
Effect of Solvents on Electrogenerated Base-Driven Transfer Hydrogenation Reactions



Abstract

1. Introduction
2. Results and Discussion
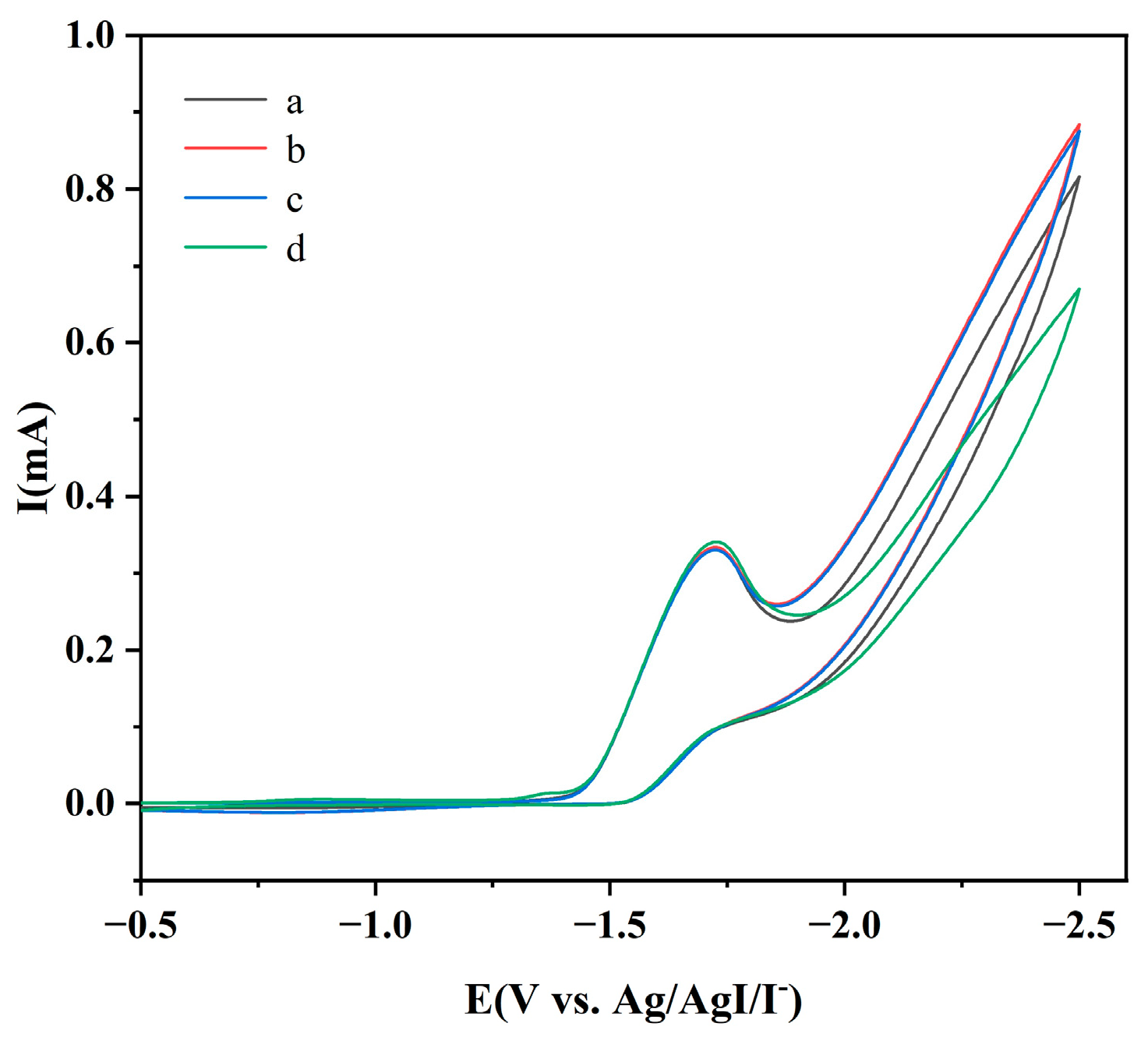
2.1. Reaction Optimization and Cyclic Voltammetry
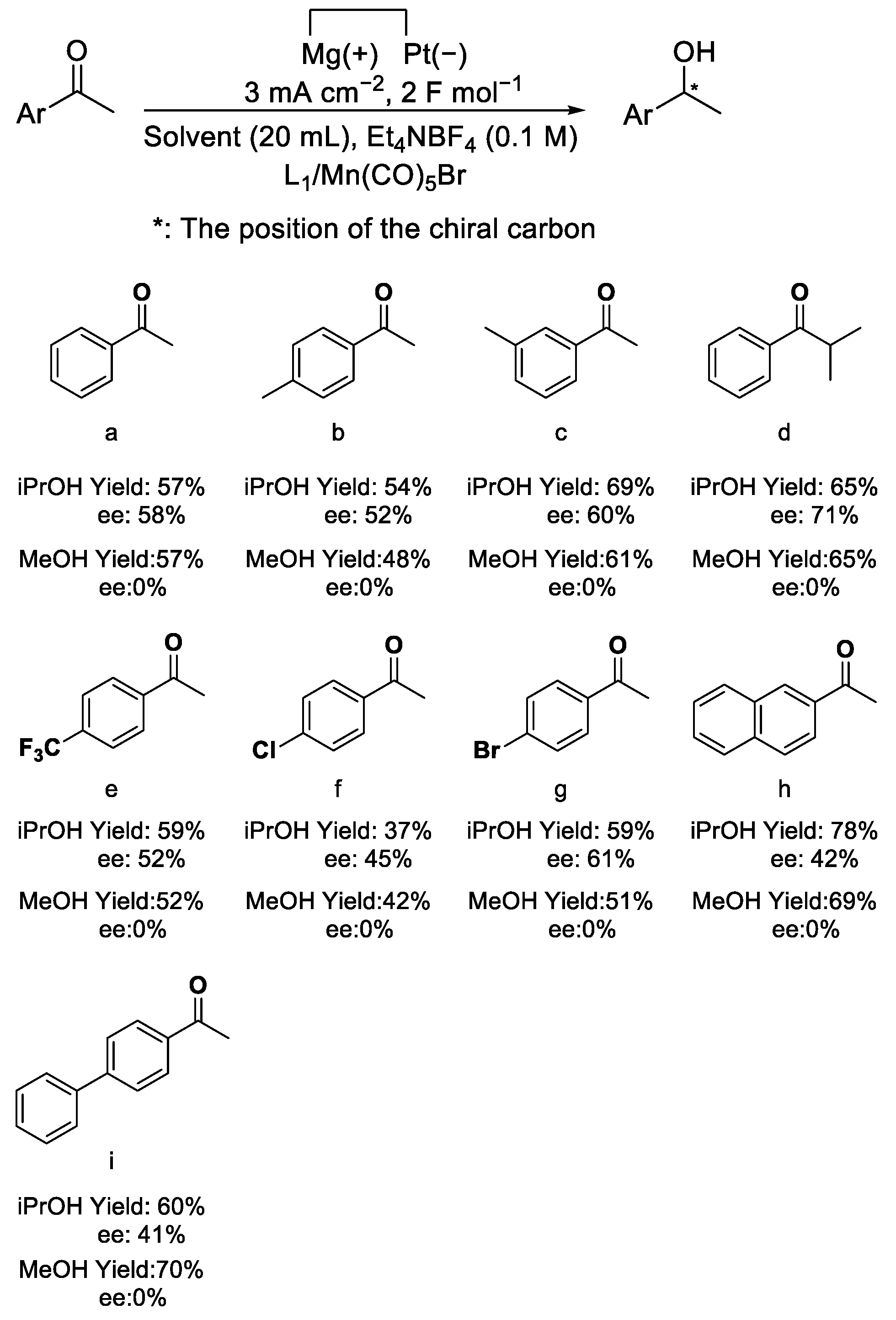
2.2. Substrate and Ligand Scopes
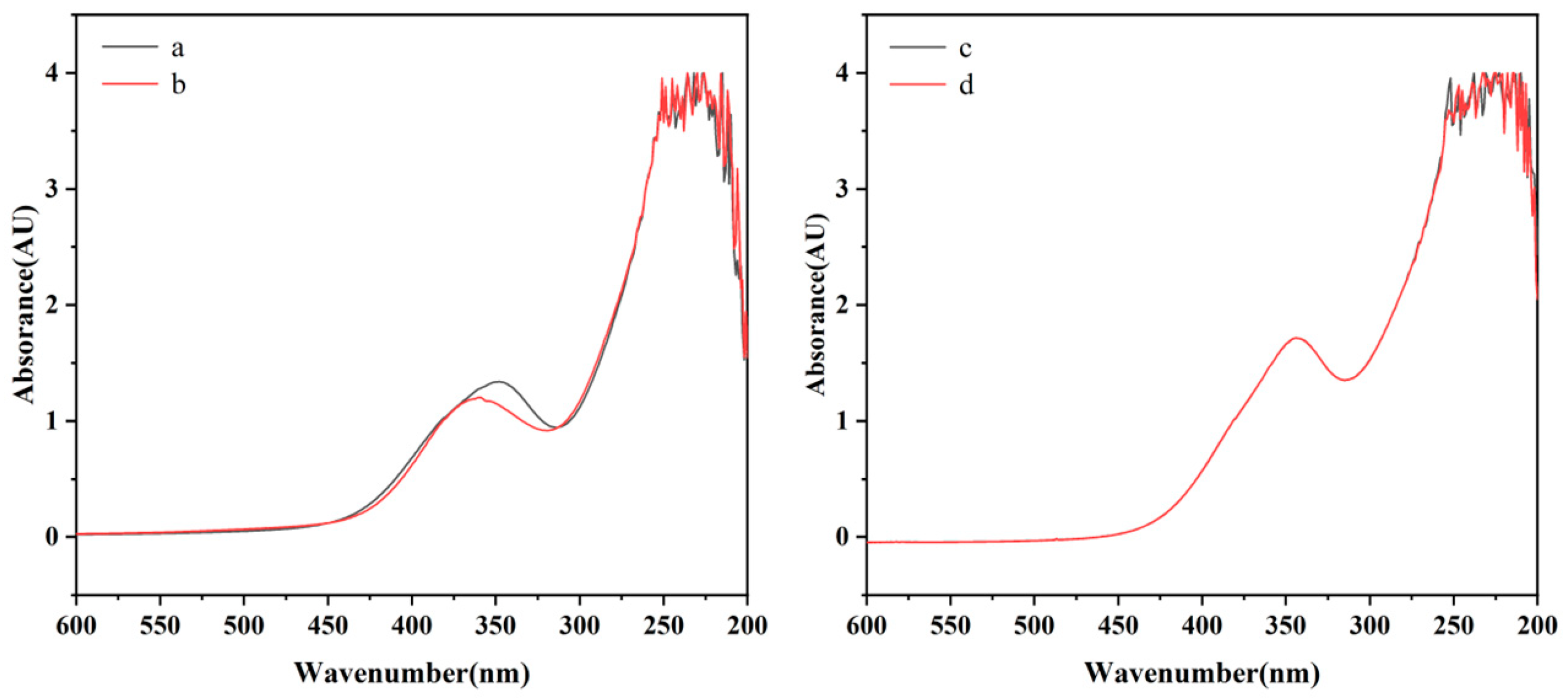
2.3. UV-Vis Absorption Spectra
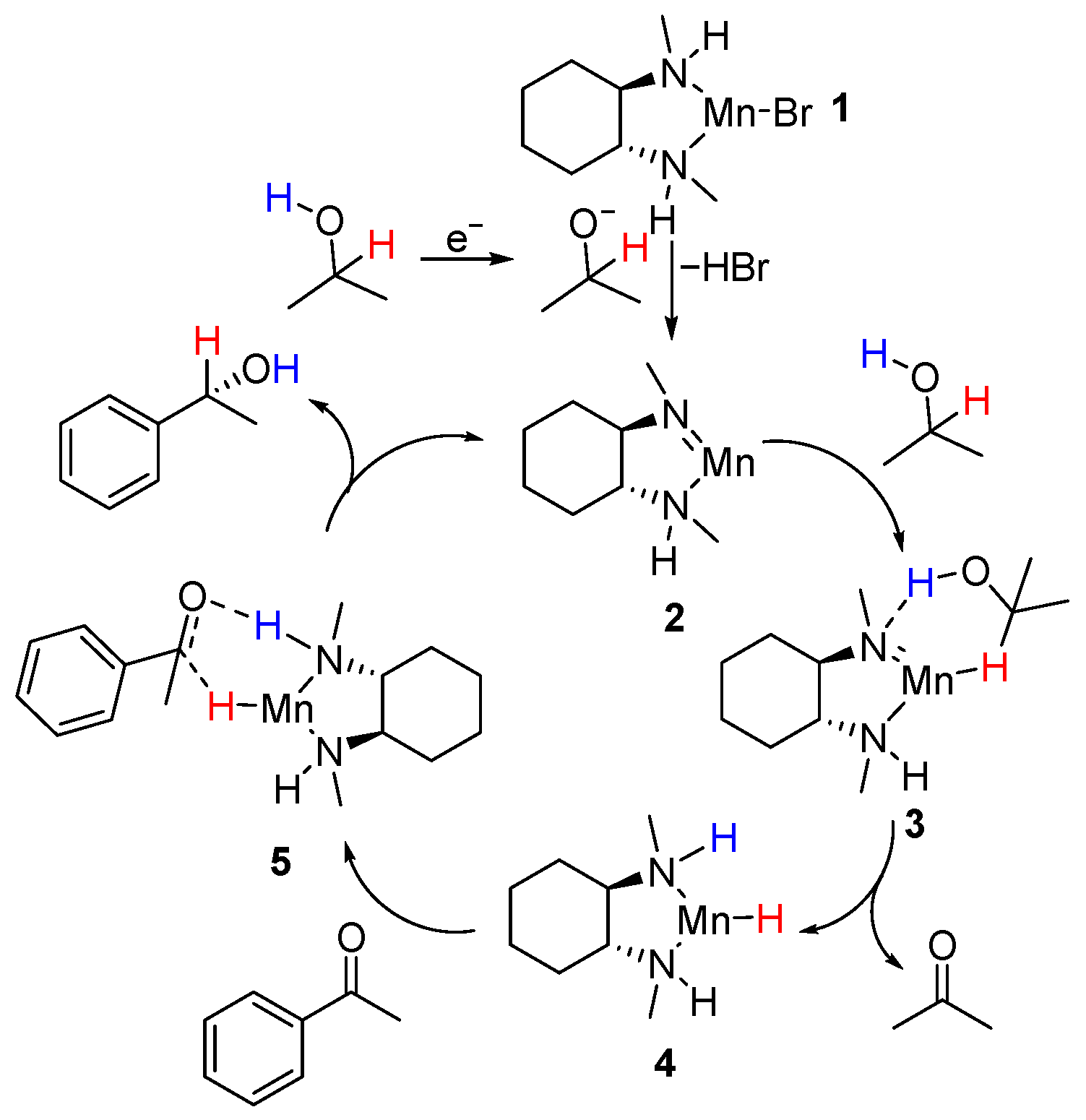
2.4. Mechanism Studies
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Electrolysis
3.3. Procedure for UV-Vis Absorption Spectra
3.4. Cyclic Voltammetry
3.5. Characterization Data for Product
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ayad, T.; Phansavath, P.; Ratovelomanana-Vidal, V. Transition-Metal-Catalyzed Asymmetric Hydrogenation and Transfer Hydrogenation: Sustainable Chemistry to Access Bioactive Molecules. Chem. Rec. 2016, 16, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Wang, H.; Zhang, W.; Chen, F.E. Asymmetric catalytic hydrogenation of imines and enamines in natural product synthesis. Green Synth. Catal. 2020, 1, 26–41. [Google Scholar] [CrossRef]
- Song, J.; Huang, Z.F.; Pan, L.; Li, K.; Zhang, X.; Wang, L.; Zou, J.J. Review on selective hydrogenation of nitroarene by catalytic, photocatalytic and electrocatalytic reactions. Appl. Catal. B- Environ. 2018, 227, 386–408. [Google Scholar] [CrossRef]
- Onisuru, O.R.; Fapojuwo, D.P.; Oseghale, C.O.; Alimi, O.A.; Meijboom, R. Transfer hydrogenation of ketone; an in situ approach toward an eco-friendly reduction. RSC Adv. 2022, 12, 19890–19900. [Google Scholar] [CrossRef]
