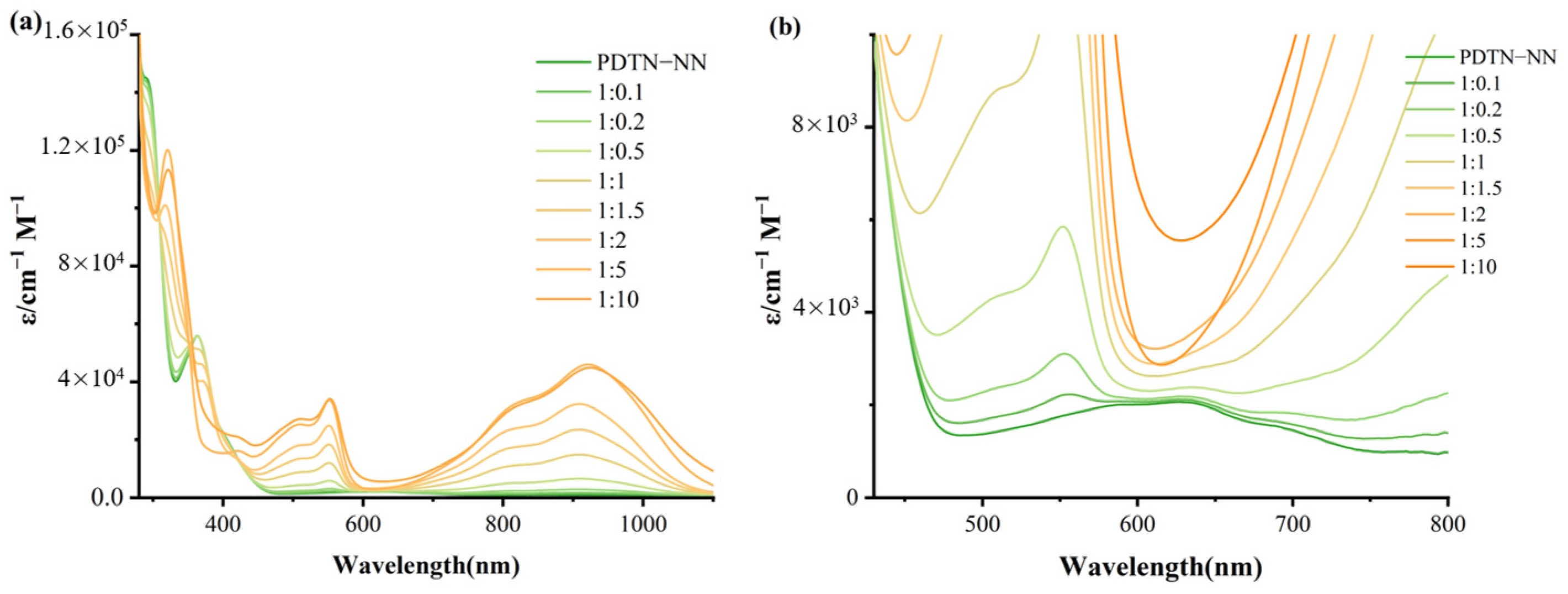
2.1. Optical Properties
2.1.1. UV-Vis Absorption Spectra of Radicals with Phenothiazine and Phenoxazine
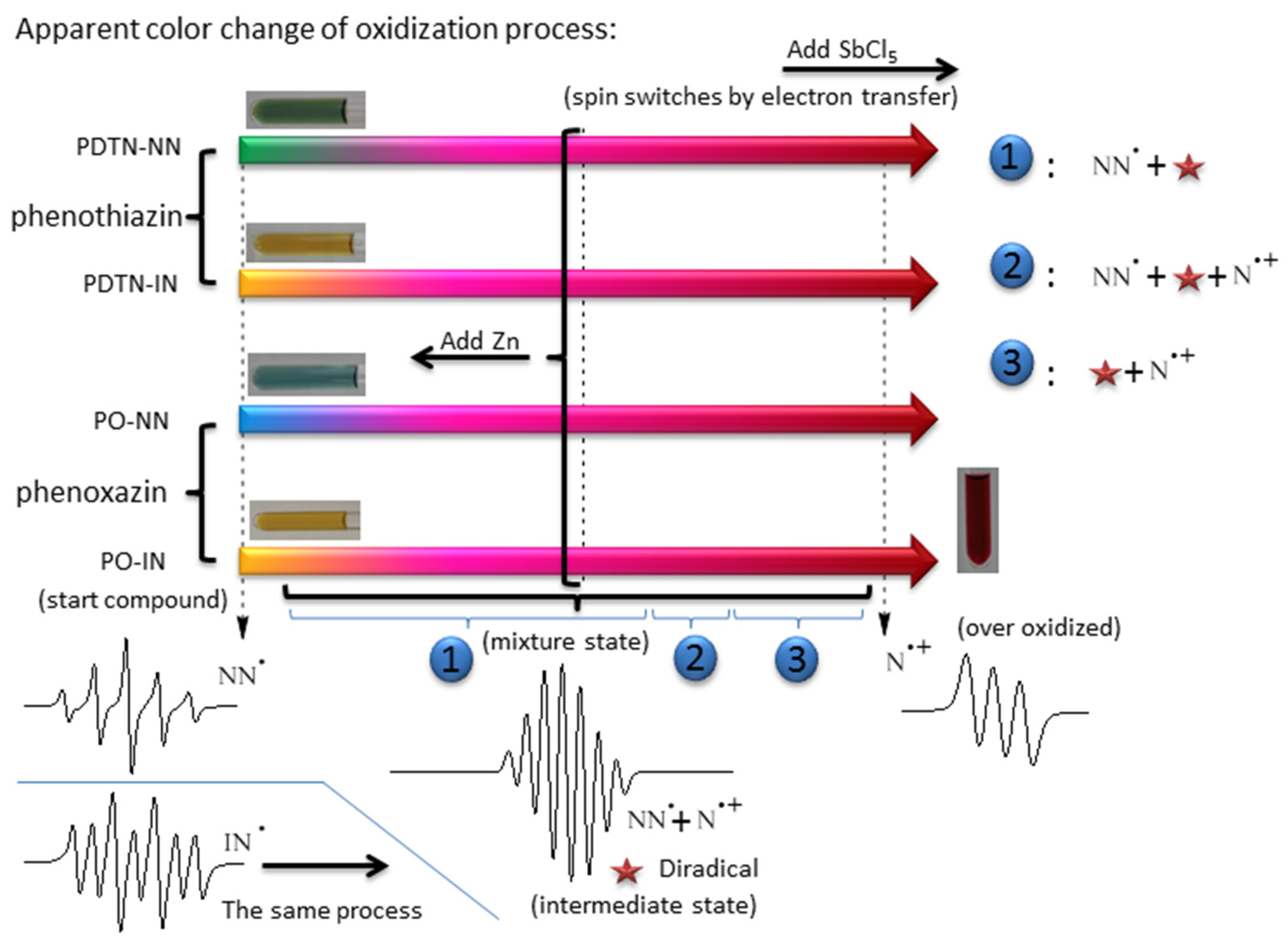
2.1.2. UV-Vis Absorption of Redox Stimuli Responsive Process
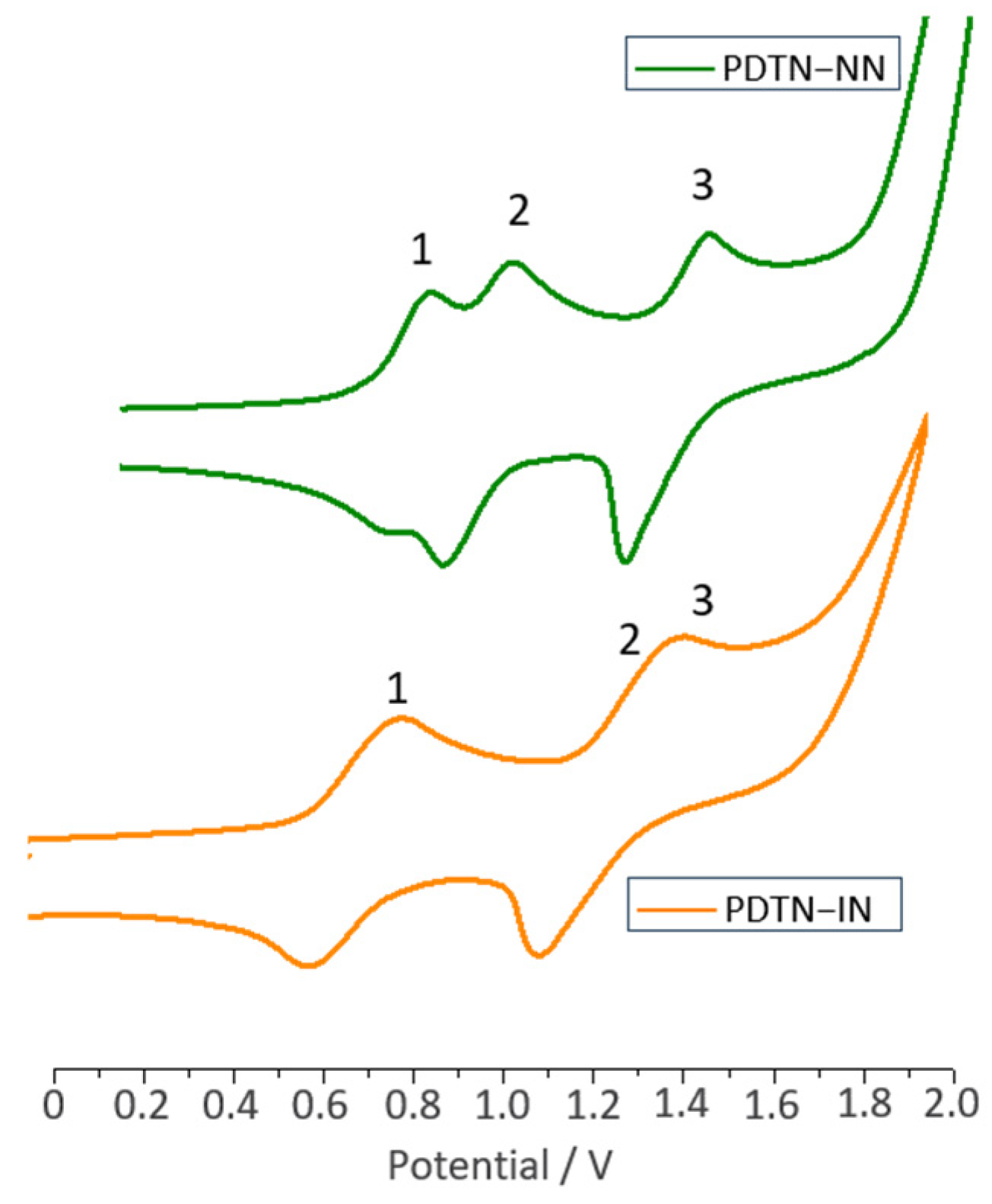
2.2. Cyclic Voltammetry Measurements
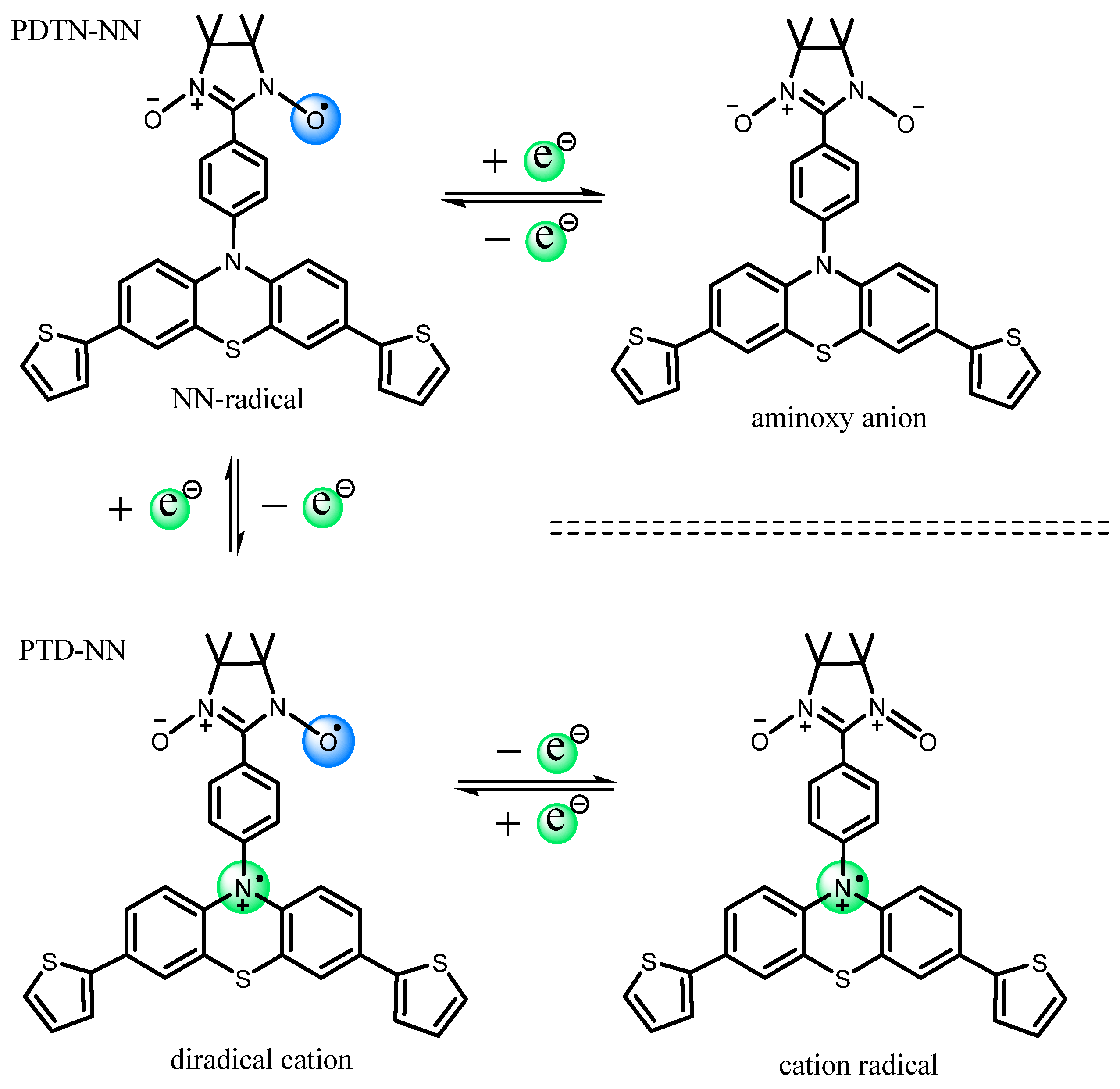
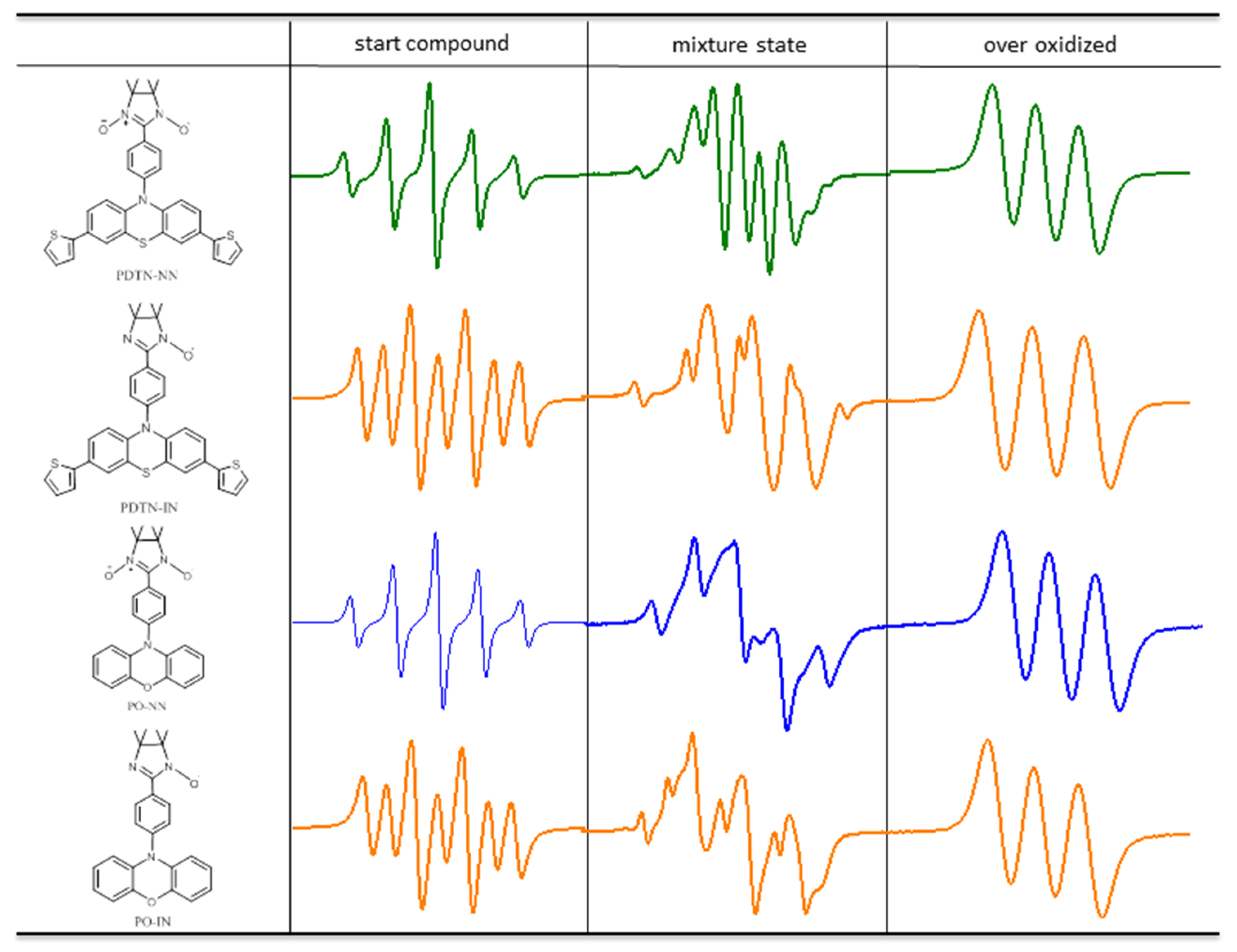
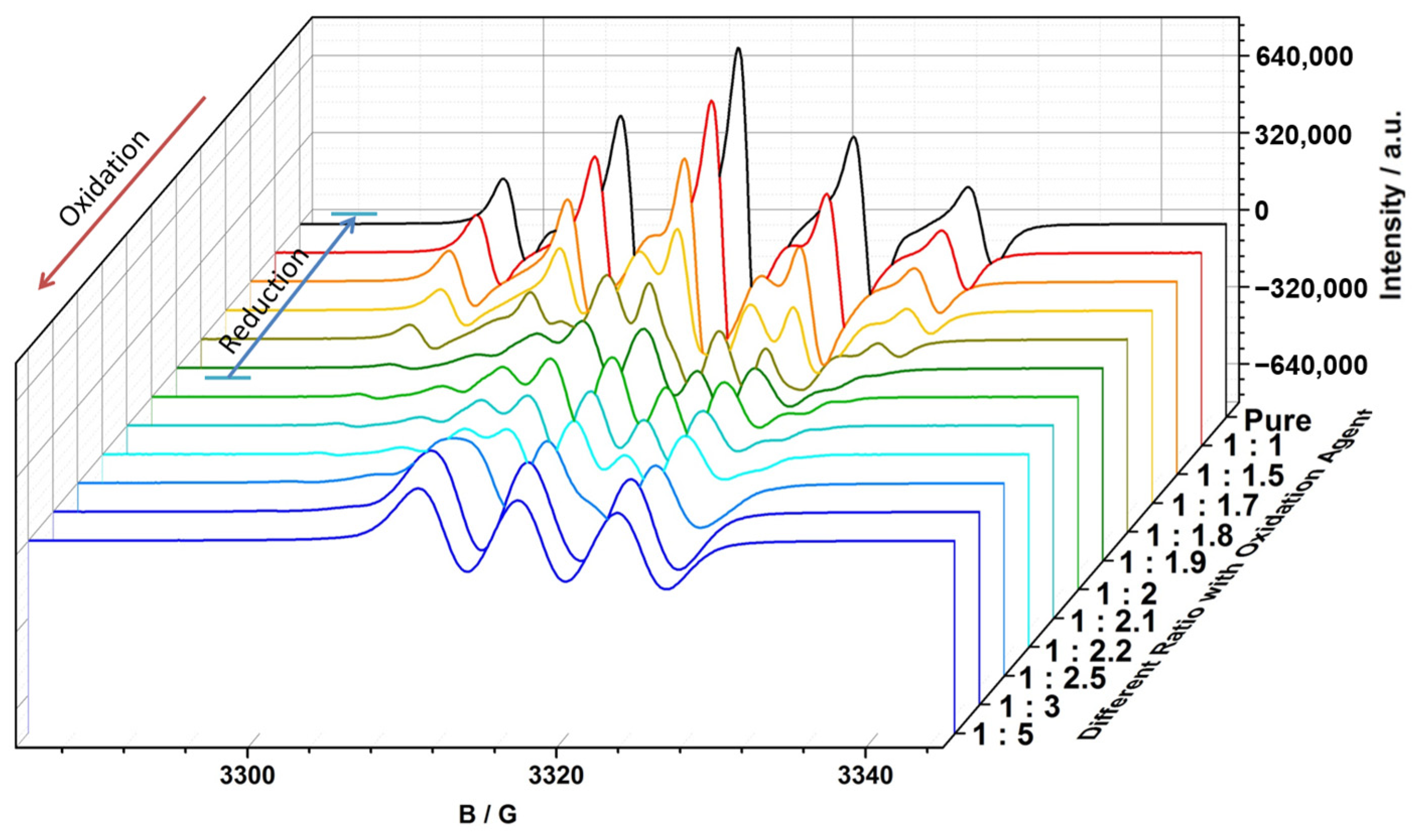
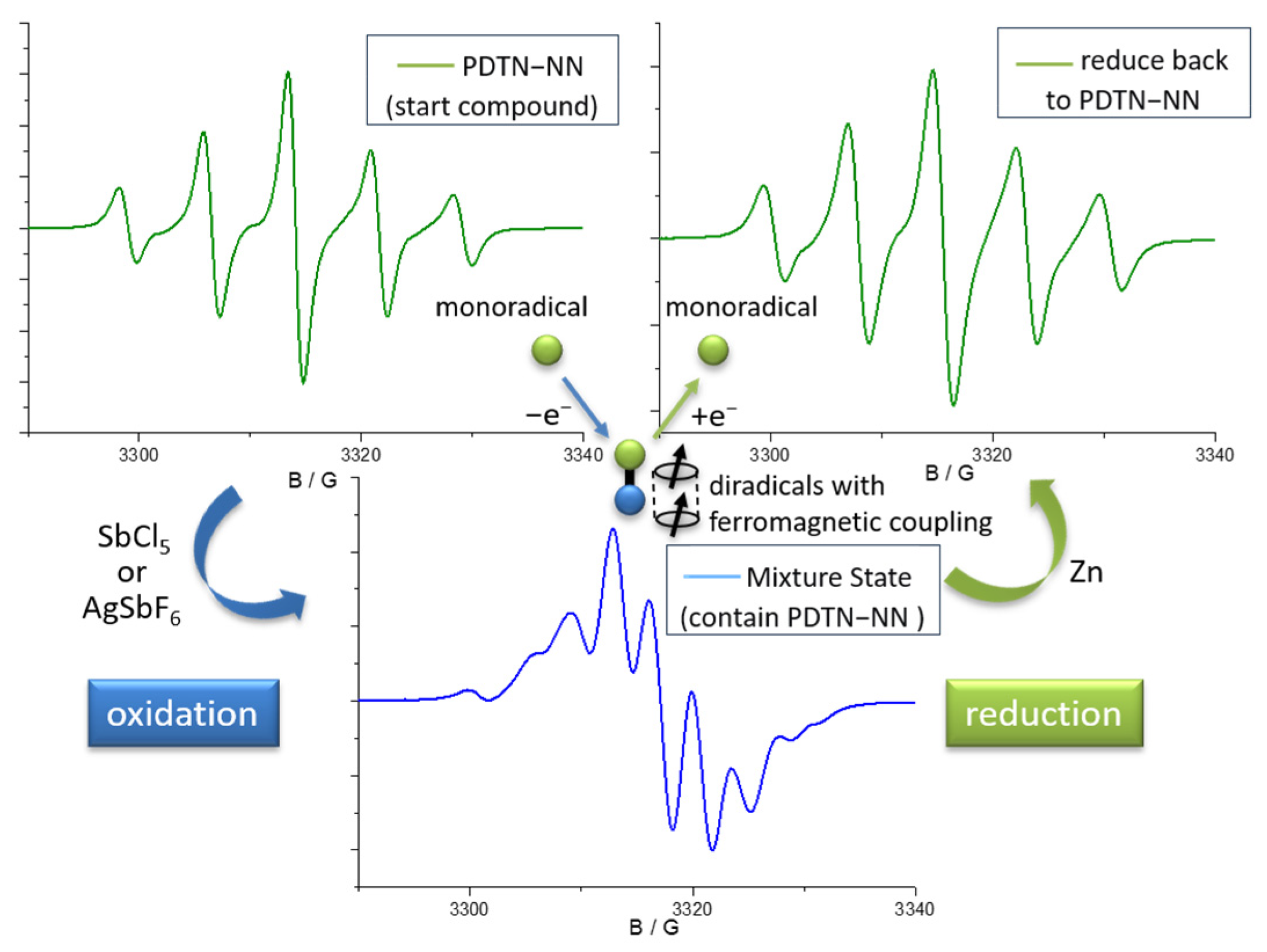
2.3. EPR Measurements and Properties
2.3.1. EPR Spectra of Redox Stimuli Responsive Process
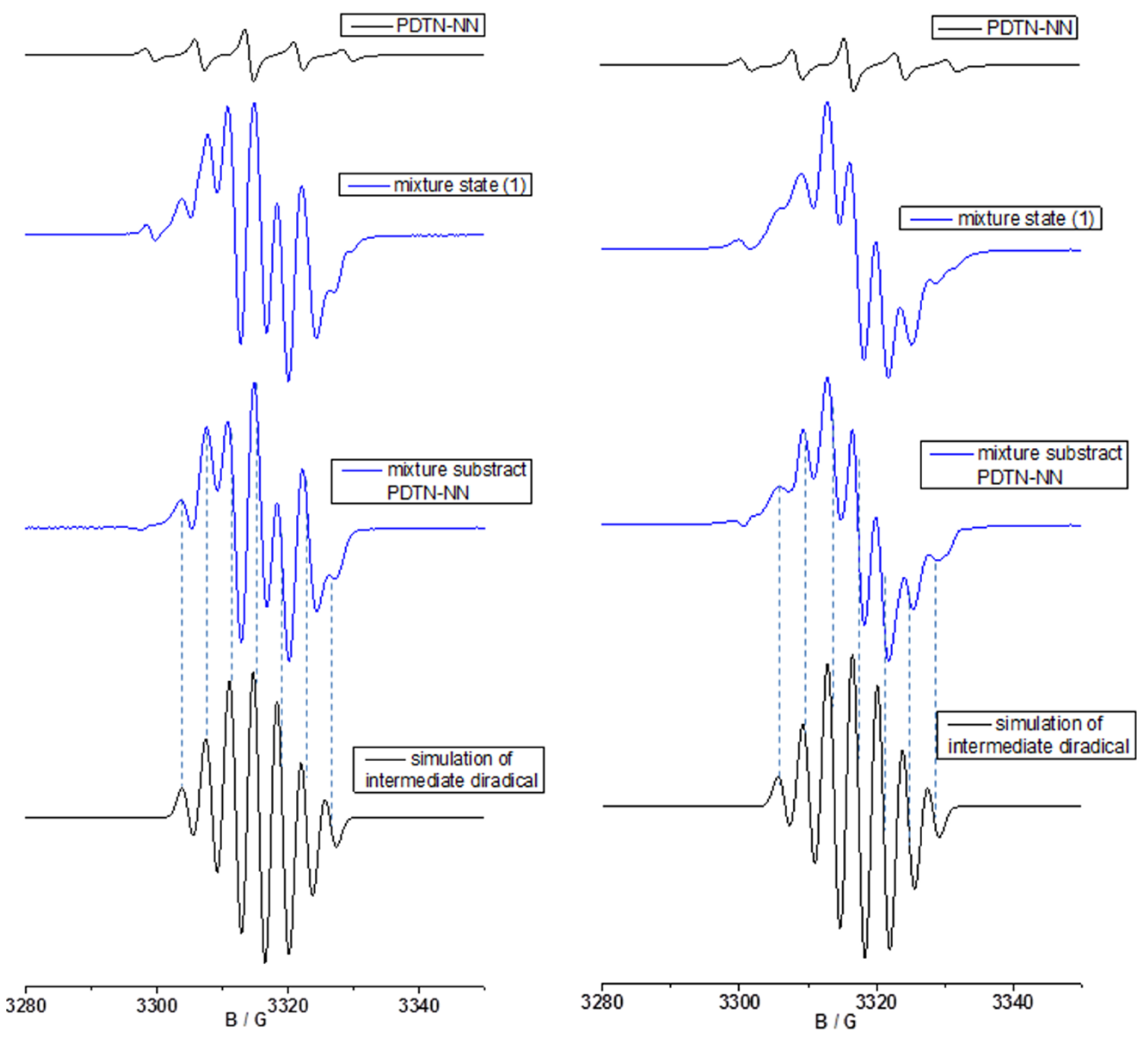
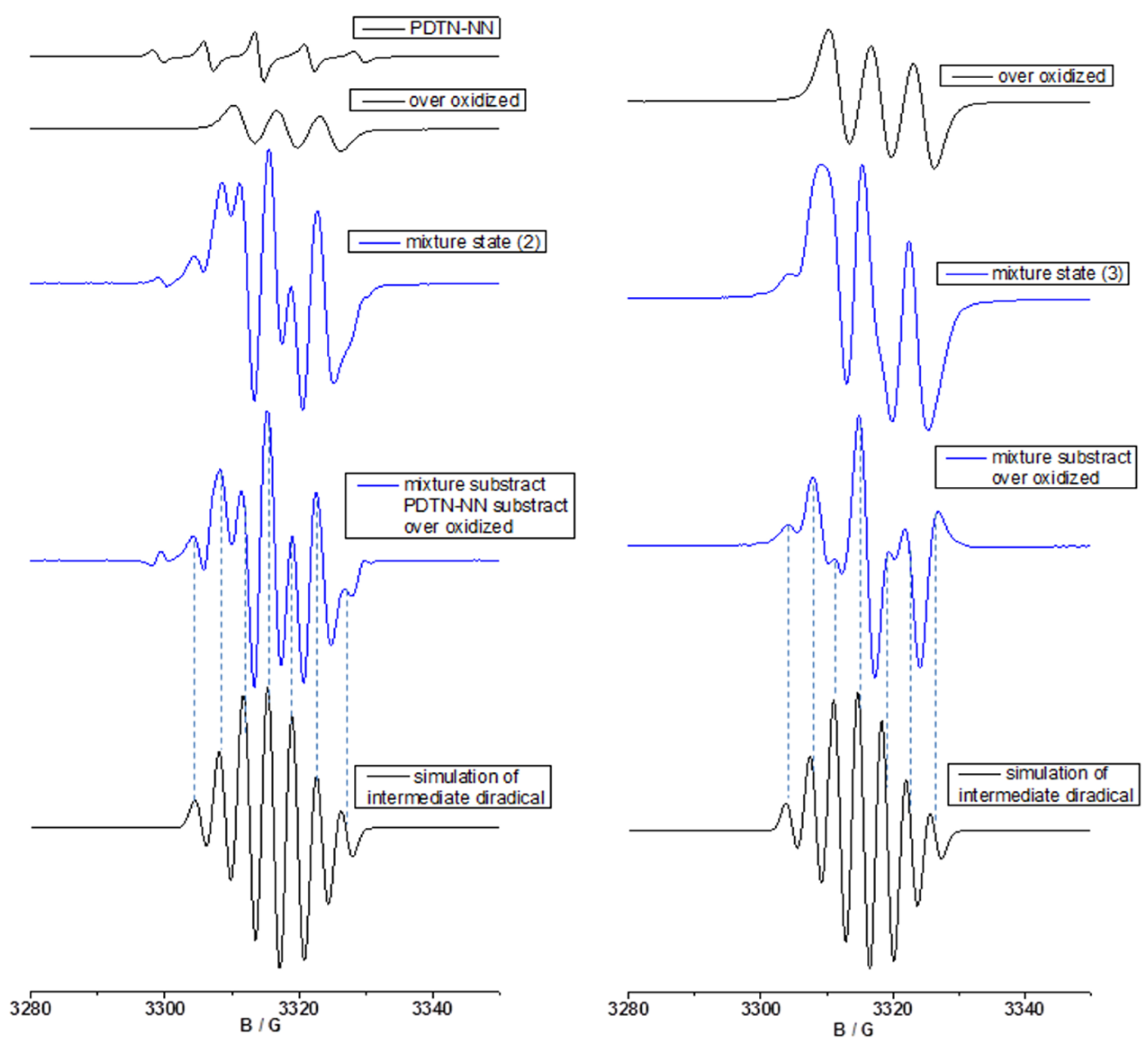
2.3.2. Fitting of Simulated EPR with New Signal Pattern of Experimental EPR Spectra
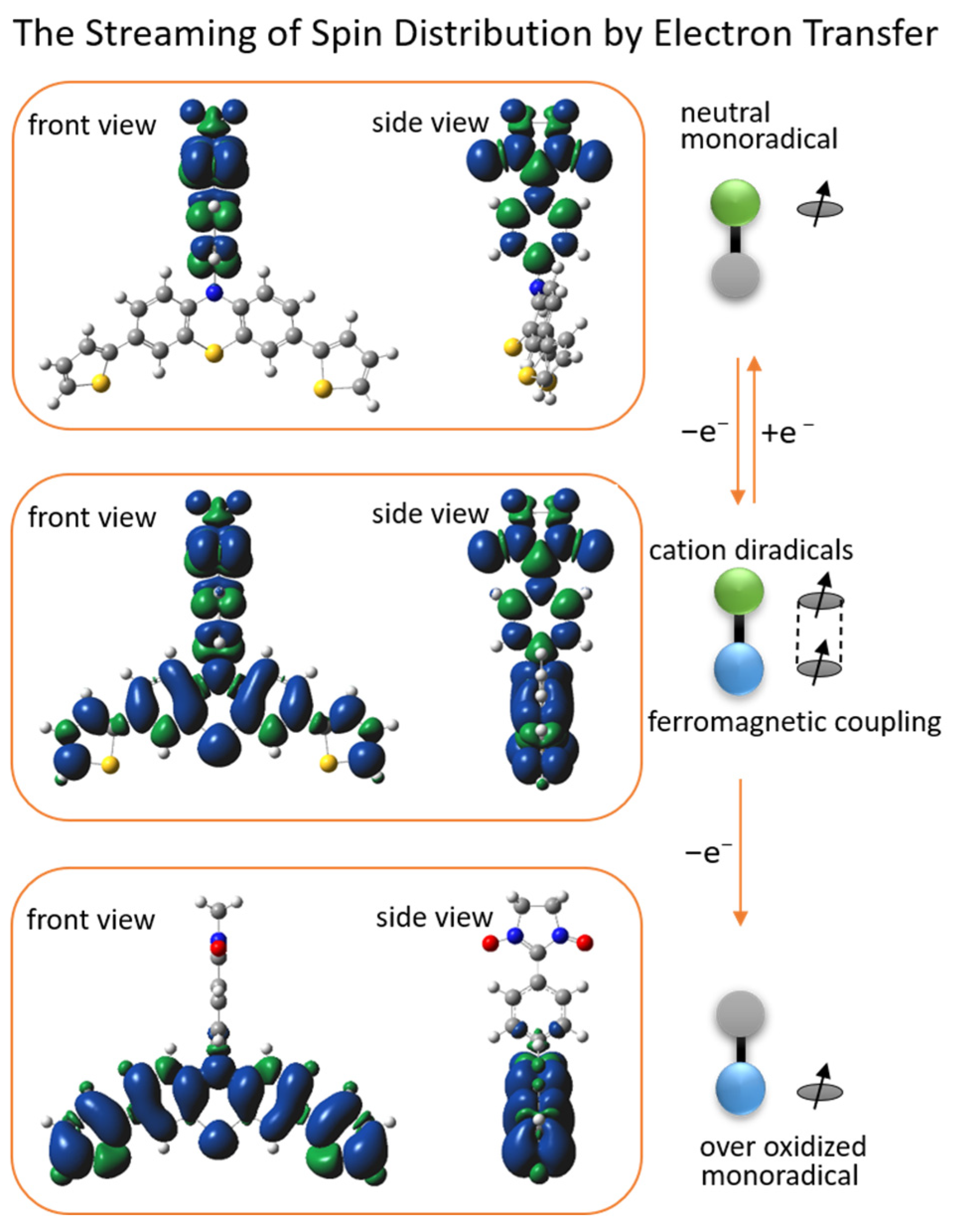
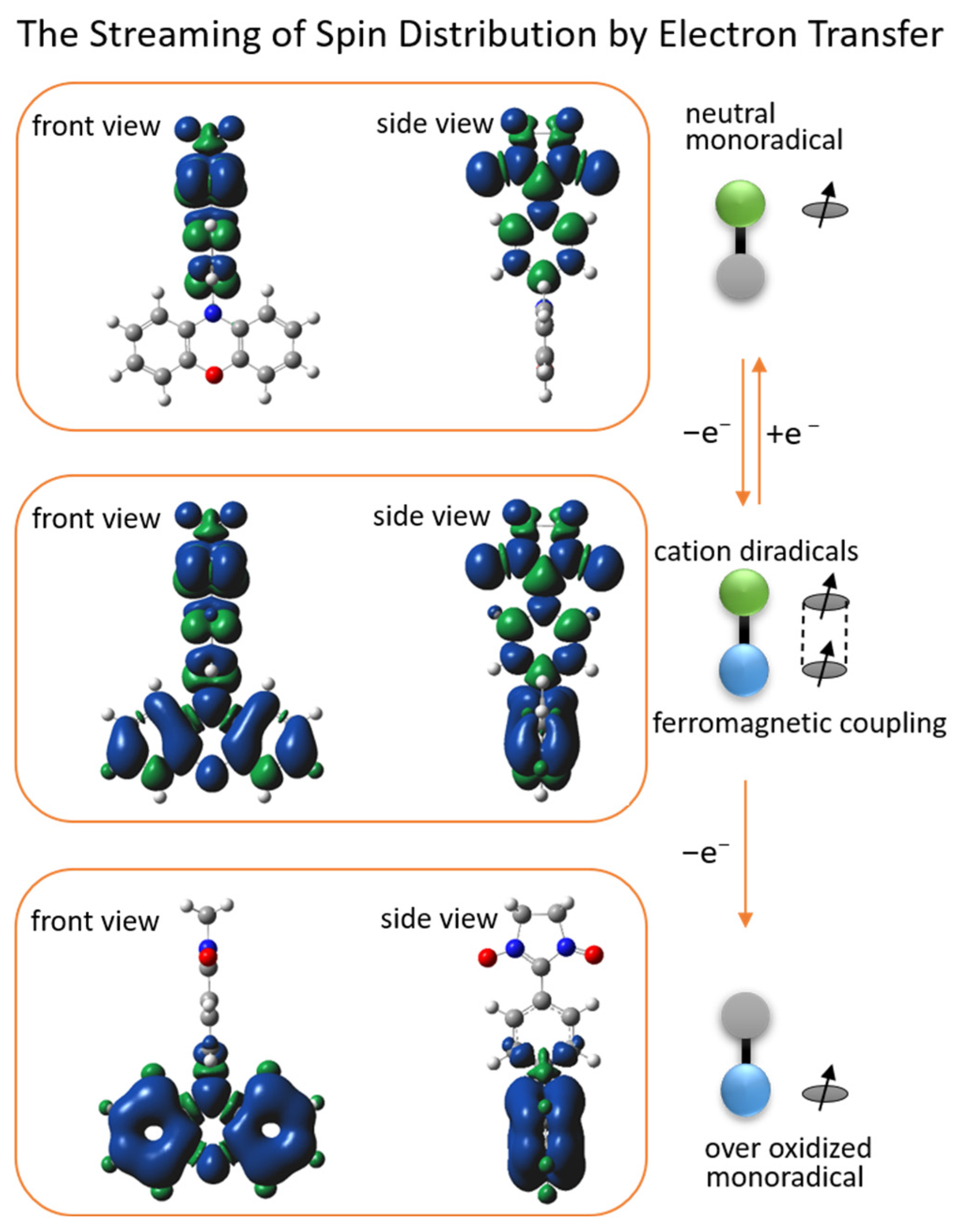
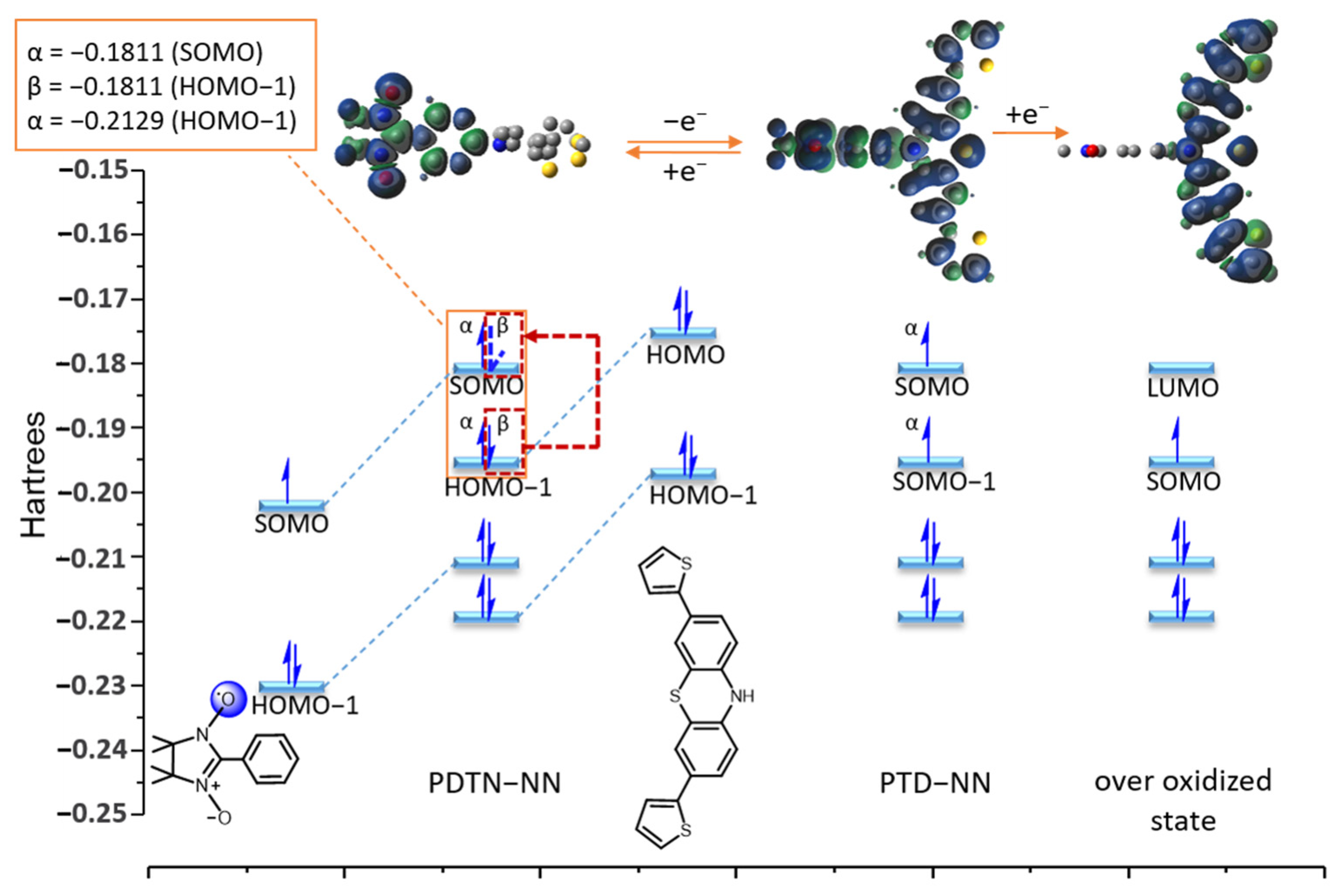
2.4. DFT Calculations of Redox Stimuli Responsive System
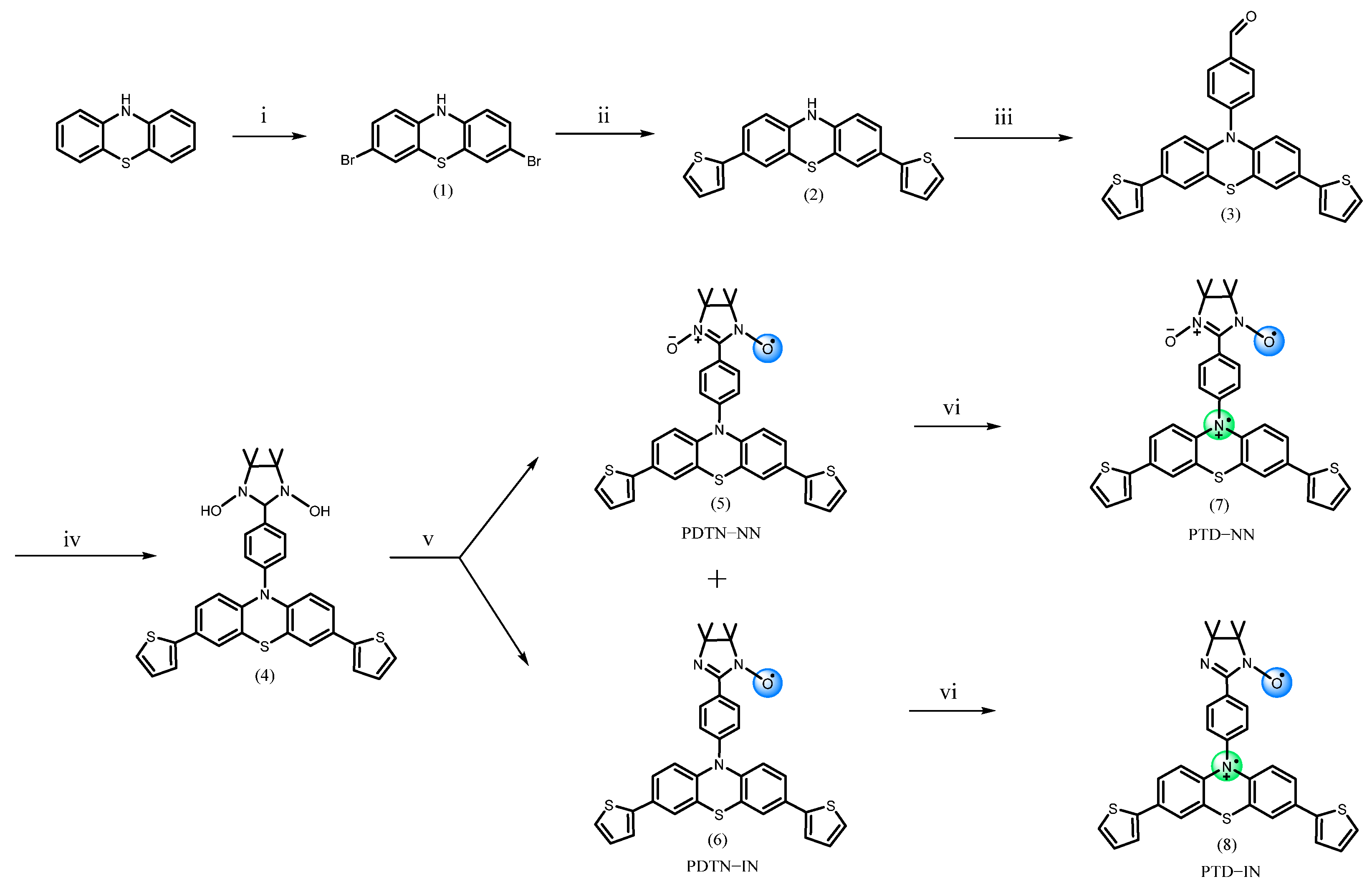
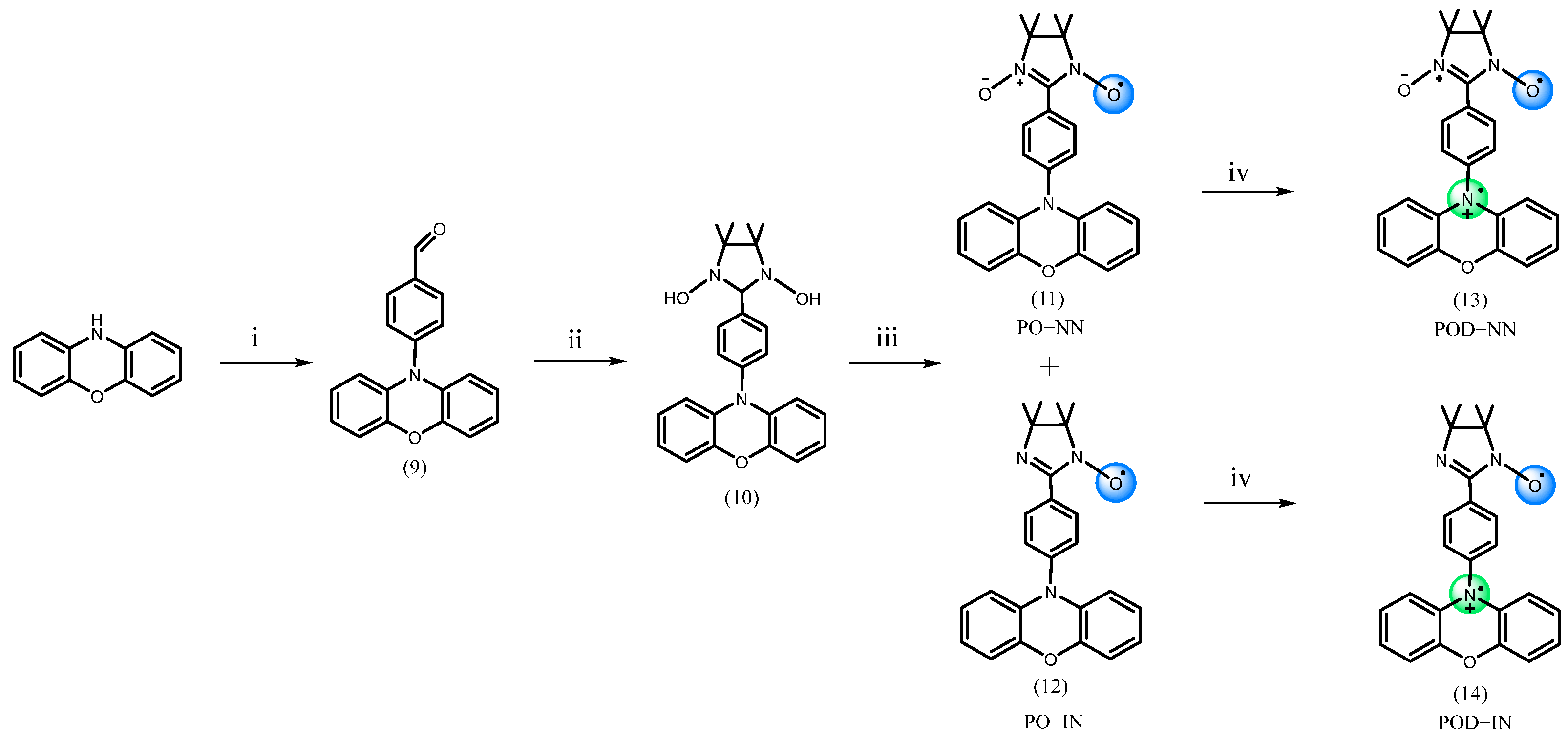
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolanji, K.; Baumgarten, M. Dithienopyrrole Derivatives with Nitronyl Nitroxide Radicals and Their Oxidation to Cation High-Spin Molecules. Chem. Eur. J. 2020, 26, 3626–3632. [Google Scholar] [CrossRef]
- Yokoyama, N.; Tanaka, N.; Fujimoto, N.; Tanaka, R.; Suzuki, S.; Shiomi, D.; Sato, K.; Takui, T.; Kozaki, M.; Okada, K. Syntheses and Properties of (Nitronyl nitroxide)-substituted Triphenylamine ortho-Bridged by Two Oxygen and Sulfur Atoms. Chem. Asian J. 2021, 16, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kuratsu, M.; Suzuki, S.; Kozaki, M.; Shiomi, D.; Sato, K.; Takui, T.; Kanzawa, T.; Hosokoshi, Y.; Lan, X.Z.; Miyazaki, Y.; et al. (Nitronyl Nitroxide)-Substituted Trioxytriphenylamine Radical Cation Tetrachlorogallate Salt: A 2p-Electron-Based Weak Ferromagnet Composed of a Triplet Diradical Cation. Chem. Asian J. 2012, 7, 1604–1609. