
From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria

 , ,
, ,
Abstract
1. Introduction
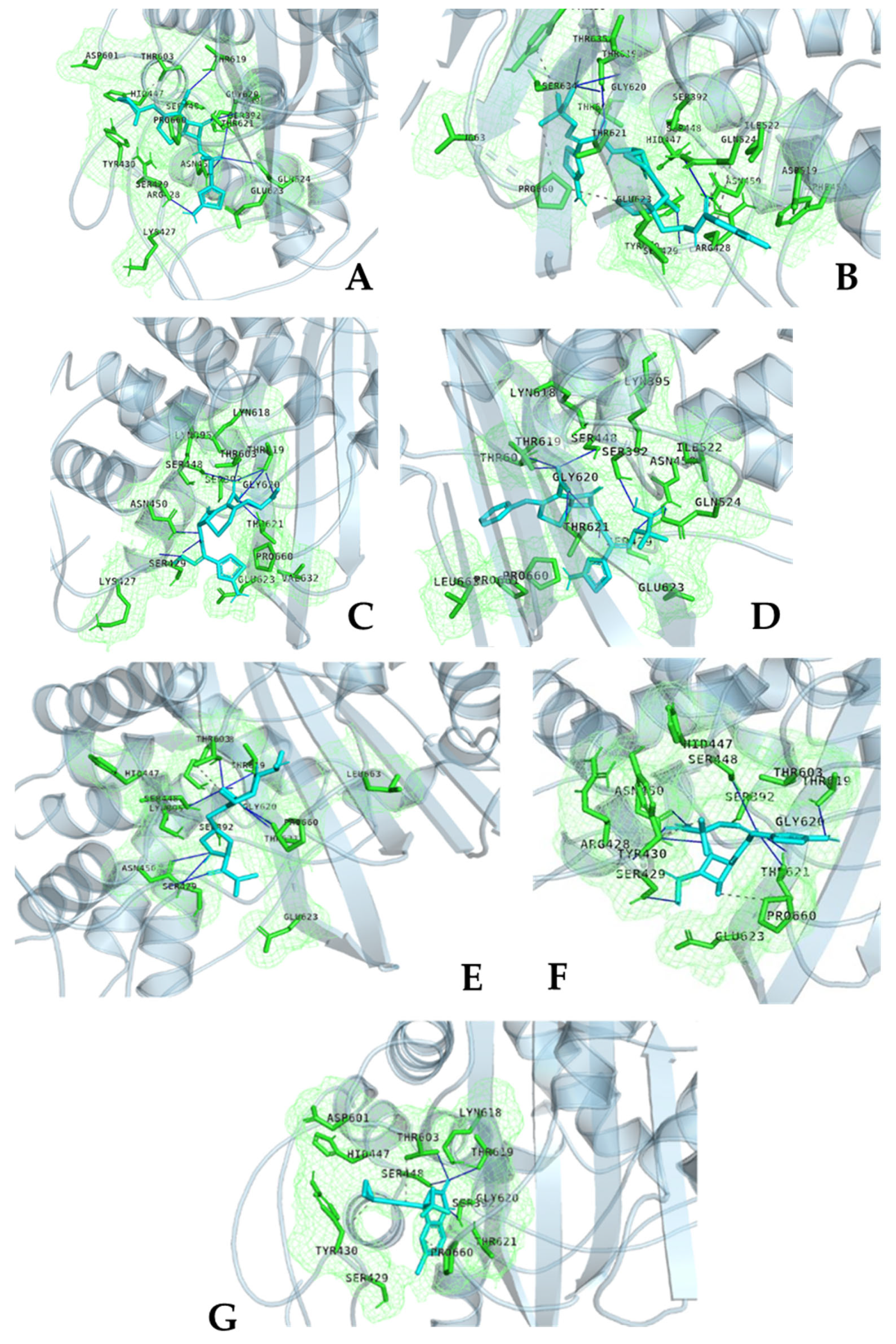
2. Results and Discussion
3. Materials and Methods
3.1. Bacterial Isolates and Culture Conditions
3.2. Susceptibility Testing Using the Disk Diffusion and Broth Microdilution Method
3.3. Formatting of Mathematical Components
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMK | Amikacin |
| ARV | Antiretroviral |
| A/S | Ampicillin/Sulbactam |
| CIP | Ciprofloxacin |
| CNN | Convolutional Neural Network |
| DMSO | Dimethyl Sulfoxide |
| DOX | Doxycycline |
| EFV | Efavirenz |
| ERY | Erythromycin |
| FOX | Cefoxitin |
| GNINA | Genetic Neural Network-Based Interaction Analyzer |
| HIV | Human Immunodeficiency Virus |
| IQR | Interquartile Range |
| MCMC | Markov Chain Monte Carlo |
| MDR | Multi-Drug Resistant |
| MIC | Minimum Inhibitory Concentration |
| MEM | Meropenem |
| MRSA | Methicillin-Resistant Staphylococcus aureus |
| MSSA | Methicillin-Sensitive Staphylococcus aureus |
| NNRTI | Non-Nucleoside Reverse Transcriptase Inhibitor |
| PBP | Penicillin-Binding Protein |
| PDB | Protein Data Bank |
| PLIP | Protein–Ligand Interaction Profiler |
| VAN | Vancomycin |
References
- Huemer, M.; Mairpady Shambat, S.; Brugger, S.D.; Zinkernagel, A.S. Antibiotic resistance and persistence—Implications for human health and treatment perspectives. EMBO Rep. 2020, 21, e51034. [Google Scholar] [CrossRef] [PubMed]
- Aira, A.; Fehér, C.; Rubio, E.; Soriano, A. The intestinal microbiota as a reservoir and a therapeutic target to fight multi-drug-resistant bacteria: A narrative review of the literature. Infect. Dis. Ther. 2019, 8, 469–482. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. 2019 Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Thomas, P.A.; Liu, H.; Umberson, D. Family relationships and well-being. Innov. Aging 2017, 1, igx025. [Google Scholar] [CrossRef] [PubMed]
- Abavisani, M.; Khoshrou, A.; Eshaghian, S.; Karav, S.; Sahebkar, A. Overcoming antibiotic resistance: The potential and pitfalls of drug repurposing. J. Drug Target. 2025, 33, 341–367. [Google Scholar] [CrossRef]
- Beachy, S.H.; Johnson, S.G.; Olson, S.; Berger, A.C. Drug Repurposing and Repositioning: Workshop Summary; National Academies Press: Washington, DC, USA, 2014. [Google Scholar]
- Li, S.X.; Armstrong, A.J.; Neff, C.P.; Shaffer, M.; Lozupone, C.A.; Palmer, B.E. Complexities of gut microbiome dysbiosis in the context of HIV infection and antiretroviral therapy. Clin. Pharmacol. Ther. 2016, 99, 600–611. [Google Scholar] [CrossRef]
- Pinto-Cardoso, S.; Klatt, N.R.; Reyes-Terán, G. Impact of antiretroviral drugs on the microbiome: Unknown answers to important questions. Curr. Opin. HIV AIDS 2018, 13, 53–60. [Google Scholar] [CrossRef]
- Shilaih, M.; Angst, D.C.; Marzel, A.; Bonhoeffer, S.; Günthard, H.F.; Kouyos, R.D. Antibacterial Effects of Antiretrovirals, Potential Implications for Microbiome Studies in HIV; SAGE Publications Sage UK: London, UK, 2018. [Google Scholar]
