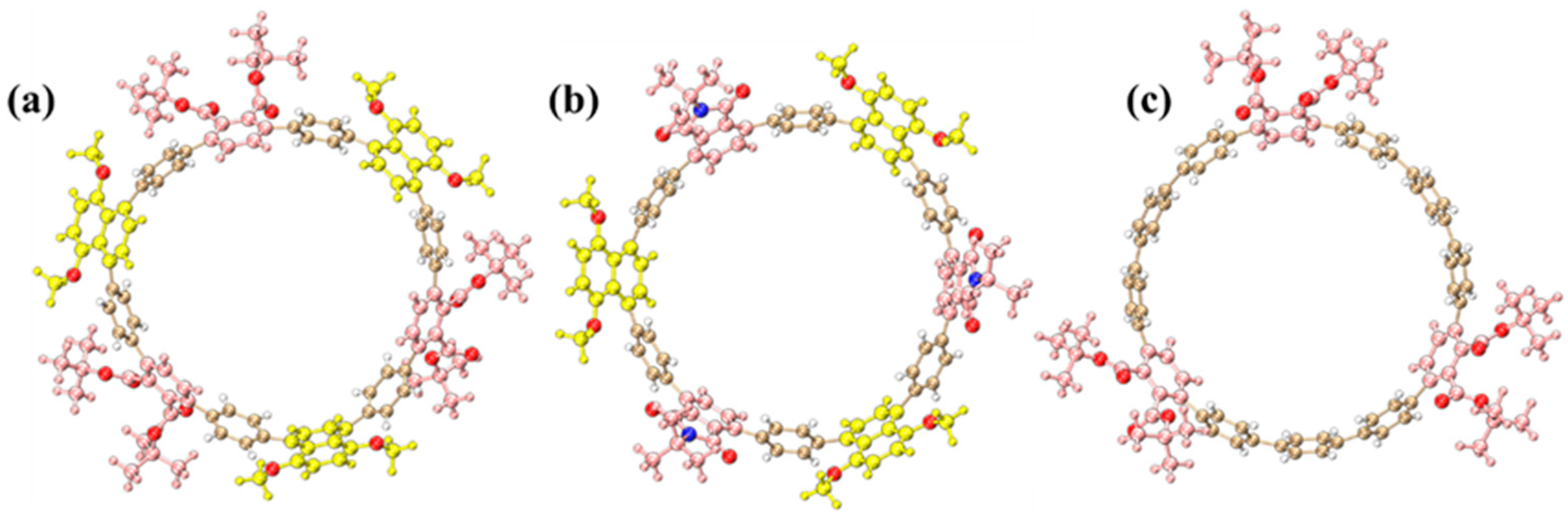
2.1. Molecular Structures
The optimized molecular structures of three [12]cycloparaphenylene ([12]CPP) derivatives exhibit distinct structural configurations (
Figure 1). The alternating donor–acceptor (D–A) [12]CPP ([12]CPP 1a) comprises 1,4-dimethoxynaphthalene (donor) and phthalate (acceptor) units alternately arranged along the cyclic CPP backbone, forming a nanoring with pronounced electron push-pull characteristics (
Figure 1a). This configuration facilitates efficient intramolecular charge transfer, substantially reducing the HOMO–LUMO energy gap and imparting unique photophysical properties.
For [12]CPP 2a, the acceptor unit is replaced with a stronger electron-withdrawing phthalimide group via imidation, leading to a more polarized D–A configuration (
Figure 1b). The introduction of the imide moiety further amplifies the acceptor’s electron affinity, leading to a narrower HOMO–LUMO gap. As a control, the symmetrically functionalized [12]CPP hexacarboxylate ([12]CPP 3a) features three phthalate groups uniformly distributed on the CPP ring, devoid of alternating D–A motifs (
Figure 1c). Structural optimizations confirm that all derivatives retain the cyclic π-conjugated framework of CPPs (
Figure 1), while the introduced functional groups modulate both electronic environments and conformational dynamics, thereby critically influencing their photophysical behaviors. The ChemDraw 19.1 schemes of the three structures are shown in
Figure S1, and the corresponding atomic coordinates are listed in
Tables S2–S4.
As a key parameter for evaluating the structural characteristics of cyclic π-conjugated molecules, the inner diameter data of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a are listed in
Table 1. For these three differently structured [12]CPP derivatives, the inner diameter is defined as the distance between the centroids of two para-positioned benzene rings within the molecule. Schematic diagrams defining the inner diameter are shown on the right-hand side of
Table 1 and
Figure S2. A comparison with the structural parameters of the parent [12]CPP and the reduced-state [12]CPP [
24] shows that their distortion parameters (D.P.) are in good agreement with that of [12]CPP (D.P. = 1.03), fluctuating within a narrow range of 0.01–0.02, and are significantly reduced by approximately 0.2 compared to the D.P. value of the reduced-state [12]CPP. This indicates that the molecular skeletons maintain planar conjugated characteristics similar to those of the parent molecule. The increased inner diameter of [12]CPP 1a suggests that the introduction of 1,4-dimethoxynaphthalene (donor) and phthalate (acceptor) leads to an expansion of the [12]CPP structure’s inner diameter.
As shown in
Figure S3, the dihedral angle is defined as the angle between the planes of two para-substituted benzene rings on the [12]CPP backbone. The dihedral angle ranges of [12]CPP 1a and [12]CPP 2a are relatively narrow with concentrated values, which implies that the interactions within their donor–acceptor structures may render the arrangement of benzene rings more regular. [12]CPP 3a, as a single-receptor structure, possesses the broadest dihedral angle range (99.96–152.63°), revealing a more diverse spatial orientation of benzene rings. This is likely due to the absence of donor interactions, which results in less constraint on the arrangement of the benzene rings. This discrepancy reflects the impact of molecular design (donor–acceptor vs. single–receptor) on the spatial configuration of benzene rings, providing crucial structural insights for a deep understanding of intermolecular interaction mechanisms and structure–function relationships.