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Ovcharenko, V.I. The Chemistry of Nitroxide Radicals in the Molecular Design of Magnets. Rus. Chem. Rev. 2009, 78, 971–1012. [Google Scholar] [CrossRef]
- Ionita, P.; Whitwood, A.C.; Gilbert, B.C. Synthesis and Characterization of Some Novel Hetero-diradicals Containing Linked Hydrazyl and Aminoxyl (Nitroxide) Moieties. J. Chem. Soc. Perkin Trans. 2001, 2, 1453–1462. [Google Scholar] [CrossRef]
- Ratera, I.; Veciana, J. Playing with Organic Radicals as Building Blocks for Functional Molecular Materials. Chem. Soc. Rev. 2012, 41, 303–349. [Google Scholar] [CrossRef]
- Wei, D.D.; Qin, Y.L.; Xu, Z.P.; Liu, J.; Chen, R.R.; Yu, Y.; Wang, D. Study of Molecular Dimer Morphology Based on Organic Spin Centers: Nitronyl Nitroxide Radicals. Molecules 2024, 29, 2042. [Google Scholar] [CrossRef]
- Yue, Z.; Liu, J.; Baumgarten, M.; Wang, D. Spirobifluorene Mediating the Spin−Spin Coupling of Nitronyl Nitroxide Diradicals. J. Phys. Chem. A 2023, 127, 1565–1575. [Google Scholar] [CrossRef]
- Wang, D.; Ma, Y.J.; Wolf, B.; Kokorin, A.I.; Baumgarten, M. Temperature-Dependent Intramolecular Spin Coupling Interactions of a Flexible Bridged Nitronyl Nitroxide Biradical in Solution. J. Phys. Chem. A 2018, 122, 574–581. [Google Scholar] [CrossRef]
- Wang, D.; Shi, C.F.; Baumgarten, M.; Wang, W.P.; Liu, J. Nonalternating π-System Mediated Spin Coupling in Azulene Nitronyl Nitroxide Diradicals. J. Org. Chem. 2024, 89, 12277–12285. [Google Scholar] [CrossRef]
- Luo, J.S.; Wan, Z.Q.; Jia, C.Y. Recent advances in phenothiazine-based dyes for dye-sensitiezed solar cells. Chin. Chem. Lett. 2016, 27, 1304–1318. [Google Scholar] [CrossRef]
