

Production of Green Diesel via the Ni/Al Mo Hydrotalcite Catalyzed Deoxygenation of Rapeseed Oil



Abstract

1. Introduction
2. Results
2.1. Catalyst Synthesis
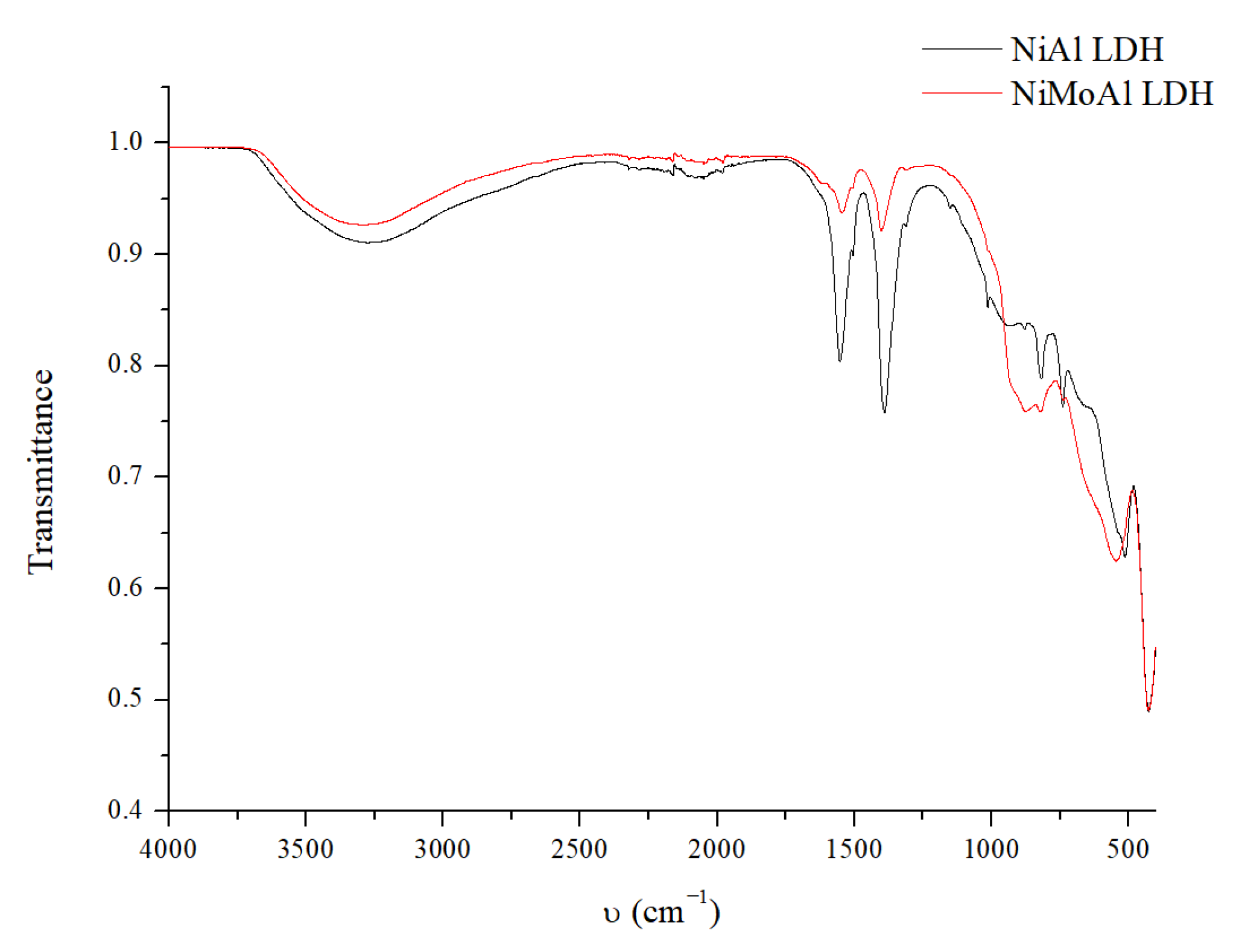
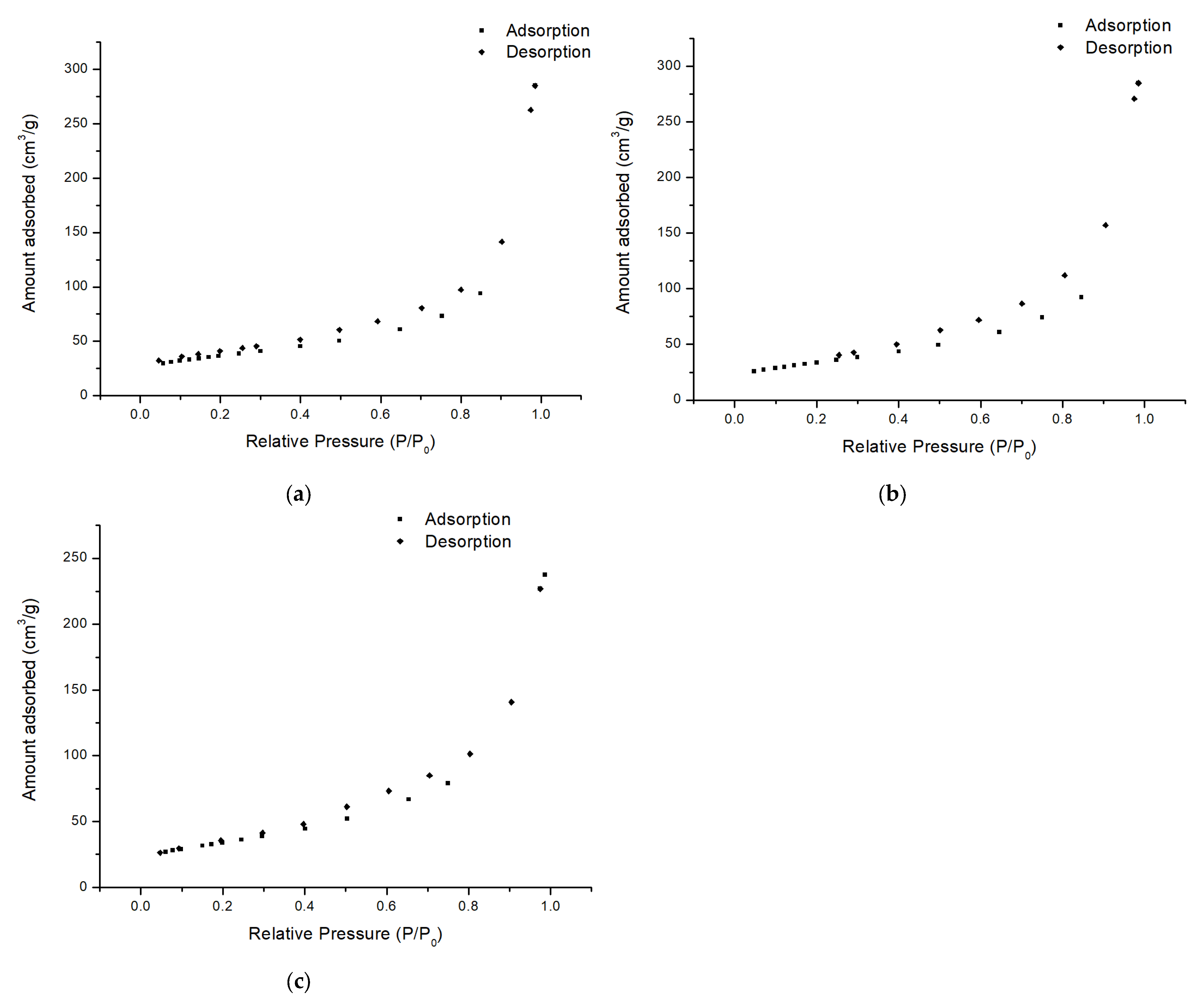
2.2. Catalyst Characterization
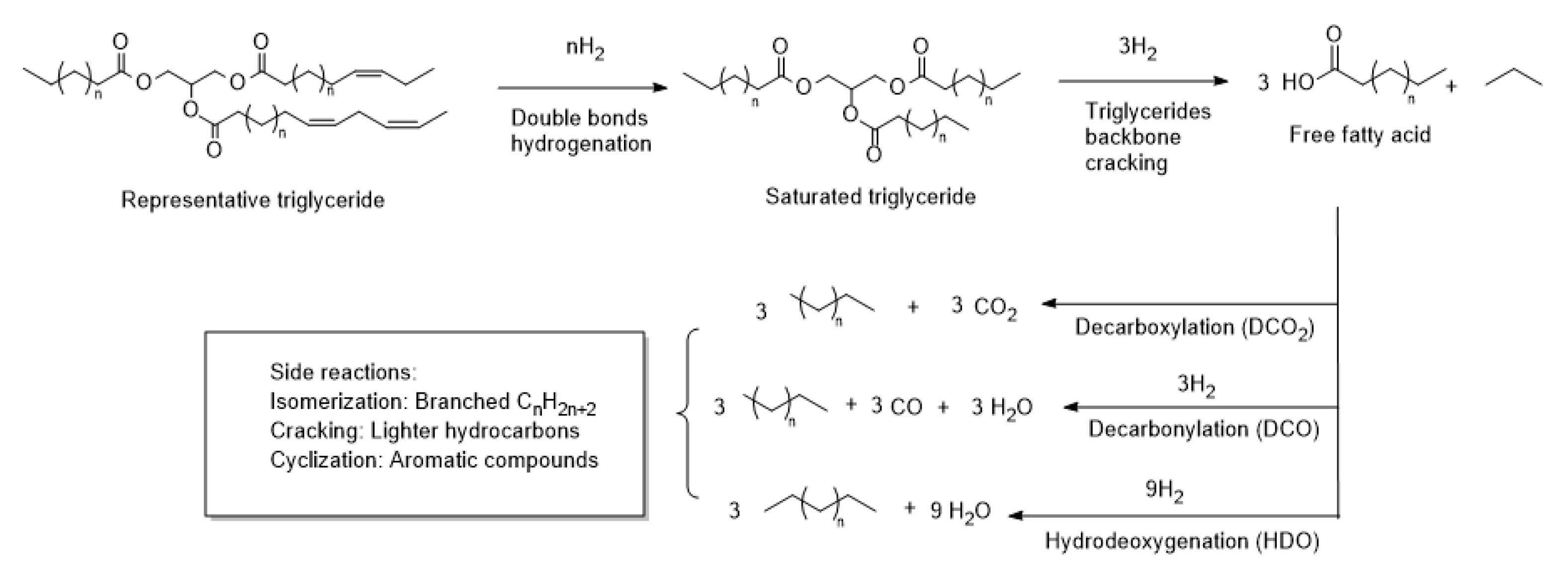
2.3. Catalytic Deoxygenation Reaction of Rapeseed Oil
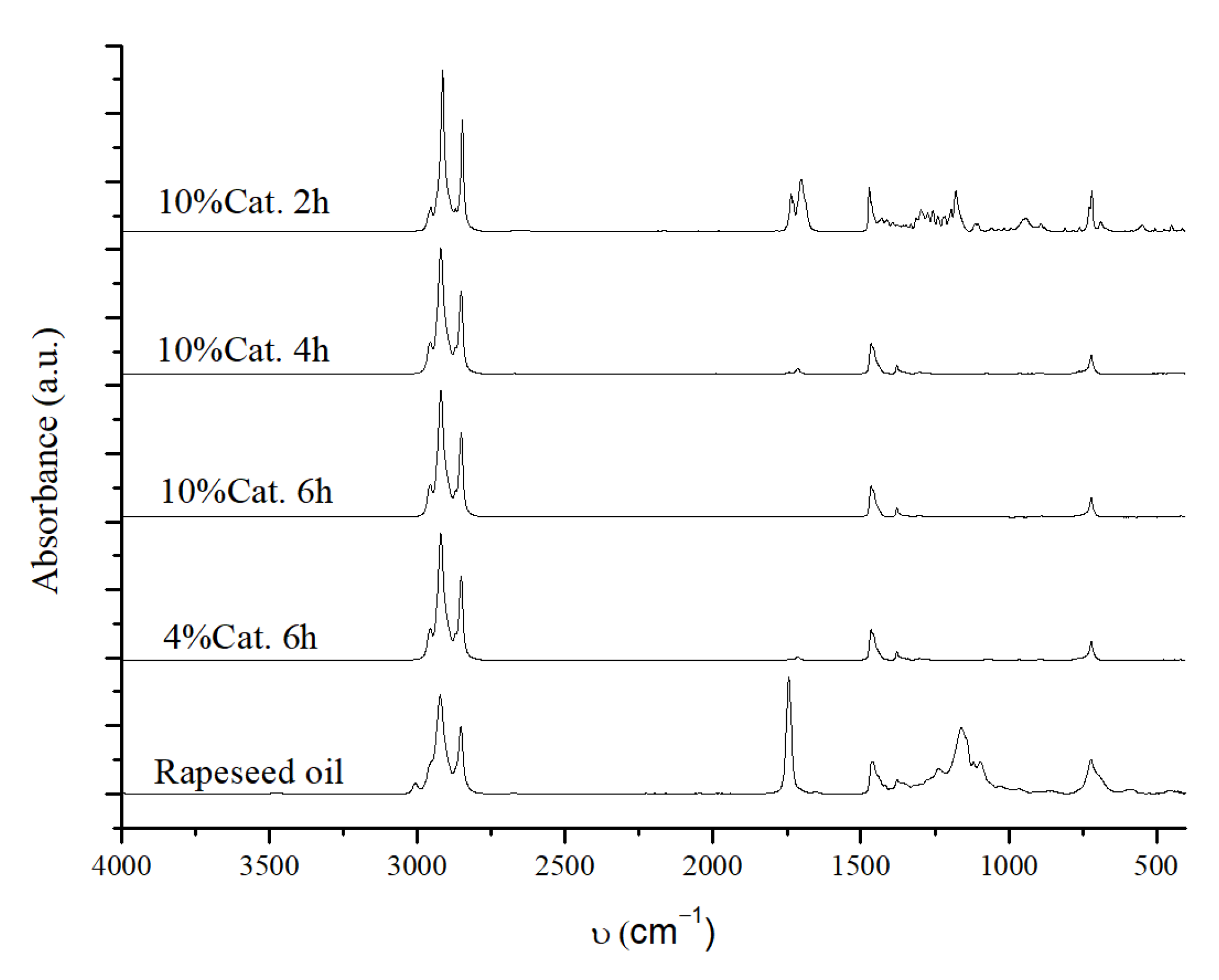
2.3.1. Reaction Conditions Screening
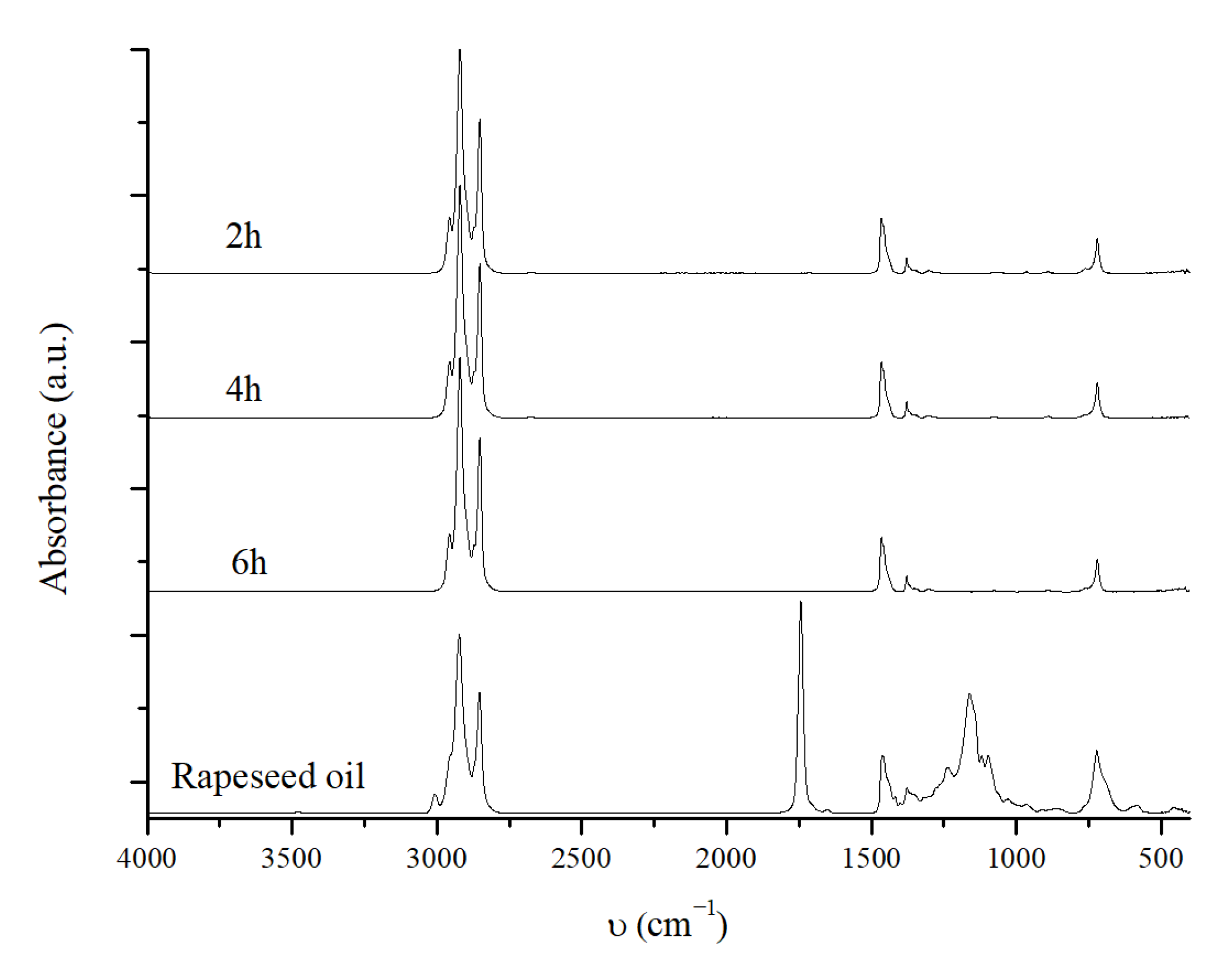
2.3.2. Reaction Time Effect
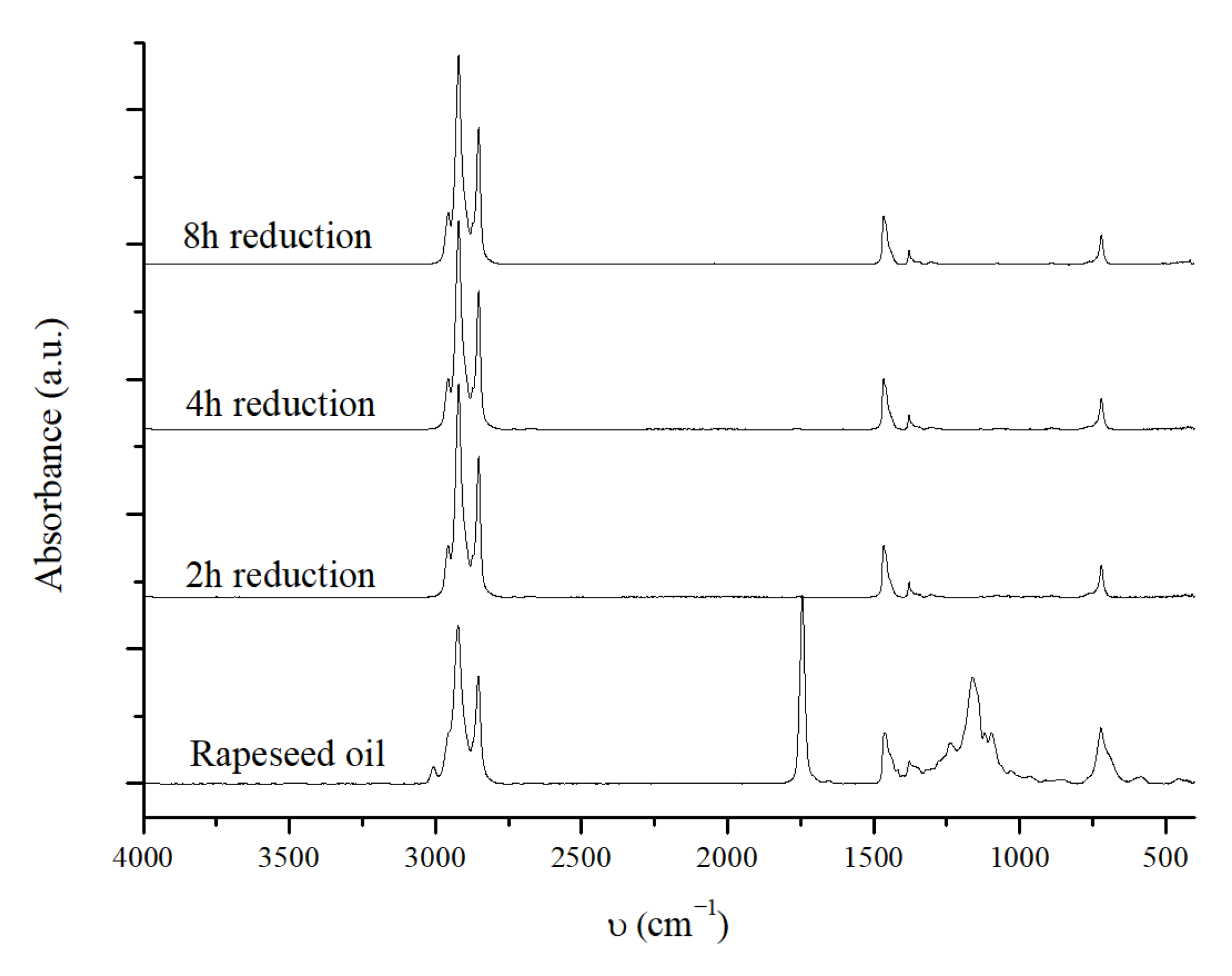
2.3.3. The Effect of the Reduction Time of the Catalyst
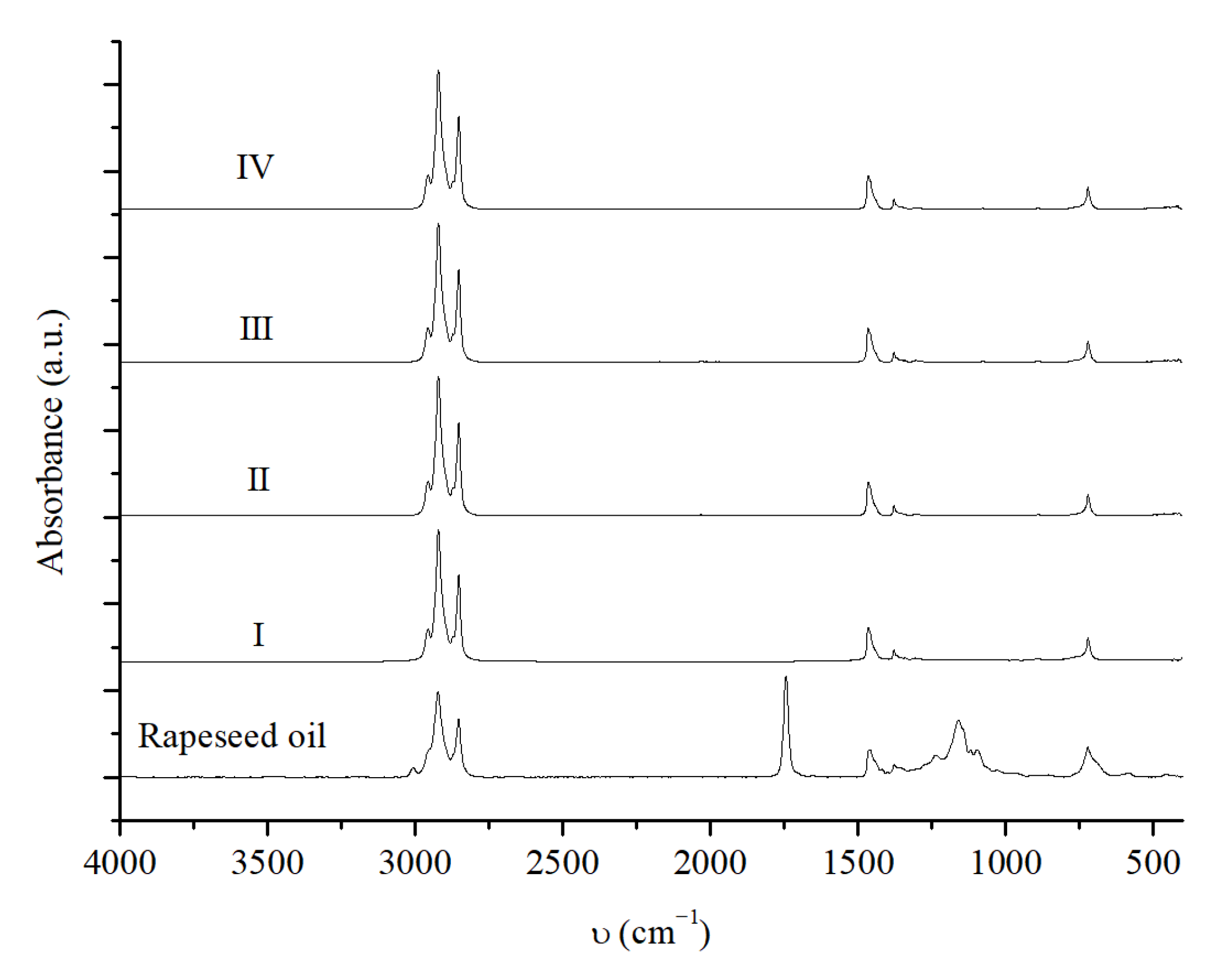
2.3.4. NiMoAl (0.6) R. Recycling Tests
2.3.5. Catalyst Activity Without the Pre-Reduction Step
2.4. Summary of Results
3. Materials and Methods
3.1. Catalyst Synthesis
3.2. Catalyst Characterization
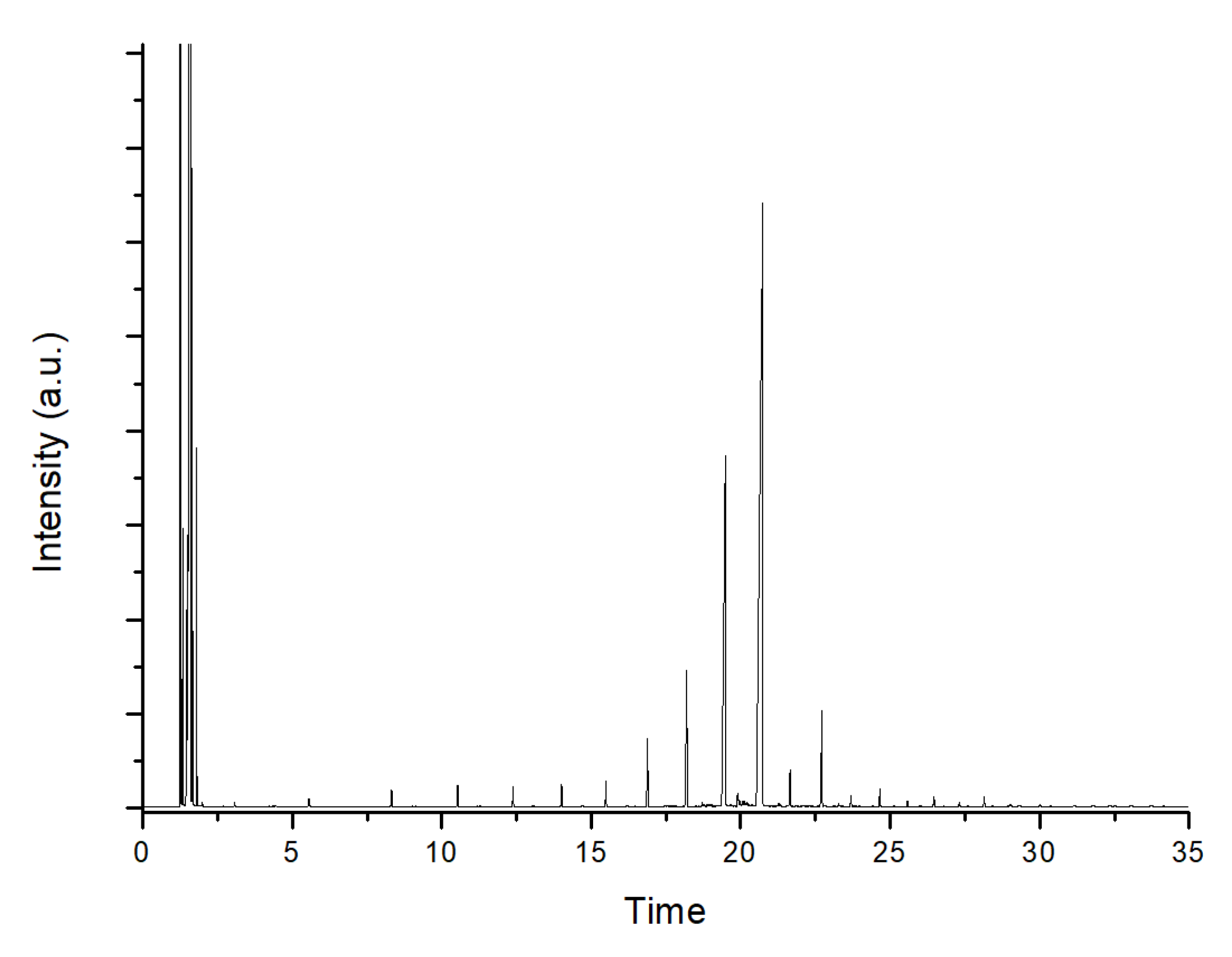
3.3. Catalytic Deoxygenation Reaction and Product Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- U.S. Energy Information Administration. International Energy Outlook 2023; U.S. Energy Information Administration: Washington, DC, USA, 2023.
- Maher, K.D.; Bressler, D.C. Pyrolysis of Triglyceride Materials for the Production of Renewable Fuels and Chemicals. Bioresour. Technol. 2007, 98, 2351–2368. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, L.; Cheng, S.; Julson, J. Review of Heterogeneous Catalysts for Catalytically Upgrading Vegetable Oils into Hydrocarbon Biofuels. Catalysts 2017, 7, 83. [Google Scholar] [CrossRef]
- Saha, B.C.; Woodward, J. (Eds.) Fuels and Chemicals from Biomass; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 666, ISBN 978-0-8412-3508-3. [Google Scholar]
- McKendry, P. Energy Production from Biomass (Part 1): Overview of Biomass. Bioresour. Technol. 2002, 83, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Chen, J.; Yang, Y.; Tian, S. Catalytic Deoxygenation of Methyl Laurate as a Model Compound to Hydrocarbons on Nickel Phosphide Catalysts: Remarkable Support Effect. Fuel Process. Technol. 2014, 118, 161–170. [Google Scholar] [CrossRef]
- Vonortas, A.; Papayannakos, N. Comparative Analysis of Biodiesel versus Green Diesel. WIREs Energy Environ. 2014, 3, 3–23. [Google Scholar] [CrossRef]
- Lucantonio, S.; Di Giuliano, A.; Rossi, L.; Gallucci, K. Green Diesel Production via Deoxygenation Process: A Review. Energies 2023, 16, 844. [Google Scholar] [CrossRef]
- Vonghia, E.; Boocock, D.G.B.; Konar, S.K.; Leung, A. Pathways for the Deoxygenation of Triglycerides to Aliphatic Hydrocarbons over Activated Alumina. Energy Fuels 1995, 9, 1090–1096. [Google Scholar] [CrossRef]
- Snåre, M.; Kubičková, I.; Mäki-Arvela, P.; Eränen, K.; Wärnå, J.; Murzin, D.Y. Production of Diesel Fuel from Renewable Feeds: Kinetics of Ethyl Stearate Decarboxylation. Chem. Eng. J. 2007, 134, 29–34. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Kubickova, I.; Snåre, M.; Eränen, K.; Murzin, D.Y. Catalytic Deoxygenation of Fatty Acids and Their Derivatives. Energy Fuels 2007, 21, 30–41. [Google Scholar] [CrossRef]