2.3. OPA Analysis
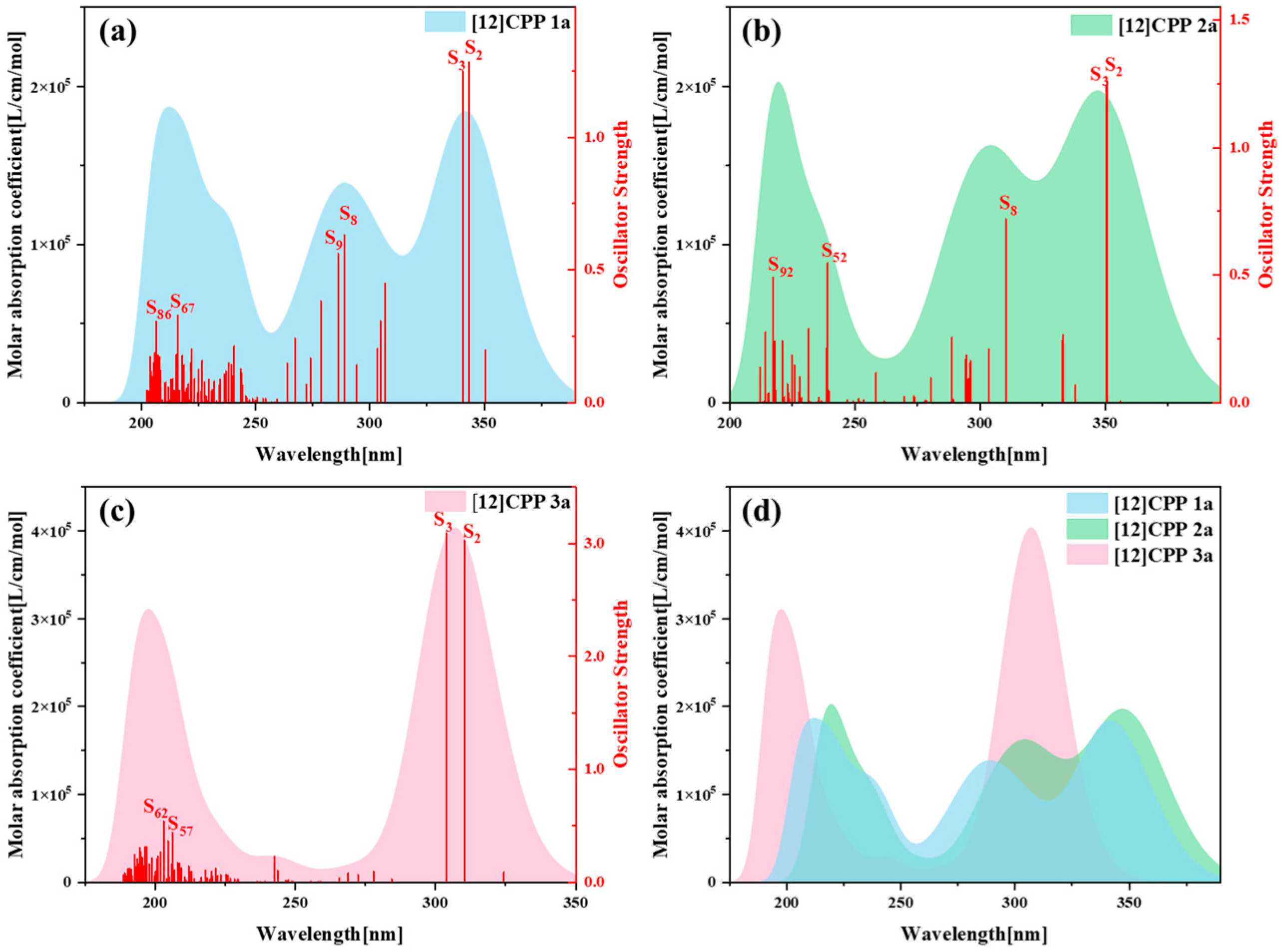
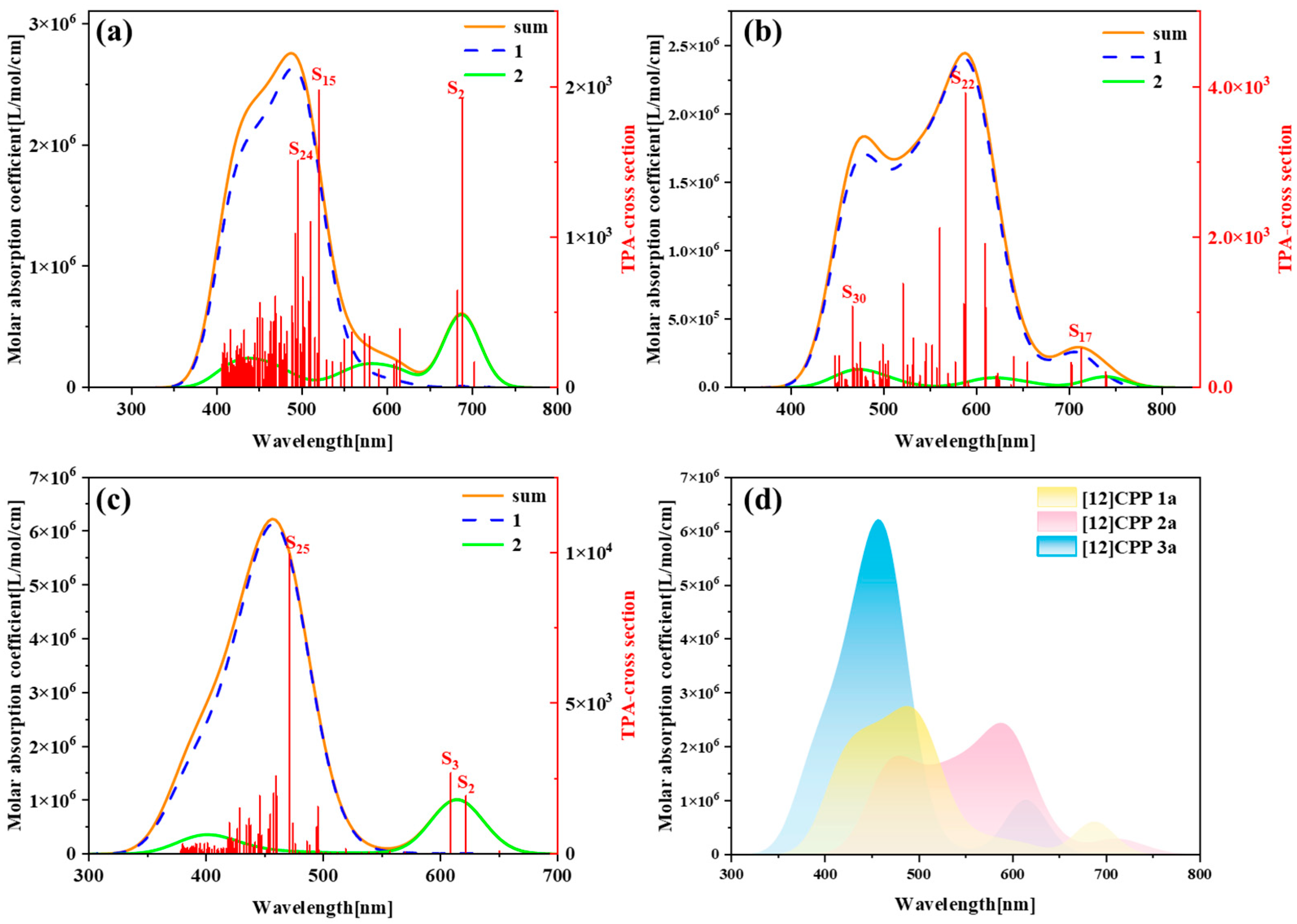
Figure 3 presents the one-photon absorption spectra of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a calculated under the chloroform implicit solvent model, revealing significant differences in their absorption intensities and peak positions.