- Wainwright, M. Tricyclic Cation Chromophores as Models for New Photoantimicrobials. J. Braz. Chem. Soc. 2015, 26, 2390–2404. [Google Scholar]
- Massie, S.P. The Chemistry of Phenotiazine. Chem. Rev. 1954, 54, 797–883. [Google Scholar] [CrossRef]
- Opperman, K.A.; Mecklenburg, S.L.; Meyer, T.J. Intramolecular, Photoinduced Electron Transfer in Ruthenium(II) Bipyridine-Quinone Complexes. Inorg. Chem. 1994, 33, 5295–5301. [Google Scholar] [CrossRef]
- Shruti; Dwivedi, J.; Kishore, D.; Sain, S. Recent Advancement in The Synthesis of Phenoxazine Derivatives and Their Analogues. Synth. Commun. 2018, 48, 1377–1402. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, S.Y.; Qiu, Y.F.; Li, Y.T.; Zhou, C.K.; Sui, Y.X.; Ma, J.; Wang, X.P. One-Electron Oxidation of an Organic Molecule by B(C6F5)3; Isolation and Structures of Stable Non-para-Substituted Triarylamine Cation Radical and Bis(triarylamine) Dication Diradicaloid. J. Am. Chem. Soc. 2013, 135, 14912–14915. [Google Scholar] [CrossRef] [PubMed]
- Oka, H. Synthesis and Magnetic Properties of Orthogonally Linked Phenothiazing Cation Radical Dimer and Tetramer. Org. Lett. 2010, 12, 448–451. [Google Scholar] [CrossRef]
- Okada, K.; Imakura, T.; Oda, M.; Murai, H. 10,10′-(m- and p-Pheny-lene)diphenothiazine Dications: Violatiion of a Topology Rule in Heterocyclic High-Spin π-Systems. J. Am. Chem. Soc. 1996, 118, 3047–3048. [Google Scholar] [CrossRef]
- Okamoto, T.; Kuratsu, M.; Kozaki, M.; Hirotsu, K.; Ichimura, A.; Matsushita, T.; Okada, K. Remarkable Structure Deformation in Phenothiazaine Trimer Radical Cation. Org. Lett. 2004, 6, 3493–3496. [Google Scholar] [CrossRef]
- Kuratsu, M.; Kozaki, M.; Okada, K. 2,2′:6′2′′:6′′,6-Trioxytriphenylamine: Synthesis and Properties of the Radical Cation and Neutral Species. Angew. Chem. Int. Ed. 2005, 44, 4056–4058. [Google Scholar] [CrossRef]