- Wu, B.; Yang, J.; Hu, S.B.; Yu, C.B.; Zhao, Z.B.; Luo, Y.; Zhou, Y.G. Diboron-mediated palladium-catalyzed asymmetric transfer hydrogenation using the proton of alcohols as hydrogen source. Sci. China Chem. 2021, 64, 1743–1749. [Google Scholar] [CrossRef]
- Ritleng, V.; de Vries, J.G. Ruthenacycles and Iridacycles as Transfer Hydrogenation Catalysts. Molecules 2021, 26, 4076. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.; Tao, Y.; Nie, Y.; Lu, T.; Wang, J.; Zhang, Y.; Lu, X.; Xu, C.C. Recent Advances in Catalytic Transfer Hydrogenation with Formic Acid over Heterogeneous Transition Metal Catalysts. ACS Catal. 2021, 11, 1071–1095. [Google Scholar] [CrossRef]
- Yao, Z.J.; Zhu, J.W.; Lin, N.; Qiao, X.C.; Deng, W. Catalytic hydrogenation of carbonyl and nitro compounds using an [N,O]-chelate half-sandwich ruthenium catalyst. Dalton. Trans. 2019, 48, 7158–7166. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Cai, X.; Liu, Y.; Ke, Z. Base-controlled NHC-Ru-catalyzed transfer hydrogenation and α-methylation/transfer hydrogenation of ketones using methanol. Chin. Chem. Lett. 2024, 35, 109323–109328. [Google Scholar] [CrossRef]
- Haddad, Y.M.Y.; Henbest, H.B.; Husbands, J. Reduction of cyclohexanones to axial alcohols via iridium containing catalysts. Proc. Chem. Soc. 1964, 361–363. [Google Scholar]
- Ma, S.; Wang, X.; Cao, J.; Chen, H.; Lu, Y.; Wang, R. Nickel-Catalyzed Efficient Transfer Hydrogenation of Ketones. ChemistrySelect 2024, 9, e202400730. [Google Scholar] [CrossRef]
- Tharra, P.R.; Svejkar, J.; Jadhav, A.S.; Necas, M.; Dub, P.A.; Halls, M.D.; Svenda, J. Enantioselective Transfer Hydrogenation of alpha-Methoxyimino-beta-keto Esters. J. Org. Chem. 2024, 89, 12902–12911. [Google Scholar] [CrossRef] [PubMed]