- Gosselink, R.W.; Hollak, S.A.W.; Chang, S.; van Haveren, J.; de Jong, K.P.; Bitter, J.H.; van Es, D.S. Reaction Pathways for the Deoxygenation of Vegetable Oils and Related Model Compounds. ChemSusChem 2013, 6, 1576–1594. [Google Scholar] [CrossRef]
- Hongloi, N.; Prapainainar, P.; Prapainainar, C. Review of Green Diesel Production from Fatty Acid Deoxygenation over Ni-Based Catalysts. Mol. Catal. 2022, 523, 111696. [Google Scholar] [CrossRef]
- Oh, M.; Jin, M.; Lee, K.; Kim, J.-C.; Ryoo, R.; Choi, M. Importance of Pore Size and Lewis Acidity of Pt/Al2O3 for Mitigating Mass Transfer Limitation and Catalyst Fouling in Triglyceride Deoxygenation. Chem. Eng. J. 2022, 439, 135530. [Google Scholar] [CrossRef]
- Di Vito Nolfi, G.; Gallucci, K.; Rossi, L. Green Diesel Production by Catalytic Hydrodeoxygenation of Vegetables Oils. Int. J. Environ. Res. Public Health 2021, 18, 13041. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sotelo-Boyás, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of Vegetable Oils to Produce Bio-Hydrogenated Diesel and Liquefied Petroleum Gas Fuel over Catalysts Containing Sulfided Ni–Mo and Solid Acids. Energy Fuels 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Kubička, D.; Kaluža, L. Deoxygenation of Vegetable Oils over Sulfided Ni, Mo and NiMo Catalysts. Appl. Catal. Gen. 2010, 372, 199–208. [Google Scholar] [CrossRef]
- Srifa, A.; Faungnawakij, K.; Itthibenchapong, V.; Viriya-empikul, N.; Charinpanitkul, T.; Assabumrungrat, S. Production of Bio-Hydrogenated Diesel by Catalytic Hydrotreating of Palm Oil over NiMoS2/γ-Al2O3 Catalyst. Bioresour. Technol. 2014, 158, 81–90. [Google Scholar] [CrossRef]
- Tiwari, R.; Rana, B.S.; Kumar, R.; Verma, D.; Kumar, R.; Joshi, R.K.; Garg, M.O.; Sinha, A.K. Hydrotreating and Hydrocracking Catalysts for Processing of Waste Soya-Oil and Refinery-Oil Mixtures. Catal. Commun. 2011, 12, 559–562. [Google Scholar] [CrossRef]
- Toba, M.; Abe, Y.; Kuramochi, H.; Osako, M.; Mochizuki, T.; Yoshimura, Y. Hydrodeoxygenation of Waste Vegetable Oil over Sulfide Catalysts. Catal. Today 2011, 164, 533–537. [Google Scholar] [CrossRef]
- Váchová, V.; Toullis, D.; Straka, P.; Šimáček, P.; Staš, M.; Gdovin, A.; Beňo, Z.; Blažek, J. Composition and Properties of Rapeseed Oil Hydrotreating Products over CoMo/Al2 O3 and NiMo/Al2 O3 Catalysts. Energy Fuels 2020, 34, 9609–9619. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Viljava, T.-R.; Krause, A.O.I. Effect of Sulphiding Agents on the Hydrodeoxygenation of Aliphatic Esters on Sulphided Catalysts. Appl. Catal. Gen. 2007, 326, 236–244. [Google Scholar] [CrossRef]
- Kubička, D.; Horáček, J. Deactivation of HDS Catalysts in Deoxygenation of Vegetable Oils. Appl. Catal. Gen. 2011, 394, 9–17. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Inaba, M.; Takahara, I. Production of Synthetic Diesel by Hydrotreatment of Jatropha Oils Using Pt−Re/H-ZSM-5 Catalyst. Energy Fuels 2010, 24, 2404–2409. [Google Scholar] [CrossRef]
- Berenblyum, A.S.; Podoplelova, T.A.; Shamsiev, R.S.; Katsman, E.A.; Danyushevsky, V.Y. On the Mechanism of Catalytic Conversion of Fatty Acids into Hydrocarbons in the Presence of Palladium Catalysts on Alumina. Pet. Chem. 2011, 51, 336–341. [Google Scholar] [CrossRef]