Figure S5 displays the comparison between the computational results and experimental data for the [12]CPP 1a and [12]CPP 2a structures. The experimental absorption spectra (c,d) in
Figure S5 are adapted from Ref. [12]. The strong agreement between the experimental and calculated values confirms the accuracy of the theoretical calculations. The spectra indicate that [12]CPP 3a exhibits a higher molar absorption coefficient. Compared to [12]CPP 3a, the absorption spectrum of [12]CPP 1a displays a distinct redshift, primarily attributed to the molecular polarization effect induced by the introduction of an alternating donor–acceptor structure. Furthermore, [12]CPP 2a exhibits an even greater redshift compared to [12]CPP 1a, which is attributed to the strong electron-withdrawing nature of the imide acceptor that significantly lowers the LUMO energy level, thereby reducing the HOMO–LUMO gap and decreasing the transition energy. In the three structures, the excited states making the largest contributions to the absorption peaks are S
2 and S
3, among which S
2 and S
3 in [12]CPP 2a represent degenerate excited states.
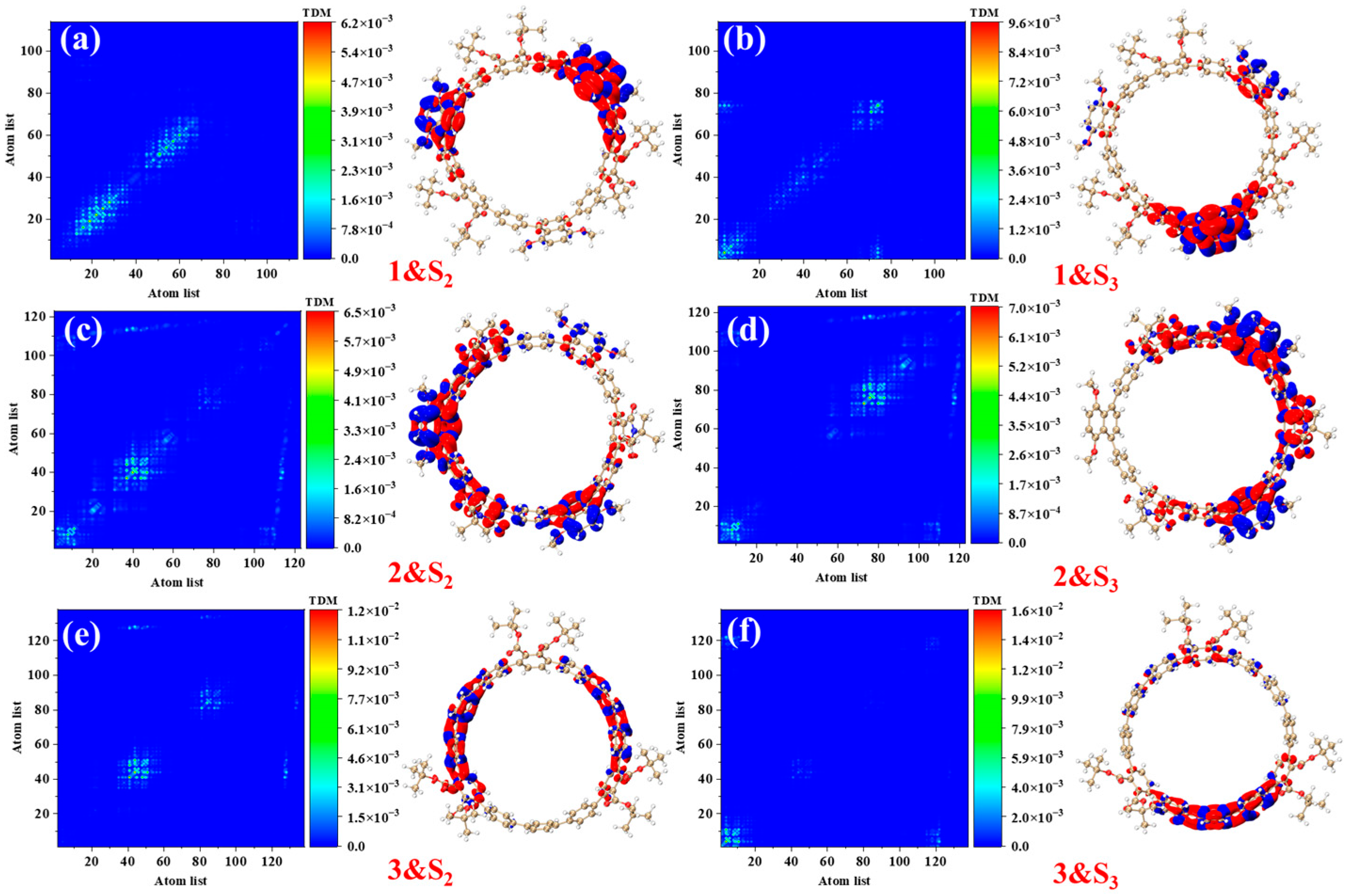
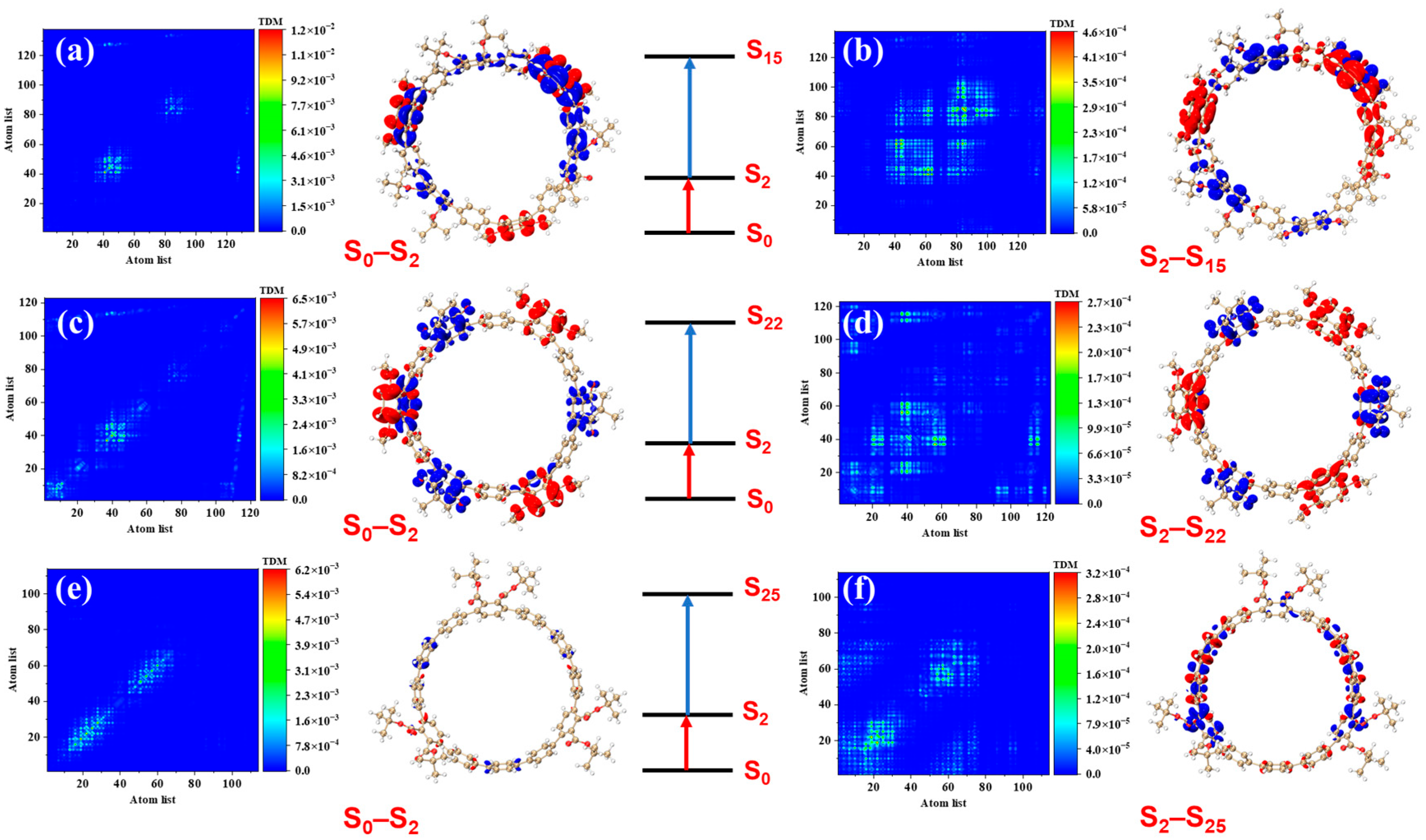
Figure 4 presents the transition density matrices (TDM) and electron–hole density distribution maps for [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a in the S
2 and S