- Suzuki, S.; Nakamura, F.; Naota, T. Direct Synthetic Method for (Nitronyl Nitroxide)-substituted π-Electronic Compounds by Palladium-catalyzed Cross-Coupling Reaction with a Zinc Complex. Mater. Chem. Front. 2018, 2, 591–596. [Google Scholar] [CrossRef]
- Tahara, T.; Suzuki, S.; Kozaki, M.; Shiomi, D.; Sugisaki, K.; Sato, K.; Takui, T.; Miyake, Y.; Hosokoshi, Y.; Nojiri, H.; et al. Triplet Diradical Cation Salts Consisting of Phenothiazine Radical Cation and Nitronyl Nitroxide. Chem. Eur. J. 2019, 25, 7201–7209. [Google Scholar] [CrossRef] [PubMed]
- Kuratsu, M.; Suzuki, S.; Kozaki, M.; Shiomi, D.; Sato, K.; Takui, T.; Okada, K. Magnetic Interaction of Tri- and Di-oxytriphenylamine Radical Cation FeCl4 Salts. Inorg. Chem. 2007, 46, 10153–10157. [Google Scholar] [CrossRef]
- Suzuki, S.; Nagata, A.; Kuratsu, M.; Kozaki, M.; Tanaka, R.; Shiomi, D.; Sugisaki, K.; Toyota, K.; Sato, K.; Takui, T.; et al. Trinitroxide-Trioxytriphenylamine: Spin-State Conversion from Triradical Doublet to Diradical Cation Triplet by Oxidative Modulation of a p-Conjugated System. Angew. Chem. Int. Ed. 2012, 51, 3193–3197. [Google Scholar] [CrossRef]
- Sato, O.; Tao, J.; Zhang, Y.Z. Control of Magnetic Properties through External Stimuli. Angew. Chem. Int. Ed. 2007, 46, 2152–2187. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, S. Recent Progress toward the Exploitation of Organic Radical Compounds with Photo-Responsive Magnetic Properties. Chem. Soc. Rev. 2004, 33, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Jalilov, A.S.; Han, L.; Nelsen, S.F.; Guzei, I.A. Oxidation Products of Doubly Trimethylene-Bridged Tetrabenzyl p-Phenylenediamine Paracyclophane. J. Org. Chem. 2013, 78, 11373–11381. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Ismagilov, R.F.; Teki, Y. Comparison of the Singlet, Triplet Energy Gap of a Symmetrical Diradical Dication with ET Parameters Derived from Its Optical Spectrum. J. Am. Chem. Soc. 1998, 120, 2200–2201. [Google Scholar] [CrossRef]