- Beaufils, A.; Melle, P.; Lentz, N.; Albrecht, M. Air-Stable Coordinatively Unsaturated Ruthenium(II) Complex for Ligand Binding and Catalytic Transfer Hydrogenation of Ketones from Ethanol. Inorg. Chem. 2024, 63, 2072–2081. [Google Scholar] [CrossRef]
- Mayakrishnan, G.; Ick Soo, K.; Min, C., III. Stepwise Construction of Ru(II)Center Containing Chiral Thiourea Ligand on Graphene Oxide: First Efficient, Reusable, and Stable Catalyst for Asymmetric Transfer Hydrogenation of Ketones. Catalysts 2020, 10, 175. [Google Scholar] [CrossRef]
- Both, N.F.; Fessler, J.; Vicenzi, A.; Andres, K.; Spannenberg, A.; Junge, K.; Beller, M. Air-Stable Manganese NNS Pincer Complexes Enable Ketone Reduction at Room Temperature. ChemCatChem 2024, 16, e202301562. [Google Scholar] [CrossRef]
- Altan, O.; Yılmaz, M.K. New phosphine-amino-alcohol tridentate ligands for ruthenium catalyzed asymmetric transfer hydrogenation of ketones. J. Organometa. Chem. 2018, 861, 252–262. [Google Scholar] [CrossRef]
- Nagalakshmi, V.; Nandhini, R.; Venkatachalam, G.; Balasubramani, K. Synthesis and characterization of new ruthenium(III) complexes derived from fluoreneamine-based Schiff base ligands and their catalytic activity in transfer hydrogenation of ketones. J. Coord. Chem. 2020, 73, 206–216. [Google Scholar] [CrossRef]
- Hackl, L.; Ho, L.P.; Bockhardt, D.; Bannenberg, T.; Tamm, M. Tetraaminocyclopentadienone Iron Complexes as Hydrogenation Catalysts. Organometallics 2022, 41, 836–851. [Google Scholar] [CrossRef]
- Huo, S.; Wang, Q.; Zuo, W. An iron variant of the Noyori hydrogenation catalyst for the asymmetric transfer hydrogenation of ketones. Dalton. Trans. 2020, 49, 7959–7967. [Google Scholar] [CrossRef]