- Lestari, S.; Mäki-Arvela, P.; Eränen, K.; Beltramini, J.; Max Lu, G.Q.; Murzin, D.Y. Diesel-like Hydrocarbons from Catalytic Deoxygenation of Stearic Acid over Supported Pd Nanoparticles on SBA-15 Catalysts. Catal. Lett. 2010, 134, 250–257. [Google Scholar] [CrossRef]
- Chen, J.; Xu, Q. Hydrodeoxygenation of Biodiesel-Related Fatty Acid Methyl Esters to Diesel-Range Alkanes over Zeolite-Supported Ruthenium Catalysts. Catal. Sci. Technol. 2016, 6, 7239–7251. [Google Scholar] [CrossRef]
- Lestari, S.; Mäki-Arvela, P.; Simakova, I.; Beltramini, J.; Lu, G.Q.M.; Murzin, D.Y. Catalytic Deoxygenation of Stearic Acid and Palmitic Acid in Semibatch Mode. Catal. Lett. 2009, 130, 48–51. [Google Scholar] [CrossRef]
- Morgan, T.; Grubb, D.; Santillan-Jimenez, E.; Crocker, M. Conversion of Triglycerides to Hydrocarbons Over Supported Metal Catalysts. Top. Catal. 2010, 53, 820–829. [Google Scholar] [CrossRef]
- Veriansyah, B.; Han, J.Y.; Kim, S.K.; Hong, S.-A.; Kim, Y.J.; Lim, J.S.; Shu, Y.-W.; Oh, S.-G.; Kim, J. Production of Renewable Diesel by Hydroprocessing of Soybean Oil: Effect of Catalysts. Fuel 2012, 94, 578–585. [Google Scholar] [CrossRef]
- Harnos, S.; Onyestyák, G.; Kalló, D. Hydrocarbons from Sunflower Oil over Partly Reduced Catalysts. React. Kinet. Mech. Catal. 2012, 106, 99–111. [Google Scholar] [CrossRef]
- Asikin-Mijan, N.; Lee, H.V.; Abdulkareem-Alsultan, G.; Afandi, A.; Taufiq-Yap, Y.H. Production of Green Diesel via Cleaner Catalytic Deoxygenation of Jatropha Curcas Oil. J. Clean. Prod. 2017, 167, 1048–1059. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, C.; Kasakov, S.; Foraita, S.; Lercher, J.A. Manipulating Catalytic Pathways: Deoxygenation of Palmitic Acid on Multifunctional Catalysts. Chem.—Eur. J. 2013, 19, 4732–4741. [Google Scholar] [CrossRef]
- Peng, B.; Yao, Y.; Zhao, C.; Lercher, J.A. Towards Quantitative Conversion of Microalgae Oil to Diesel-Range Alkanes with Bifunctional Catalysts. Angew. Chem. Int. Ed. 2012, 51, 2072–2075. [Google Scholar] [CrossRef] [PubMed]
- Zulkepli, S.; Lee, H.V.; Rahman, N.A.; Chuan, L.T.; Show, P.L.; Chen, W.-H.; Juan, J.C. Highly Active Iron-Promoted Hexagonal Mesoporous Silica (HMS) for Deoxygenation of Triglycerides to Green Hydrocarbon-like Biofuel. Fuel 2022, 308, 121860. [Google Scholar] [CrossRef]
- Liu, J.; Liu, C.; Zhou, G.; Shen, S.; Rong, L. Hydrotreatment of Jatropha Oil over NiMoLa/Al2O3 Catalyst. Green Chem. 2012, 14, 2499. [Google Scholar] [CrossRef]
- Gousi, M.; Kordouli, E.; Bourikas, K.; Symianakis, E.; Ladas, S.; Kordulis, C.; Lycourghiotis, A. Green Diesel Production over Nickel-Alumina Nanostructured Catalysts Promoted by Copper. Energies 2020, 13, 3707. [Google Scholar] [CrossRef]
- Krár, M.; Kasza, T.; Kovács, S.; Kalló, D.; Hancsók, J. Bio Gas Oils with Improved Low Temperature Properties. Fuel Process. Technol. 2011, 92, 886–892. [Google Scholar] [CrossRef]