- Barlow, S.; Risko, C.; Odom, S.A.; Zheng, S.J.; Coropceanu, V.; Beverina, L.; Brédas, J.L.; Marder, S.R. Tuning Delocalization in the Radical Cations of 1,4-Bis[4(diarylamino)styryl]benzenes, 2,5-Bis[4(diarylamino)styryl]thiophenes, and 2,5-Bis[4(diarylamino)styryl]pyrroles through Substituent Effects. J. Am. Chem. Soc. 2012, 134, 10146–10155. [Google Scholar] [CrossRef]
- Sato, K.; Yano, M.; Furuichi, M.; Shiomi, D.; Takui, T.; Abe, K.; Itoh, K.; Higuchi, A.; Katsuma, K.; Shirota, Y. Polycation High-Spin States of One- and Two-Dimensional (Diarylamino)benzenes, Prototypical Model Units for Purely Organic Ferromagnetic Metals As Studied by Pulsed ESR/ Electron Spin Transient Nutation Spectroscopy. J. Am. Chem. Soc. 1997, 119, 6607–6613. [Google Scholar] [CrossRef]
- Stickley, K.R.; Blackstock, S.C. Triplet Dication and Quartet Trication of a Triaminobenzene. J. Am. Chem. Soc. 1994, 116, 11576–11577. [Google Scholar] [CrossRef]
- Bushby, R.J.; McGill, D.R.; Ng, K.M.; Taylor, N. Disjoint and coextensive diradical diions. J. Chem. Soc. Perkin Trans. 2 1997, 2, 1405–1414. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Sakamaki, D.; Ito, A.; Tanaka, K.; Shiro, M. A Triphenylamine Double-Decker: From a Delocalized Radical Cation to a Diradical Dication with an Excited Triplet State. Angew. Chem. Int. Ed. 2012, 51, 9403–9406. [Google Scholar] [CrossRef] [PubMed]
- Fatila, E.M.; Clérac, R.; Rouzières, M.; Soldatov, D.V.; Jennings, M.; Preuss, K.E. High-Spin Ribbons and Antiferromagnetic Ordering of a MnII-Biradical-MnII Complex. J. Am. Chem. Soc. 2013, 135, 13298–13301. [Google Scholar] [CrossRef] [PubMed]
- Karpinska, J.; Starczewska, B.; Tarasiewicz, H.P. Analytical Properties of 2- and 10-Disubstituted Phenothiazine Derivatives. Anal. Sci. 1996, 12, 161–170. [Google Scholar] [CrossRef]