- Kathuria, L.; Din Reshi, N.U.; Samuelson, A.G. N-Heterocyclic Carbene (NHC)-Stabilized Ru(0) Nanoparticles: In Situ Generation of an Efficient Transfer Hydrogenation Catalyst. Chemistry 2020, 26, 7622–7630. [Google Scholar] [CrossRef]
- Xu, X.; You, Y.; Jin, M.Y.; Meng, F.J.; Xu, C.; Xing, X. Pincer Ru with a single stereogenic identity for highly efficient asymmetric transfer hydrogenation of ketones. Sci. China Chem. 2023, 66, 1443–1449. [Google Scholar] [CrossRef]
- Perez, M.; Elangovan, S.; Spannenberg, A.; Junge, K.; Beller, M. Molecularly Defined Manganese Pincer Complexes for Selective Transfer Hydrogenation of Ketones. ChemSusChem 2017, 10, 83–86. [Google Scholar] [CrossRef]
- Su, Y.; Ma, Z.; Wang, J.; Li, L.; Yan, X.; Ma, N.; Liu, Q.; Solan, G.A.; Wang, Z. Asymmetric Transfer Hydrogenation of Ketones Improved by PNN-Manganese Complexes. J. Org. Chem. 2024, 89, 12318–12325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Ma, Z.; Li, L.; Yan, X.; Cao, Q.; Su, Y.; Ma, N.; Wang, Z. Cycloheptyl-fused-PNN-manganese catalyzed asymmetric transfer hydrogenation of ketones. Mol. Catal. 2024, 564, 114274–114285. [Google Scholar] [CrossRef]
- Zhou, J.; Ye, J.; Zhang, Y.; Li, Z.; Li, J.; Liu, D.; Zhang, W. Synthesis of Chiral Diaryl Methanols via RuPHOX-Ru Catalyzed Asymmetric Hydrogenation. Adv. Synth. Catal. 2023, 365, 1275–1286. [Google Scholar] [CrossRef]
- Liang, Q.; Zhang, C.; Wang, F.; Luo, Z.; Yang, W.; Zhang, G.; Ding, D.; Zhang, G. Triazole backbone ligand in an unprecedented efficient manganese catalyst for use in transfer hydrogenation. Sci. China Chem. 2023, 66, 2028–2036. [Google Scholar] [CrossRef]