- Yenumala, S.R.; Maity, S.K.; Shee, D. Hydrodeoxygenation of Karanja Oil over Supported Nickel Catalysts: Influence of Support and Nickel Loading. Catal. Sci. Technol. 2016, 6, 3156–3165. [Google Scholar] [CrossRef]
- Wang, H.; Yan, S.; Salley, S.O.; Simon Ng, K.Y. Support Effects on Hydrotreating of Soybean Oil over NiMo Carbide Catalyst. Fuel 2013, 111, 81–87. [Google Scholar] [CrossRef]
- Horáček, J.; Akhmetzyanova, U.; Skuhrovcová, L.; Tišler, Z.; De Paz Carmona, H. Alumina-Supported MoNx, MoCx and MoPx Catalysts for the Hydrotreatment of Rapeseed Oil. Appl. Catal. B Environ. 2020, 263, 118328. [Google Scholar] [CrossRef]
- Du, X.; Zhou, K.; Zhou, L.; Lei, X.; Yang, H.; Li, D.; Hu, C. Efficient Catalytic Conversion of Jatropha Oil to High Grade Biofuel on Ni-Mo2C/MCM-41 Catalysts with Tuned Surface Properties. J. Energy Chem. 2021, 61, 425–435. [Google Scholar] [CrossRef]
- Monnier, J.; Sulimma, H.; Dalai, A.; Caravaggio, G. Hydrodeoxygenation of Oleic Acid and Canola Oil over Alumina-Supported Metal Nitrides. Appl. Catal. Gen. 2010, 382, 176–180. [Google Scholar] [CrossRef]
- Wang, H.; Yan, S.; Salley, S.O.; Ng, K.Y.S. Hydrocarbon Fuels Production from Hydrocracking of Soybean Oil Using Transition Metal Carbides and Nitrides Supported on ZSM-5. Ind. Eng. Chem. Res. 2012, 51, 10066–10073. [Google Scholar] [CrossRef]
- Skuhrovcová, L.; De Paz Carmona, H.; Tišler, Z.; Svobodová, E.; Michálková, M.; Strejcová, K.; Velvarská, R.; Akhmetzyanova, U. Synthesis of Sulfur-Free Co-Mo Nitride Catalysts for the Hydrotreating of Atmospheric Gasoil and Co-Processing of Rapeseed Oil. Mol. Catal. 2023, 537, 112930. [Google Scholar] [CrossRef]
- Song, Y.; Beaumont, S.K.; Zhang, X.; Wilson, K.; Lee, A.F. Catalytic Applications of Layered Double Hydroxides in Biomass Valorisation. Curr. Opin. Green Sustain. Chem. 2020, 22, 29–38. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-Type Anionic Clays: Preparation, Properties and Applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Liao, C.; Liu, X.; Ren, Y.; Gong, D.; Zhang, Z. Catalytic Deoxygenation of Vanillin over Layered Double Hydroxide Supported Pd Catalyst. J. Ind. Eng. Chem. 2018, 68, 380–386. [Google Scholar] [CrossRef]
- Do Nascimento, L.A.; Barroso-Martín, L.; Peçanha, S.R.S.; Arias, S.; Santos, B.S.; Pacheco, J.G.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Barros, I.D.C.L. NiAlCe Mixed Oxides Obtained from Layered Double Hydroxides Applied to Anisole Hydrodeoxygenation. Catal. Today 2022, 394–396, 282–294. [Google Scholar] [CrossRef]
- Rizescu, C.; Sun, C.; Popescu, I.; Urdă, A.; Da Costa, P.; Marcu, I.-C. Hydrodeoxygenation of Benzyl Alcohol on Transition-Metal-Containing Mixed Oxides Catalysts Derived from Layered Double Hydroxide Precursors. Catal. Today 2021, 366, 235–244. [Google Scholar] [CrossRef]
- Zeng, Y.; Lin, L.; Hu, D.; Jiang, Z.; Saeed, S.; Guo, R.; Ashour, I.; Yan, K. Highly Dispersed Ru Nanoparticles Anchored on NiAl Layered Double Oxides Catalyst for Selective Hydrodeoxygenation of Vanillin. Catal. Today 2023, 423, 114252. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, A.; Yang, L.; Fan, G.; Li, F. Supported Ru Nanocatalyst over Phosphotungstate Intercalated Zn-Al Layered Double Hydroxide Derived Mixed Metal Oxides for Efficient Hydrodeoxygenation of Guaiacol. Mol. Catal. 2022, 528, 112503. [Google Scholar] [CrossRef]