- Baumgarten, M.; Gügel, A. EPR and Optical Absorption Spectra of Reduced Buckminsterfullerene. Adv. Mater. 1993, 5, 458–461. [Google Scholar] [CrossRef]
- Rathore, R.; Burns, C.L. A Practical One-Pot Synthesis of Soluble Hexa-peri-hexabenzocoronene and Isolation of Its Cation-Radical Salt. J. Org. Chem. 2003, 68, 4071–4074. [Google Scholar] [CrossRef]
- Rosspeintner, A.; Griesser, M.; Matsumoto, I.; Teki, Y.; Li, G.Q.; Nelsen, S.F.; Gescheidt, G. EPR and ENDOR Studies of Dimeric Paracyclophane Radical Cations and Dications Containing Tri- and Pentamethylene-Bridged p-Phenylene Diamine Units. J. Phys. Chem. A. 2010, 114, 6487–6492. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Ismagilov, R.F.; Powell, D.R. Charge-Localized p-Phenylenedihydrazine Radical Cations: ESR and Optical Studies of Intramolecular Electron Transfer Rates. J. Am. Chem. Soc. 1997, 119, 10213–10222. [Google Scholar] [CrossRef]
- Maurel, V.; Skorka, L.; Onofrio, N.; Szewczyk, E.; Djurado, D.; Dubois, L.; Mouesca, J.M.; Kulszewicz-Bajer, I. Ferromagnetic Spin Coupling through the 3,4′-Biphenyl Moiety in Arylamine Oligomers Experimental and Computational Study. J. Phys. Chem. B 2014, 118, 7657–7667. [Google Scholar] [CrossRef]
- Fang, S.; Lee, M.S.; Hrovat, D.A.; Borden, W.T. Ab Initio Calculations Show Why m-Phenylene Is Not Always a Ferromagnetic Coupler. J. Am. Chem. Soc. 1995, 117, 6727–6731. [Google Scholar] [CrossRef]
- Mañeru, D.R.; Pal, A.K.; Moreira, I.P.R.; Datta, S.N.; Illas, F. The Triplet−Singlet Gap in the m-Xylylene Radical: A Not So Simple One. J. Chem. Theory Comput. 2014, 10, 335–345. [Google Scholar] [CrossRef]