- Wang, D.; Bruneau-Voisine, A.; Sortais, J.B. Practical (asymmetric) transfer hydrogenation of ketones catalyzed by manganese with (chiral) diamines ligands. Catal. Commun. 2018, 105, 31–36. [Google Scholar] [CrossRef]
- van Putten, R.l.; Filonenko, G.A.; Gonzalez de Castro, A.; Liu, C.; Weber, M.; Muller, C.; Lefort, L.; Pidko, E. Mechanistic Complexity of Asymmetric Transfer Hydrogenation with Simple Mn-Diamine Catalysts. Organometallics 2019, 38, 3187–3196. [Google Scholar] [CrossRef]
- Paşa, S.; Arslan, N.; Merīç, N.; Kayan, C.; Bingül, M.; Durap, F.; Aydemir, M. Boron containing chiral Schiff bases: Synthesis and catalytic activity in asymmetric transfer hydrogenation (ATH) of ketones. J. Mol. Struct. 2020, 1200, 127064–127071. [Google Scholar] [CrossRef]
- Chang, K.; Chou, R.; Yu, P.; Zuo, L.; Liu, Q.; Zhang, X.; Yin, C.; Zhou, H. Rhodium-Catalyzed Asymmetric Transfer Hydrogenation of Aryl Ketones Involving Free Phenolic Hydroxyl Group(s). Chemistry 2024, 30, e202403055. [Google Scholar] [CrossRef]
- Yu, P.; Chen, D.; Liu, Y.; Yin, C.; Liu, Q.; Zhou, H. Synthesis of Chiral 2-Oxazolidinones by Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of 2-Oxazolones. Adv. Synth. Catal. 2023, 366, 1199–1204. [Google Scholar] [CrossRef]
- Jie, L.H.; Xu, H.C. Electrocatalytic Cyclopropanation of Active Methylene Compounds. J. Electrochem. 2024, 30, 2313001–2313006. [Google Scholar]
- Kazemi-Rad, R.; Azizian, J.; Kefayati, H. Improved Synthesis of 2,2-Arylmethylene Bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) and 1,8-Dioxo-octahydroxanthene Derivatives Catalyzed by Electrogenerated Base and Sulfuric Acid. J. Chin. Chem. Soc. 2015, 62, 311–315. [Google Scholar] [CrossRef]