- Hu, H.; Ma, D.; Yi, M.; Li, H.; Yuan, Z.; Liu, J.; Luo, Q.; Zhong, Q.; Wei, J.; Liu, G.; et al. Unveiling the Enhancement of Catalytic Activity and Stability by Single-Atom Mo Anchored to Cationic Vacancy for the Catalytic Hydrodeoxygenation of Lignin to Naphthenes. Chem. Eng. J. 2025, 505, 159303. [Google Scholar] [CrossRef]
- Na, J.-G.; Yi, B.E.; Kim, J.N.; Yi, K.B.; Park, S.-Y.; Park, J.-H.; Kim, J.-N.; Ko, C.H. Hydrocarbon Production from Decarboxylation of Fatty Acid without Hydrogen. Catal. Today 2010, 156, 44–48. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, R.; Chen, J. Catalytic Deoxygenation of Methyl Laurate as a Model Compound to Hydrocarbons on Hybrid Catalysts Composed of Ni–Zn Alloy and HY Zeolite. J. Chem. Technol. Biotechnol. 2019, 94, 1777–1787. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, F.; Jiang, J.; Zhu, H.; Du, Y.; Feng, J.; Li, H.; Jiang, X. LDH Derived Co-Al Nanosheet for Lipid Hydrotreatment to Produce Green Diesel. Fuel 2023, 333, 126341. [Google Scholar] [CrossRef]
- Qian, Z.; Wang, L.; Da, W.; Zhang, G.; Tian, Z.; Zou, Q.; Yang, Y.; Wei, M. Highly-Efficient NiCo Alloy Catalysts towards Hydrodeoxygenation of Fatty Acids to Prepare Alkanes. Mol. Catal. 2025, 572, 114754. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Morgan, T.; Loe, R.; Crocker, M. Continuous Catalytic Deoxygenation of Model and Algal Lipids to Fuel-like Hydrocarbons over Ni–Al Layered Double Hydroxide. Catal. Today 2015, 258, 284–293. [Google Scholar] [CrossRef]
- Morgan, T.; Santillan-Jimenez, E.; Harman-Ware, A.E.; Ji, Y.; Grubb, D.; Crocker, M. Catalytic Deoxygenation of Triglycerides to Hydrocarbons over Supported Nickel Catalysts. Chem. Eng. J. 2012, 189–190, 346–355. [Google Scholar] [CrossRef]
- Arias, S.; Eon, J.G.; San Gil, R.A.S.; Licea, Y.E.; Palacio, L.A.; Faro, A.C. Synthesis and Characterization of Terephthalate-Intercalated NiAl Layered Double Hydroxides with High Al Content. Dalton Trans 2013, 42, 2084–2093. [Google Scholar] [CrossRef]
- Arias, S.; Licea, Y.E.; Palacio, L.A.; Faro, A.C. Unsupported NiMoAl Hydrotreating Catalysts Prepared from NiAl-Terephthalate Hydrotalcites Exchanged with Heptamolybdate. Catal. Today 2013, 213, 198–205. [Google Scholar] [CrossRef]
- Li, C.; Zhang, X.; Chen, X.; Peng, Z.; Tsang, C.-W.; Liang, C. NiMoAl Catalysts Derived from Heptamolybdate-Intercalated Layered Double Hydroxides for Hydrodeoxygenation of Anisole. BMC Chem. Eng. 2019, 1, 14. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Arias, S.; Licea, Y.E.; Soares, D.; Eon, J.G.; Palacio, L.A.; Faro, A.C. Mixed NiMo, NiW and NiMoW Sulfides Obtained from Layered Double Hydroxides as Catalysts in Simultaneous HDA and HDS Reactions. Catal. Today 2017, 296, 187–196. [Google Scholar] [CrossRef]
- Firestone, D.; Yurawecz, M. AOAC Official Methods of Analysis; AOAC International: Rockville, MD, USA, 2002; Volume 41. [Google Scholar]