3 excited states, offering a visual comparison of their one-photon absorption characteristics. In the TDM maps, electrons and holes in [12]CPP 1a and 2a exhibit pronounced localization features. The transition density of [12]CPP 1a is predominantly concentrated in the donor region, while that of [12]CPP 2a is distributed across both the donor and acceptor regions, reflecting strong electronic coupling between the donor–acceptor units. In the electron–hole density maps, both electrons and holes in [12]CPP 1a and [12]CPP 2a are mainly localized in the donor region. A clear separation is observed: electrons are concentrated on the CPP ring, while holes are localized on the upper functional groups, suggesting significant electron–hole separation. The [12]CPP 2a structure also exhibits some electron–hole density in the donor region. The excitation types of [12]CPP 1a and [12]CPP 2a are characterized by a combination of charge transfer and localized excitation. Moreover, in the S
2 and S
3 states, electron–hole distributions are localized in distinct acceptor regions, suggesting that the donor–acceptor coupling mode depends on the excitation state. For [12]CPP 3a, due to the absence of a donor–acceptor alternating structure, its TDM map exhibits a relatively uniform transition distribution, suggesting that the electronic transitions are primarily confined within the [12]CPP host structure. The electron–hole density maps reveal that, in the S
2 and S
3 excited states, the electron and hole densities are uniformly distributed across the [12]CPP ring and exhibit a certain spatial complementarity, lacking significant polarization characteristics.