- Chiarotto, I.; Mattiello, L.; Feroci, M. The Electrogenerated Cyanomethyl Anion: An Old Base Still Smart. Acc. Chem. Res. 2019, 52, 3297–3308. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Wang, L.T.; Wang, Y.L.; Chen, L.B.; Lu, J.X.; Wang, H. Electrochemically Driven Asymmetric Hydrogenation of Aromatic Ketones by Noyori-Ikariya Catalyst. Appl. Organomet. Chem. 2024, 39, 7773–7780. [Google Scholar] [CrossRef]
- Shen, Y.; Zhan, Y.; Li, S.; Ning, F.; Du, Y.; Huang, Y.; He, T.; Zhou, X. Hydrogen generation from methanol at near-room temperature. Chem. Sci. 2017, 8, 7498–7504. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Sarkar, A.; Sundararaju, B. Recent developments on methanol as liquid organic hydrogen carrier in transfer hydrogenation reactions. Coord. Chem. Rev. 2021, 433, 213728–213746. [Google Scholar] [CrossRef]
- Palo, D.R. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Heimann, J.E.; Bernskoetter, W.H.; Hazari, N. Understanding the Individual and Combined Effects of Solvent and Lewis Acid on CO(2) Insertion into a Metal Hydride. J. Am. Chem. Soc. 2019, 141, 10520–10529. [Google Scholar] [CrossRef] [PubMed]
- Connelly Robinson, S.J.; Zall, C.M.; Miller, D.L.; Linehan, J.C.; Appel, A.M. Solvent influence on the thermodynamics for hydride transfer from bis(diphosphine) complexes of nickel. Dalton Trans. 2016, 45, 10017–10023. [Google Scholar] [CrossRef]
- Dey, S.; Thakur, S.D.; Sau, A.; Panja, D.; Roy, T.; Zhang, J.; Annadata, H.V.; Kundu, S. Cobalt catalyzed condensation interrupted selective transfer hydrogenation using methanol. J. Catal. 2024, 439, 115759–115769. [Google Scholar] [CrossRef]