- Kaewchada, A.; Akkarawatkhoosith, N.; Bunpim, D.; Bangjang, T.; Ngamcharussrivichai, C.; Jaree, A. Production of Bio-Hydrogenated Diesel from Palm Oil Using Rh/HZSM-5 in a Continuous Mini Fixed-Bed Reactor. Chem. Eng. Process.-Process Intensif. 2021, 168, 108586. [Google Scholar] [CrossRef]
- Wang, C.; Tian, Z.; Wang, L.; Xu, R.; Liu, Q.; Qu, W.; Ma, H.; Wang, B. One-Step Hydrotreatment of Vegetable Oil to Produce High Quality Diesel-Range Alkanes. ChemSusChem 2012, 5, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.E.; Mohamed, A.R.; Kay Lup, A.N.; Koh, M.K. Palm Fatty Acid Distillate Derived Biofuels via Deoxygenation: Properties, Catalysts and Processes. Fuel Process. Technol. 2022, 236, 107394. [Google Scholar] [CrossRef]
- Kiméné, A.; Wojcieszak, R.; Paul, S.; Dumeignil, F. Catalytic Decarboxylation of Fatty Acids to Hydrocarbons over Non-noble Metal Catalysts: The State of the Art. J. Chem. Technol. Biotechnol. 2019, 94, 658–669. [Google Scholar] [CrossRef]
- Satyarthi, J.K.; Srinivas, D. Fourier Transform Infrared Spectroscopic Method for Monitoring Hydroprocessing of Vegetable Oils To Produce Hydrocarbon-Based Biofuel. Energy Fuels 2011, 25, 3318–3322. [Google Scholar] [CrossRef]
- Šimáček, P.; Kubička, D.; Šebor, G.; Pospíšil, M. Hydroprocessed Rapeseed Oil as a Source of Hydrocarbon-Based Biodiesel. Fuel 2009, 88, 456–460. [Google Scholar] [CrossRef]
- Madsen, A.T.; Ahmed, E.H.; Christensen, C.H.; Fehrmann, R.; Riisager, A. Hydrodeoxygenation of Waste Fat for Diesel Production: Study on Model Feed with Pt/Alumina Catalyst. Fuel 2011, 90, 3433–3438. [Google Scholar] [CrossRef]
- Krár, M.; Kovács, S.; Kalló, D.; Hancsók, J. Fuel Purpose Hydrotreating of Sunflower Oil on CoMo/Al2O3 Catalyst. Bioresour. Technol. 2010, 101, 9287–9293. [Google Scholar] [CrossRef]
- Yang, Y.; Gilbert, A.; Xu, C. Hydrodeoxygenation of Bio-Crude in Supercritical Hexane with Sulfided CoMo and CoMoP Catalysts Supported on MgO: A Model Compound Study Using Phenol. Appl. Catal. Gen. 2009, 360, 242–249. [Google Scholar] [CrossRef]
- Xu, C.; Hamilton, S.; Mallik, A.; Ghosh, M. Upgrading of Athabasca Vacuum Tower Bottoms (VTB) in Supercritical Hydrocarbon Solvents with Activated Carbon-Supported Metallic Catalysts. Energy Fuels 2007, 21, 3490–3498. [Google Scholar] [CrossRef]
- Sotelo-Boyás, R.; Liu, Y.; Minowa, T. Renewable Diesel Production from the Hydrotreating of Rapeseed Oil with Pt/Zeolite and NiMo/Al2 O3 Catalysts. Ind. Eng. Chem. Res. 2011, 50, 2791–2799. [Google Scholar] [CrossRef]
- Lee, S.-P.; Ramli, A. Methyl Oleate Deoxygenation for Production of Diesel Fuel Aliphatic Hydrocarbons over Pd/SBA-15 Catalysts. Chem. Cent. J. 2013, 7, 149. [Google Scholar] [CrossRef]
- Nimkarde, M.R.; Vaidya, P.D. Toward Diesel Production from Karanja Oil Hydrotreating over CoMo and NiMo Catalysts. Energy Fuels 2016, 30, 3107–3112. [Google Scholar] [CrossRef]