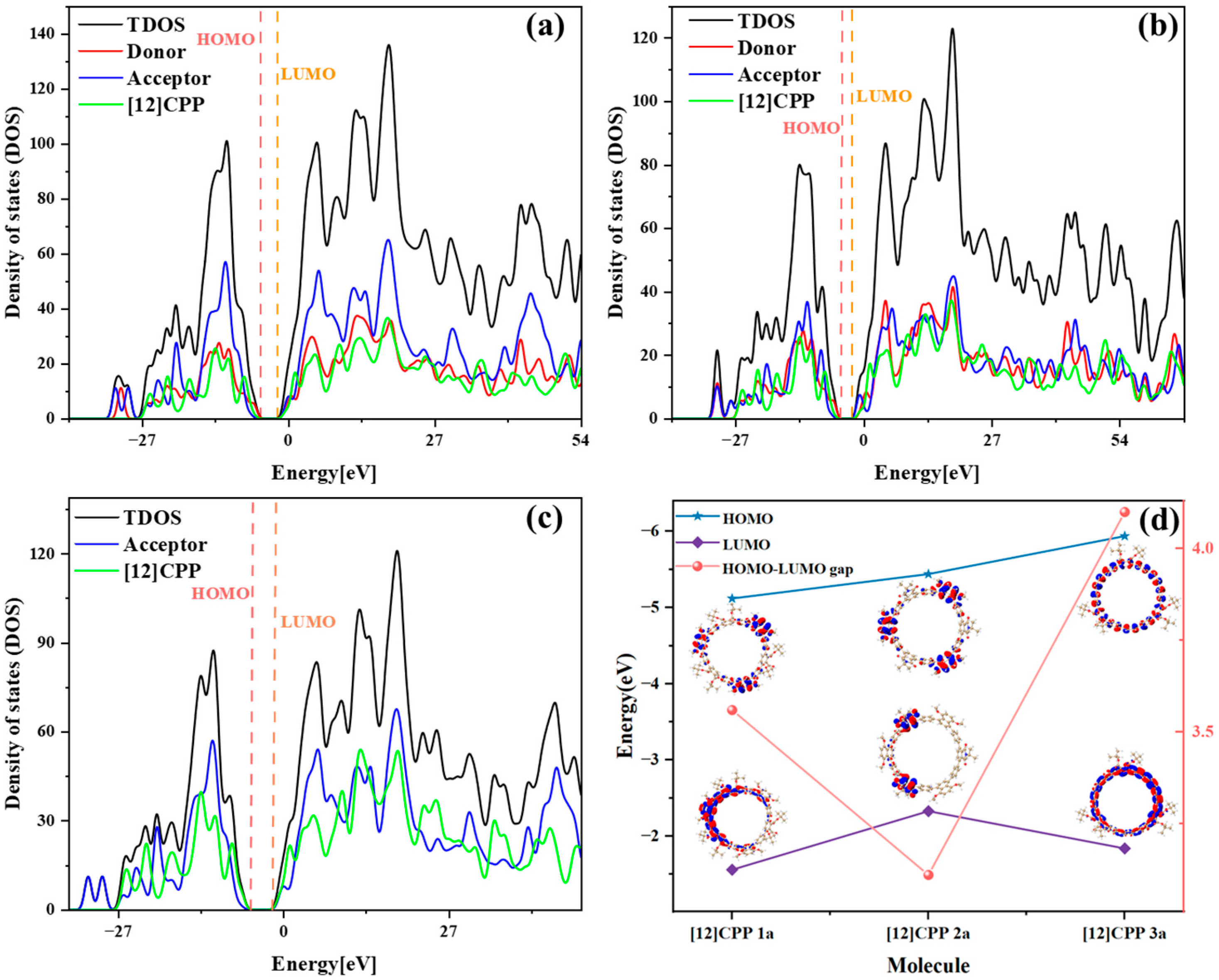
Based on a systematic analysis of key excitation parameters (D/Sr/H/t indices, Ecoul, and HDI/EDI), the electronic excitation characteristics of [12]CPP derivatives exhibit a clear configuration-dependent behavior, as shown in
Table 2. The D index measures the spatial distance between the centroids of the electron and the hole in the excited state; a larger D index indicates greater spatial separation, typically reflecting stronger charge transfer (CT) character. The H index describes the overall average spatial distribution breadth of the electron and hole, while the t index specifically quantifies the degree of electron–hole separation and serves as a key parameter for evaluating exciton dissociation. The Sr index represents the geometric average overlap between the electron and hole distributions, where lower Sr values correspond to weaker spatial overlap. In addition, the hole delocalization index (HDI) and electron delocalization index (EDI) provide a quantitative measure of the degree of delocalization of the hole and electron in the excited state. [12]CPP 2a exhibits partial CT characteristics in its electronic excitation. The relatively large D index suggests pronounced spatial separation between electron and hole distributions. However, its negative t index indicates that localized excitation (LE) features are also significant. Thus, [12]CPP 2a can be classified as a CT-LE hybrid excitation mode. In contrast, [12]CPP 3a has the smallest D value and the most negative Coulomb interaction energy (Ecoul), indicating a stronger exciton binding and a predominantly localized excitation nature. The Sr also supports this observation: the electron–hole overlap in [12]CPP 2a is lower than that in 1a and 3a under both excited states. [12]CPP 3a shows distinct LE features, characterized by the smallest D value and the highest Sr value, implying that the electronic transitions are mainly confined within the CPP framework. [12]CPP 1a displays intermediate values across all parameters, indicating a CT-LE hybrid excitation mechanism as well, though with a dominant LE character. This conclusion is consistent with the transition density distribution patterns shown in
Figure 4.