- Sau, A.; Mahapatra, D.; Dey, S.; Panja, D.; Saha, S.; Kundu, S. Utilization of methanol for condensation interrupted chemoselective transfer hydrogenation of C=C, C=O, and C=N bonds under low catalyst loading. Org. Chem. Front. 2023, 10, 2274–2286. [Google Scholar] [CrossRef]
- Aboo, A.H.; Begum, R.; Zhao, L.; Farooqi, Z.H.; Xiao, J.L. Methanol as hydrogen source: Chemoselective transfer hydrogenation of α,β-unsaturated ketones with a rhodacycle. Chin. J. Catal. 2019, 40, 1795–1799. [Google Scholar] [CrossRef]
- Mandal, A.; Ganguli, K.; Pradhan, M.; Gorai, A.; Kundu, S. Selective Transfer Hydrogenation of C=O and Conjugated C=C Bonds Using An NHC-Based Pincer (CNC)MnI Complex in Methanol. Chemsuschem. 2023, 16, e202300683. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-groupthermochemistry, transition metal bonding, thermochemicalkinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194120. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Schleyer, P.v.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comp. Chem. 1983, 4, 294–302. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Kuechle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—the IRC approach. Acc. Chem. Res. 1981, 14, 363. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B. 2009, 113, 6378–6397. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
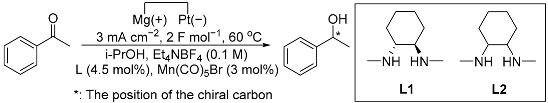 | ||||
|---|---|---|---|---|
| Entry | Deviation from Standard Conditions | Conversion/% | Selectivity/% | ee/% a |
| 1 | None | 58 | 99 | 63 (R) |
| 2 | At 40 °C | 50 | 99 | 62 (R) |
| 3 | At 20 °C | 7 | 98 | 72 (R) |
| 4 | No L1 and Mn(CO)5Br | trace | - | NA. |