- Anand, M.; Sinha, A.K. Temperature-Dependent Reaction Pathways for the Anomalous Hydrocracking of Triglycerides in the Presence of Sulfided Co–Mo-Catalyst. Bioresour. Technol. 2012, 126, 148–155. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Morgan, T.; Lacny, J.; Mohapatra, S.; Crocker, M. Catalytic Deoxygenation of Triglycerides and Fatty Acids to Hydrocarbons over Carbon-Supported Nickel. Fuel 2013, 103, 1010–1017. [Google Scholar] [CrossRef]
- Vázquez-Garrido, I.; López-Benítez, A.; Guevara-Lara, A.; Berhault, G. Synthesis of NiMo Catalysts Supported on Mn-Al2O3 for Obtaining Green Diesel from Waste Soybean Oil. Catal. Today 2021, 365, 327–340. [Google Scholar] [CrossRef]
- Arora, P.; Ojagh, H.; Woo, J.; Lind Grennfelt, E.; Olsson, L.; Creaser, D. Investigating the Effect of Fe as a Poison for Catalytic HDO over Sulfided NiMo Alumina Catalysts. Appl. Catal. B Environ. 2018, 227, 240–251. [Google Scholar] [CrossRef]
- Cheah, Y.W.; Salam, M.A.; Sebastian, J.; Ghosh, S.; Arora, P.; Öhrman, O.; Olsson, L.; Creaser, D. Upgrading of Triglycerides, Pyrolysis Oil, and Lignin over Metal Sulfide Catalysts: A Review on the Reaction Mechanism, Kinetics, and Catalyst Deactivation. J. Environ. Chem. Eng. 2023, 11, 109614. [Google Scholar] [CrossRef]
- Arora, P.; Abdolahi, H.; Cheah, Y.W.; Salam, M.A.; Grennfelt, E.L.; Rådberg, H.; Creaser, D.; Olsson, L. The Role of Catalyst Poisons during Hydrodeoxygenation of Renewable Oils. Catal. Today 2021, 367, 28–42. [Google Scholar] [CrossRef]
- Wang, H.; Li, G.; Rogers, K.; Lin, H.; Zheng, Y.; Ng, S. Hydrotreating of Waste Cooking Oil over Supported CoMoS Catalyst—Catalyst Deactivation Mechanism Study. Mol. Catal. 2017, 443, 228–240. [Google Scholar] [CrossRef]
- Akhmetzyanova, U.; Tišler, Z.; Sharkov, N.; Skuhrovcová, L.; Pelíšková, L.; Kikhtyanin, O.; Mäki-Arvela, P.; Opanasenko, M.; Peurla, M.; Murzin, D.Y. Molybdenum Nitrides, Carbides and Phosphides as Highly Efficient Catalysts for the (Hydro)Deoxygenation Reaction. ChemistrySelect 2019, 4, 8453–8459. [Google Scholar] [CrossRef]
- De Paz Carmona, H.; Akhmetzyanova, U.; Tišler, Z.; Vondrová, P. Hydrotreating Atmospheric Gasoil and Co-Processing with Rapeseed Oil Using Supported Ni-Mo and Co-Mo Carbide Catalysts. Fuel 2020, 268, 117363. [Google Scholar] [CrossRef]
- Qin, Y.; He, L.; Duan, J.; Chen, P.; Lou, H.; Zheng, X.; Hong, H. Carbon-Supported Molybdenum-Based Catalysts for the Hydrodeoxygenation of Maize Oil. ChemCatChem 2014, 6, 2698–2705. [Google Scholar] [CrossRef]
- Domínguez-Barroso, M.V.; Herrera, C.; Larrubia, M.A.; Alemany, L.J. Diesel Oil-like Hydrocarbon Production from Vegetable Oil in a Single Process over Pt–Ni/Al2O3 and Pd/C Combined Catalysts. Fuel Process. Technol. 2016, 148, 110–116. [Google Scholar] [CrossRef]
- Zharova, P.A.; Chistyakov, A.V.; Shapovalov, S.S.; Pasynskii, A.A.; Tsodikov, M.V. Original Pt-Sn/Al2O3 Catalyst for Selective Hydrodeoxygenation of Vegetable Oils. Energy 2019, 172, 18–25. [Google Scholar] [CrossRef]
- Srifa, A.; Faungnawakij, K.; Itthibenchapong, V.; Assabumrungrat, S. Roles of Monometallic Catalysts in Hydrodeoxygenation of Palm Oil to Green Diesel. Chem. Eng. J. 2015, 278, 249–258. [Google Scholar] [CrossRef]
- UNI 2331-1:1980; Tele Metalliche per Vagliatura. Fili Metallici. ISO: Geneva, Switzerland, 1980.