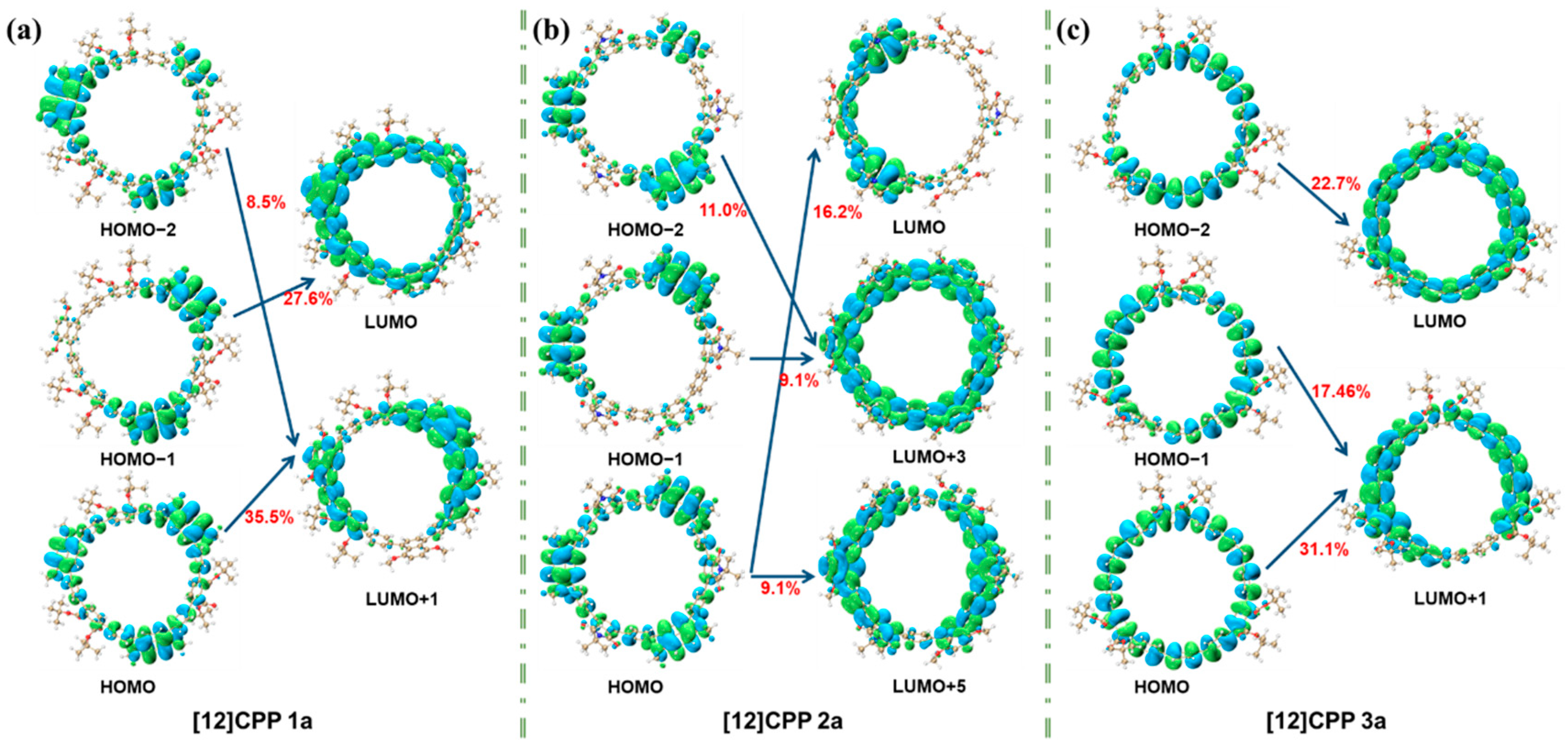
Quantitative exciton binding energy data effectively characterize Coulomb interaction intensities across different excited states, while orbital contribution analysis (
Figure 5) combined with these metrics fully elucidates the donor–acceptor interaction modes.
Figure 5 illustrates the orbital contributions of [12]CPP 1a, 2a, and 3a in the S
2 excited state. For [12]CPP 1a, transitions like HOMO-2→LUMO + 1 (8.5%), HOMO→LUMO + 1 (35.5%), and HOMO-1→LUMO (27.8%) highlight concentrated contributions on specific paths. [12]CPP 2a shows dispersed contributions, reflecting complex orbital interactions. [12]CPP 3a features dominant contributions from HOMO-2→LUMO (22.7%) and HOMO→LUMO + 1 (31.1%). These distinct patterns reveal unique orbital participation in each structure’s electronic excitation, offering microscopic insights into their photophysical properties.
To better elucidate the electron transfer process during electronic excitation, the electron transfer amounts between the donor, acceptor, and [12]CPP fragments in the S
2 excited state were calculated using the IFCT (Inter-Fragment Charge Transfer) method. The IFCT analysis is based on the contributions of fragments to holes and electrons. In this method, the electron transfer amount from fragment R to fragment S during the electronic excitation process is calculated as follows:
In Equation (1), represents the proportion of R in the excited electrons, and represents the proportion of S in the destination of the electrons.
The net electron transfer amount between two fragments, that is, the difference between the electron transfers in two directions, is shown in Equation (2):
The net change in the number of electrons of a certain fragment, which is the sum of the net electron transfer amounts between this fragment and all other fragments, is shown in Equation (3).
Table 3 presents the net electron transfer amounts between different fragments in the three molecules. From the table, it is evident that in [12]CPP 1a, electrons are transferred overall from the donor to other regions, with a net transfer of 0.024 electrons from the donor to the acceptor and 0.089 electrons from the donor to [12]CPP. Additionally, [12]CPP transfers a small number of electrons to the acceptor. Combined with the data in
Table 4, the donor loses −0.114 electrons, the acceptor gains 0.027 electrons, and [12]CPP gains 0.087 electrons, indicating that a significant portion of electrons is transferred from the donor to [12]CPP.
Table 5 further reveals that, during excitation, electrons in the acceptor and CPP fragments show negligible intra-fragment redistribution, whereas the donor exhibits substantial redistribution (value = 0.56). This indicates that electronic excitation strongly perturbs the donor’s electronic structure, underscoring its pivotal role in the excitation process. In [12]CPP 2a, the net electron transfer from the donor to the acceptor is significantly higher than that from the donor to [12]CPP. Additionally, electrons are transferred from both the donor and CPP fragment to the acceptor, leading to a marked increase in its net charge. The electrons on the donor of [12]CPP also exhibit significant redistribution. In the case of [12]CPP 3a, which lacks a donor, electrons are only transferred from [12]CPP to the acceptor region, and the electrons on [12]CPP undergo substantial redistribution, making the electronic excitation particularly prominent in this part. According to
Table 6, the excitation modes of [12]CPP 1a and [12]CPP 2a are characterized by a combination of charge transfer and localized excitation, with [12]CPP 2a exhibiting more pronounced charge transfer characteristics, whereas [12]CPP 3a primarily exhibits localized excitation. This conclusion is entirely consistent with the electron–hole density maps.