| 5 | No Mn(CO)5Br | trace | - | NA. |
| 6 | No L1 | trace | - | NA. |
| 7 | No electricity | trace | - | NA. |
| 8 | L2 instead of L1 | 54 | 98 | NA. |
| 9 | PhCOCH3, L1, and Mn(CO)5Br added After electrolysis | 57 | 97 | 62 (R) |
| 10 | t-BuOK (0.2 M), no electricity | 61 | 98 | 50 (R) |
 | ||||
|---|---|---|---|---|
| Entry | Deviation from Standard Conditions | Conversion/% | Selectivity/% | ee/% a |
| 1 | None | 65 | 60 | NA. |
| 2 | at 40 °C | 64 | 64 | NA. |
| 3 | L1 (9 mol%) + Mn(CO)5Br (6 mol%) | 68 | 79 | NA. |
| 4 | L1 (13.5 mol%) + Mn(CO)5Br (9 mol%) | 71 | 77 | NA. |
| 5 | no electricity | - | - | NA. |
| 6 | t-BuOK (0.2 M), no electricity | Trace | - | NA. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.-W.; Li, M.-H.; Zhang, F.; Wang, Y.-L.; Lu, J.-X.; Wang, H. Effect of Solvents on Electrogenerated Base-Driven Transfer Hydrogenation Reactions. Molecules 2025, 30, 910. https://doi.org/10.3390/molecules30040910
Zhu J-W, Li M-H, Zhang F, Wang Y-L, Lu J-X, Wang H. Effect of Solvents on Electrogenerated Base-Driven Transfer Hydrogenation Reactions. Molecules. 2025; 30(4):910. https://doi.org/10.3390/molecules30040910
Chicago/Turabian StyleZhu, Jing-Wei, Meng-Han Li, Feng Zhang, Ya-Li Wang, Jia-Xing Lu, and Huan Wang. 2025. "Effect of Solvents on Electrogenerated Base-Driven Transfer Hydrogenation Reactions" Molecules 30, no. 4: 910. https://doi.org/10.3390/molecules30040910
APA StyleZhu, J.-W., Li, M.-H., Zhang, F., Wang, Y.-L., Lu, J.-X., & Wang, H. (2025). Effect of Solvents on Electrogenerated Base-Driven Transfer Hydrogenation Reactions. Molecules, 30(4), 910. https://doi.org/10.3390/molecules30040910