- ISO 565:1990; Test Sieves—Metal Wire Cloth, Perforated Metal Plate and Electroformed Sheet—Nominal Sizes of Openings. ISO: Geneva, Switzerland, 1990.
- Maryutina, T.A.; Savonina, E.Y.; Fedotov, P.S.; Smith, R.M.; Siren, H.; Hibbert, D.B. Terminology of Separation Methods (IUPAC Recommendations 2017). Pure Appl. Chem. 2018, 90, 181–231. [Google Scholar] [CrossRef]
- Kordouli, E.; Lycourghiotis, S.; Bourikas, K.; Lycourghiotis, A.; Kordulis, C. Renewable Diesel Synthesis by Hydro-Processing in Green Solvents. Curr. Opin. Green Sustain. Chem. 2024, 48, 100936. [Google Scholar] [CrossRef]
- Hongloi, N.; Rahman, T.; Feyzbar-Khalkhali-Nejad, F.; Prapainainar, C.; Wongsurakul, P.; Aransiola, E.; Zhang, L.; Bargiela, P.; Baltrusaitis, J.; Prapainainar, P.; et al. Palm Oil Deoxygenation with Glycerol as a Hydrogen Donor for Renewable Fuel Production Using Nickel-Molybdenum Catalysts: The Effect of Support. Fuel Process. Technol. 2025, 270, 108196. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Al (wt%) | Ni (wt%) | Mo (wt%) | Ni/Al 1 (Exp.) | Ni/Al 1 (Theor.) | Ni/Mo 1 (Exp.) | Ni/Mo (Theor.) | Experimental Formula | Theoretical Formula |
|---|---|---|---|---|---|---|---|---|---|
| NiMoAl C. | 15.4 ± 0.03 | 21.1 ± 0.2 | 21.3 ± 0.01 | 0.63 | 0.63 | 1.63 | 0.57 | Ni16Al26Mo10O85 | Ni13Al20Mo22O109 |
| Sample | BET 1 (m2/g) | BJH 2 (cm3/g) | Average Pore Diameter 3 (nm) |
|---|---|---|---|
| NiAl LDH | 130 | 0.43 | 13.2 |
| NiMoAl LDH C. | 120 | 0.44 | 14.5 |
| NiMoAl LDH R. | 122 | 0.37 | 12.2 |
| Entry | Reaction Conditions (T °C/Pbar/wt% Cat) | Olefines (%) | n-C8-C14 (%) | n-C15-C18 (%) | >C18 (%) | Branch (%) | FAME (%) | Other (%) | S | χ (%) | OLP Yield (wt%) | GD Yield (wt%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 320/40/10 | -- | 1.7 | 94.1 | 2.0 | 2.2 | -- | -- | 1.8 | 100 | 69.1 | 65.7 |
| 2 | 320/20/10 | 8.7 | 7.0 | 70.5 | 6.9 | 4.5 | -- | 2.4 | 2.4 | 100 | 75.7 | 53.4 |
| 3 | 320/40/4 | -- | 3.1 | 92.0 | 2.8 | 2.1 | -- | -- | 2.7 | 100 | 72.9 | 67.1 |
| 4 | 320/20/4 | 18 | 6.6 | 66.5 | 2.8 | 1.8 | -- | 4.3 | 2.3 | 100 | 74.7 | 49.7 |
| 5 | 280/30/7 | 1.9 | 3.1 | 86.4 | 4.0 | 0.6 | 4.0 | -- | 1.3 | 97 | 88.3 | 76.3 |
| 6 | 240/40/10 | -- | 0.1 | 88.2 | 2.4 | 0.1 | 7.2 | 2.0 | 0.7 | 94 | 85.6 | 75.6 |
| 7 | 240/20/10 | -- | 0.2 | 37.8 | 0.6 | -- | 57.1 | 4.3 | 1.0 | 45 | 95.7 | 36.0 |
| 8 | 240/40/4 | 0.1 | -- | 30.3 | 0.8 | -- | 64.3 | 4.5 | 0.8 | 37 | 97.4 | 29.5 |
| 9 | 240/20/4 | 0.1 | -- | 14.6 | 1.0 | -- | 77.2 | 7.1 | 0.8 | 25 | 97.1 | 14.1 |
| Entry | Reaction Time (h) | n-C8-C14 (%) | n-C15-C18 (%) | >C18 (%) | Branch (%) | S | χ (%) | OLP Yield (wt%) | GD Yield (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.0 2 | 2.3 | 91.0 | 2.9 | 1.5 | 2.2 | 100 | 81.2 | 73.6 |
| 2 | 4.0 | 2.3 | 93.1 | 3.2 | 1.3 | 2.1 | 100 | 76.9 | 71.6 |
| 3 | 6.0 | 3.1 | 92.0 | 3.8 | 2.1 | 2.7 | 100 | 72.9 | 67.1 |
| Entry | Reduction Time (h) | n-C8-C14 (%) | n-C15-C18 (%) | >C18 (%) | Branch (%) | S | χ (%) | OLP Yield (wt%) | GD Yield (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.0 | 2.6 | 92.1 | 4.1 | 1.2 | 1.6 | 100 | 81.1 | 75.5 |
| 2 | 4.0 | 2.4 | 93.2 | 2.8 | 1.6 | 2.1 | 100 | 76.0 | 70.8 |
| 3 | 8.0 | 3.1 | 92.0 | 2.8 | 2.1 | 2.7 | 100 | 72.9 | 67.1 |
| Entry | Cycle | n-C8-C14 (%) | n-C15-C18 (%) | >C18 (%) | Branch (%) | S | χ (%) | OLP Yield (wt%) | GD Yield (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | I 1 | 1.6 | 93.7 | 3.7 | 1 | 1.4 | 100 | 82 | 76.6 |
| 2 | II 2 | 1 | 91.7 | 3.3 | 3.5 | 2.2 | 100 | 83.2 | 76.0 |
| 3 | III 2 | 1.1 | 90.6 | 3.3 | 3.3 | 1.9 | 100 | 84.1 | 76.0 |
| 4 | IV 2 | 1.1 | 91.5 | 3.4 | 3.4 | 1.7 | 100 | 81.9 | 74.8 |
| Entry | Catalyst wt% | Reaction Time (h) | n-C8-C14 (%) | n-C15-C18 (%) | >C18 (%) | Branch (%) | S | χ (%) | OLP Yield (wt%) | GD Yield (wt%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10 | 6 | 3.1 | 87.0 | 3.2 | 4.2 | 0.4 | 100 | 69.6 | 60.6 |
| 2 2 | 10 | 4 | 4.5 | 83.6 | 3.8 | 2.8 | 0.3 | 98.0 | 76.4 | 63.9 |
| 3 3 | 10 | 2 | 2.8 | 56.2 | 2.5 | 1.6 | 0.3 | 72.0 | 84.7 | 47.6 |
| 4 4 | 4 | 6 | 4.1 | 86.3 | 3.6 | 1.9 | 0.4 | 98.0 | 70.4 | 60.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Vito Nolfi, G.; Gallucci, K.; Mucciante, V.; Rossi, L. Production of Green Diesel via the Ni/Al Mo Hydrotalcite Catalyzed Deoxygenation of Rapeseed Oil. Molecules 2025, 30, 1699. https://doi.org/10.3390/molecules30081699
Di Vito Nolfi G, Gallucci K, Mucciante V, Rossi L. Production of Green Diesel via the Ni/Al Mo Hydrotalcite Catalyzed Deoxygenation of Rapeseed Oil. Molecules. 2025; 30(8):1699. https://doi.org/10.3390/molecules30081699
Chicago/Turabian StyleDi Vito Nolfi, Giuseppe, Katia Gallucci, Vittoria Mucciante, and Leucio Rossi. 2025. "Production of Green Diesel via the Ni/Al Mo Hydrotalcite Catalyzed Deoxygenation of Rapeseed Oil" Molecules 30, no. 8: 1699. https://doi.org/10.3390/molecules30081699
APA StyleDi Vito Nolfi, G., Gallucci, K., Mucciante, V., & Rossi, L. (2025). Production of Green Diesel via the Ni/Al Mo Hydrotalcite Catalyzed Deoxygenation of Rapeseed Oil. Molecules, 30(8), 1699. https://doi.org/10.3390/molecules30081699