- Hiraoka, S.; Okamoto, T.; Kozaki, M.; Shiomi, D.; Sato, K.; Takui, T.; Okada, K. A Stable Radical-Substituted Radical Cation with Strongly Ferromagnetic Interaction: Nitronyl Nitroxide-Substituted 5,10-Diphenyl-5,10-dihydrophenazine Radical Cation. J. Am. Chem. Soc. 2004, 126, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, V.I.; Fokin, S.V.; Romanenko, G.V.; Korobkov, I.V.; Rey, P. Synthesis of vicinal bishydroxylamine. Russ. Chem. Bull. 1999, 48, 1519–1525. [Google Scholar] [CrossRef]
- Rao, K.S.; Wu, T.S. Chan-Lam Coupling Reactions: Synthesis of Heterocycles. Tetrahedron 2012, 68, 7735–7754. [Google Scholar]
- Chan, D.M.T.; Monaco, K.L.; Wang, R.P.; Winters, M.P. New N- and O-Arylations with Phenylboronic Acids and Cupric Acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar] [CrossRef]
- Wexler, R.P.; Nuhant, P.; Senter, T.J.; Gale-Day, Z.J. Electrochemically Enabled Chan–Lam Couplings of Aryl Boronic Acids and Anilines. Org. Lett. 2019, 21, 4540–4543. [Google Scholar] [CrossRef]
- Guo, H.Q.; Peng, Q.M.; Chen, X.K.; Gu, Q.Y.; Dong, S.Z.; Evans, E.W.; Gillett, A.J.; Ai, X.; Zhang, M.; Credgington, D.; et al. High stability and luminescence efficiency in donor–acceptor neutral radicals not following the Aufbau principle. Nature Materials 2019, 18, 977–984. [Google Scholar] [CrossRef]
- Dong, X.; Luo, Q.C.; Zhao, Y.; Wang, T.; Sun, Q.C.; Pei, R.B.; Zhao, Y.; Zheng, Y.Z.; Wang, X. A Dynamic Triradical: Synthesis, Crystal Structure, and Spin Frustration. J. Am. Chem. Soc. 2023, 145, 17292–17298. [Google Scholar] [CrossRef]
- Bras, T.; Hsu, C.; Baum, T.Y.; Vogel, D.; Mayor, M.; van der Zant, H.S.J. Mechanically Stable Kondo Resonance in an Organic Radical Molecular Junction. J. Phys. Chem. C 2025, 129, 3152–3157. [Google Scholar] [CrossRef]
- Lang, Y.R.; Hei, Y.X.; Yu, T.R.; Yang, Y.T.; Chen, M.; Zhang, J.R.; Zhao, F.G.; Zheng, Y.H.; Liu, X.S. A donor–acceptor coupling unit modulates the spin coupling effect of a stable diradical. J. Mater. Chem. C 2024, 12, 8846–8851. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential [V] | PTP | PDTN | BP-NN | PDTN-NN | PDTN-IN | PO-NN | PO-IN |