Table 7 highlights distinct charge distribution characteristics among the three structures as revealed by NPA analysis [
25]. For [12]CPP 1a, the donor moiety and [12]CPP framework exhibit positive charges, while the acceptor group carries a negative charge, indicating that the donor and [12]CPP scaffold act as electron loss centers to facilitate electron transfer to the acceptor. In [12]CPP 2a, the donor bears a higher positive charge than in 1a, while the acceptor exhibits a slightly less negative charge, and the CPP ring carries a lower positive charge. This charge redistribution signifies a more pronounced donor-to-acceptor electron transfer intensity in [12]CPP 2a relative to [12]CPP 1a. For [12]CPP 3a, the absence of a donor group results in opposing charges between the acceptor (−0.099) and [12]CPP framework (0.099), reflecting direct electron transfer between the [12]CPP scaffold and the acceptor moiety. These NPA charge distributions demonstrate that the presence of donor units in 1a and 2a significantly modulates charge allocation compared to 3a, providing critical insights into the electron interaction mechanisms within these systems.
2.4. Two-Photon Absorption
Figure 6 presents the two-photon absorption (TPA) spectra of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a, along with their comparative overlay, revealing the differences in TPA properties and intramolecular transition mechanisms among the three molecules. TPA is a nonlinear optical process defined as a phenomenon in which a material simultaneously absorbs two low-energy photons (typically near-infrared or infrared light), undergoes excitation transitions and relaxation processes, and subsequently emits fluorescence photons via spontaneous radiation to return to the ground state. The absorption probability is proportional to the square of the incident light intensity. The underlying mechanism involves a two-step excitation pathway mediated by a virtual intermediate state, as illustrated in the Jablonski diagram (
Figure S6). It can be observed that the absorption peaks of [12]CPP 2a exhibit a significant redshift, with the absorption peaks located in the range of 400–800 nm. This indicates that the introduction of the imide acceptor effectively lowers the LUMO energy level of the molecule, resulting in a notable reduction in transition energy. Furthermore, the main absorption peaks of [12]CPP 2a originate predominantly from the first-step transition. In contrast, although the absorption peaks of [12]CPP 3a exhibit a significant blueshift, their absorption intensity is the strongest among the three, which is closely related to its symmetric structure and the electronic effects of the hexacarboxylate groups. Simultaneously, in the spectrum of [12]CPP 3a, the small absorption peak on the right side is mainly contributed by the second-step transition, while the main absorption peak is contributed by the first-step transition, indicating that the symmetric structure exhibits different transition modes in the excited-state transitions. The absorption peak distribution and spectral characteristics of [12]CPP 1a exhibit intermediate properties between [12]CPP 2a and [12]CPP 3a: the main absorption peak originates from the first-step transition, while the minor peak on the right side predominantly arises from second-step excited-state transitions, consistent with the dual photon absorption mechanism observed in [12]CPP 1a. This suggests that the donor–acceptor structure of [12]CPP 1a provides a certain degree of polarization, albeit less pronounced than that of [12]CPP 2a.
To more precisely analyze the excitation characteristics in the two-photon transitions, Transition Density Matrix (TDM) and electron–hole density analyses were employed. These results clearly elucidate the regulatory effects of the donor–acceptor structure and symmetry on the two-photon absorption process.
From
Figure 7a,b, it can be observed that [12]CPP 1a exhibits localized excitation and charge transfer characteristics in the S
0→S
2 transition, with electrons localized in the lower donor (1,4-dimethoxynaphthalene) and the upper donor’s functional group regions, while holes are concentrated on the [12]CPP ring. In the second-step transition (S
2→S
15), the TDM map reveals that the transition density expands from the molecular skeleton to a broader region between the donor and acceptor, with corresponding diffusion of electron and hole distributions. Electrons are concentrated on the donor, while holes are localized on the benzene ring of the acceptor. This transition pattern indicates that the two-photon absorption process of [12]CPP 1a is primarily driven by donor–acceptor interactions, with a gradual transition from localized excitation to charge transfer excitation. In contrast, [12]CPP 2a exhibits more pronounced charge-transfer characteristics in the S
0→S
2 transition, with electron density almost entirely localized on the donor (1,4-dimethoxynaphthalene) and hole density on the acceptor (phthalimide), indicating stronger donor–acceptor polarization. In the S
2→S
22 transition, the TDM distribution further expands, spanning a larger region of the molecule, and the separation between electrons and holes becomes more significant. This trend indicates that the introduction of the imide acceptor significantly enhances the charge transfer effect of the molecule, enabling [12]CPP 2a to exhibit superior optical performance during the two-photon transition process, as shown in
Figure 7c,d. In comparison, [12]CPP 3a contributes very little to the S
0→S
2 transition, with minimal electron and hole densities, representing charge transfer excitation. In the S
2→S
25 transition, the TDM distribution is confined to the molecular skeleton, with uniform electron and hole distributions, as illustrated in
Figure 7e,f.
Building on the above analysis, we delved deeper into the two-photon absorption mechanism by systematically calculating the transition dipole moment matrix elements for the three target molecules during two-photon absorption, with results presented in
Table 8. The two steps of the two-photon transition differ significantly in transition probability. The first-step transition, from the ground state to the intermediate state, generally exhibits a higher probability, whereas the second-step transition between excited states is relatively weak. Comparing the computational results across the three molecules, 3a displays particularly striking characteristics: in the first step of the two-photon transition, its transition dipole moment values are the largest, and the corresponding integral value of two-photon excitation is also significantly higher than those of the other molecules, indicating that 3a possesses the strongest two-photon absorption capability. By contrast with 3a, upon introducing donor structures in 1a and 2a, the transition dipole moment values decreased in magnitude, leading to weakened two-photon absorption intensity. Furthermore, in the two-photon absorption processes of 1a and 2a, the transition dipole moments of the first-step transitions dominate, confirming that two-photon absorption is primarily governed by the initial excitation.
2.5. Chiral Analysis
To investigate the influence of donors and acceptors on the molecular chirality,
Figure 8 presents the electronic circular dichroism (ECD) spectra of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a, revealing significant differences in chiroptical activity among the three molecules and their structural regulation mechanisms.
Figure S7 depicts the enantiomers of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a.