|---|---|---|---|---|---|---|---|
| EOxi-1 | 0.63 | 0.61 | 0.91 | 0.78 | 0.68 | 0.77 | 0.62 |
| EOxi-2 | 1.42 | 1.25 | — | 0.93 | 1.25 | 0.91 | 1.23 |
| EOxi-3 | — | — | — | 1.37 | 1.28 | — | — |
| G Factor | Starting Compound | Mixture State | Over-Oxidized | Diradical Cation |
|---|---|---|---|---|
| PDTN-NN | 2.0071 | 2.0067 | 2.0053 | 2.0063 |
| PDTN-IN | 2.0064 | 2.0062 | 2.0052 | 2.0059 |
| PO-NN | 2.0071 | 2.0064 | 2.0034 | 2.0056 |
| PO-IN | 2.0063 | 2.0058 | 2.0035 | 2.0052 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Yao, D.; Li, X.; Shi, L.; Wang, C.; Li, J.; Kong, W.; Qin, Y.; Baumgarten, M. Oxidation-Triggered Formation of Diradical Cations from Paramagnetic Molecules and Their Spin Density Evolution. Molecules 2025, 30, 1931. https://doi.org/10.3390/molecules30091931
Wang D, Yao D, Li X, Shi L, Wang C, Li J, Kong W, Qin Y, Baumgarten M. Oxidation-Triggered Formation of Diradical Cations from Paramagnetic Molecules and Their Spin Density Evolution. Molecules. 2025; 30(9):1931. https://doi.org/10.3390/molecules30091931
Chicago/Turabian StyleWang, Di, Dan Yao, Xinyu Li, Lingli Shi, Chunyuan Wang, Jie Li, Weili Kong, Yongliang Qin, and Martin Baumgarten. 2025. "Oxidation-Triggered Formation of Diradical Cations from Paramagnetic Molecules and Their Spin Density Evolution" Molecules 30, no. 9: 1931. https://doi.org/10.3390/molecules30091931
APA StyleWang, D., Yao, D., Li, X., Shi, L., Wang, C., Li, J., Kong, W., Qin, Y., & Baumgarten, M. (2025). Oxidation-Triggered Formation of Diradical Cations from Paramagnetic Molecules and Their Spin Density Evolution. Molecules, 30(9), 1931. https://doi.org/10.3390/molecules30091931