Figure S8 shows the chiral spectra of the three enantiomers, which are compared with those of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a. They exhibit optical signals that are mirror-image symmetrical (with opposite absorbance differences). In
Figure 8, the spectrum of [12]CPP 2a exhibits a notable redshift, with its chiral signals predominantly located in the longer wavelength region (200–500 nm), although the overall optical rotation intensity is the weakest. In contrast, the chiral activity of [12]CPP 1a is strongest in the lower wavelength region (190–270 nm), and its pronounced Cotton effect indicates that the electronic transitions between the donor and acceptor significantly contribute to the chiroptical activity. Meanwhile, the chiral signals of [12]CPP 3a are concentrated in the higher wavelength region (270–450 nm). Despite the absence of donor–acceptor polarization effects in [12]CPP 3a, its chiral signals are more intense in the higher wavelength region. These results demonstrate that the introduction of donor–acceptor structures plays a regulatory role in the intensity and distribution of chiral signals.
Figure 9 presents the density distribution maps of transition electric dipole moments (TEDM) and transition magnetic dipole moments (TMDM) for [12]CPP 1a, 2a, and 3a in various excited states. By decomposing the electromagnetic interactions during the excited-state transitions, this figure provides an in-depth understanding of the mechanisms underlying chiral electromagnetic interactions and their relationship with molecular structures. Additionally, by correlating the positive and negative peak information from the electronic circular dichroism (ECD) spectra, the correspondence between chiral characteristics and the distribution of TEDM/TMDM is further analyzed.
For [12]CPP 1a, the S32 excited state corresponds to the positive peak in the ECD spectrum. From the TEDM and TMDM maps, it can be observed that the TEDM distribution is primarily concentrated on the left and right sides (X-direction) and the upper and lower sides (Y-direction) of the [12]CPP ring, while the Z-direction component is relatively weak. The TMDM distribution exhibits significant complementarity to the TEDM, with notable features in both the X and Y directions. In the S67 excited state (corresponding to the negative peak in the ECD spectrum), the TEDM and TMDM distributions are more dispersed compared to the S32 excited state, but their complementary relationship remains pronounced. For [12]CPP 2a, its S75 excited state corresponds to the positive peak in the ECD spectrum, while the S72 excited state corresponds to the negative peak. From the TEDM and TMDM maps, it is evident that the TEDM/TMDM distribution characteristics of [12]CPP 2a are generally consistent with those of [12]CPP 1a. However, due to the introduction of the imide acceptor, there are also distinct positive and negative isosurface distributions on the acceptor and donor, indicating that the polarization effect between the donor and acceptor modulates the electromagnetic interactions. For [12]CPP 3a, its S62 excited state corresponds to the positive peak in the ECD spectrum. From the TEDM and TMDM distributions, it can be seen that the TEDM is mainly concentrated around the [12]CPP host ring, exhibiting high symmetry. The TMDM distribution overlaps significantly with the positive and negative regions of the TEDM, and its intensity is relatively strong. In the S63 excited state (corresponding to the negative peak in the ECD spectrum), the negative isosurfaces of the TMDM increase significantly, directly leading to the negative peak in the ECD spectrum.
The absolute value of the molecular tensor product represents the intensity of photoexcitation; the larger the absolute value of the tensor product, the stronger the excitation intensity. The sign of the tensor product (positive or negative) indicates the chirality (positive or negative).
Table 9 presents the TEDM/TMDM values and the characteristic values of the total tensor product for the highest excited states corresponding to the positive and negative peaks of [12]CPP 1a, [12]CPP 2a, and [12]CPP 3a. From the table, it can be observed that the absolute value of the tensor product is highest for [12]CPP 3a and lowest for [12]CPP 1a, which is entirely consistent with the intensity and direction of the ECD spectra.
The intrinsic characteristics of the transitions at the positive absorption peaks in the ECD spectra (
Table 10) were analyzed by quantifying the TEDM and TMDM contributions from individual donor and acceptor fragments. In the S
32 excited state of 1a, the Z-component of the transition electric dipole moment is dominated by a negative value from the donor, while the acceptor contributes a smaller positive value in the Z-direction; the positive TMDM value arises primarily from the Z-direction contributions of the acceptor and [12]CPP. Notably, the magnitude of the transition magnetic dipole moment in 1a is significantly larger than that of the electric dipole moment, resulting in 1a exhibiting positive chirality in the S32 state. For the 2a structure in the S
75 excited state, the transition dipole moment also shows dominant contribution along the positive
Z-axis: its negative electric dipole moment stems from the donor’s Z-component, and the positive magnetic dipole moment similarly originates from the combined Z-direction contributions of the acceptor and [12]CPP—with the acceptor’s contribution being more prominent in 2a—indicating that its chirality is primarily governed by the positive direction of the magnetic dipole moment. In the S
62 excited state of 3a, the transition dipole moment is dominated by the positive
Z-axis component: although [12]CPP contributes to both positive and negative Z-directions, the positive contribution is more pronounced, leading to positive chirality in the S
62 state. This characteristic correlation between electric and magnetic dipole moment orientations highlights both the commonalities and differences in transition mechanisms among the structures.
The analysis of the contributions of donor/acceptor fragments to the TEDM and TMDM in the negative absorption peaks of the ECD spectra, as shown in
Table S1, reveals the following: In the S
67 excited state of 1a, the TEDM and TMDM contributions from [12]CPP and the acceptor in the Z direction jointly constitute the major contributions. Notably, the acceptor exhibits the largest negative contribution to the TMDM in the Z-direction. Similarly, the characteristics of the negative absorption peaks of 2a exhibit a highly consistent contribution pattern with those of 1a. That is, there is a synergistic effect of the electromagnetic dipole moments of the [12]CPP and acceptor fragments in the
Z-axis direction, and the contribution of the TMDM is significantly higher than that of the TEDM. In contrast, the negative absorption mechanism of 3a in the S
63 state differs significantly. The main driving force is the negative contribution of the TMDM of [12]CPP along the Z axis, while the contribution degree of the electromagnetic dipole moment of the acceptor is relatively low. These fragment contribution differences that are dependent on the molecular structure reveal the composition characteristics of the transition dipole moments in the negative absorption peaks. For 1a and 2a, the electromagnetic dipole moments are characterized by the synergistic contribution among fragments, and the magnetic dipole moment plays a dominant role. In contrast, 3a shows the dominant role of the [12]CPP fragment in the dimension of TMDM, reflecting the differential influence mechanism of functional fragments in different molecular structures on the characteristics of negative absorption.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