- Ray, S.; Narayanan, A.; Giske, C.G.; Neogi, U.; Sönnerborg, A.; Nowak, P. Altered gut microbiome under antiretroviral therapy: Impact of efavirenz and zidovudine. ACS Infect. Dis. 2020, 7, 1104–1115. [Google Scholar] [CrossRef]
- Mbuagbaw, L.; Mursleen, S.; Irlam, J.H.; Spaulding, A.B.; Rutherford, G.W.; Siegfried, N. Efavirenz or nevirapine in three-drug combination therapy with two nucleoside or nucleotide-reverse transcriptase inhibitors for initial treatment of HIV infection in antiretroviral-naïve individuals. Cochrane Database Syst. Rev. 2016, 2016, CD004246. [Google Scholar] [CrossRef]
- Rubio-Garcia, E.; Ferrando, N.; Martin, N.; Ballesté-Delpierre, C.; Miró, J.M.; Paredes, R.; Casals-Pascual, C.; Vila, J. In vitro antibacterial activity of antiretroviral drugs on key commensal bacteria from the human microbiota. Front. Cell. Infect. Microbiol. 2024, 13, 1306430. [Google Scholar] [CrossRef]
- Wang, H.; Shi, Y.; Chen, J.; Wang, Y.; Wang, Z.; Yu, Z.; Zheng, J.; Shang, Y. The antiviral drug efavirenz reduces biofilm formation and hemolysis by Staphylococcus aureus. J. Med. Microbiol. 2021, 70, 001433. [Google Scholar] [CrossRef]
- Ambade, S.S.; Gupta, V.K.; Bhole, R.P.; Khedekar, P.B.; Chikhale, R.V. A review on five and six-membered heterocyclic compounds targeting the penicillin-binding protein 2 (PBP2A) of methicillin-resistant Staphylococcus aureus (MRSA). Molecules 2023, 28, 7008. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.G.; Sarkar, A.; Ghosh, T.A.; Samanta, S.; Panja, A.S. In silico and in vitro screening of selected antimicrobial compounds for inhibiting drug efflux pumps to combat threatening MRSA. Pharmacol. Res.-Nat. Prod. 2024, 4, 100070. [Google Scholar] [CrossRef]
- Sundaramoorthy, N.S.; Mitra, K.; Ganesh, J.S.; Makala, H.; Lotha, R.; Bhanuvalli, S.R.; Ulaganathan, V.; Tiru, V.; Sivasubramanian, A.; Nagarajan, S. Ferulic acid derivative inhibits NorA efflux and in combination with ciprofloxacin curtails growth of MRSA in vitro and in vivo. Microb. Pathog. 2018, 124, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, J.-L.; Morel, V.; Fourgeaud, M.; Cremniter, J.; Blanot, D.; Legrand, R.; Fréhel, C.; Arthur, M.; van Heijenoort, J.; Gutmann, L. Balance between two transpeptidation mechanisms determines the expression of β-lactam resistance in Enterococcus faecium. J. Biol. Chem. 2002, 277, 35801–35807. [Google Scholar] [CrossRef]
- Rice, L.B. Mechanisms of resistance and clinical relevance of resistance to β-lactams, glycopeptides, and fluoroquinolones. Mayo Clin. Proc. 2012, 87, 198–208. [Google Scholar] [CrossRef]
- Gaca, A.O.; Lemos, J.A. Adaptation to adversity: The intermingling of stress tolerance and pathogenesis in enterococci. Microbiol. Mol. Biol. Rev. 2019, 83, e00074-18. [Google Scholar] [CrossRef]
- Gowda, B.J.; Nechipadappu, S.K.; Shankar, S.; Chavali, M.; Paul, K.; Ahmed, M.G.; HK, S. Pharmaceutical cocrystals of Efavirenz: Towards the improvement of solubility, dissolution rate and stability. Mater. Today Proc. 2022, 51, 394–402. [Google Scholar] [CrossRef]
- Dubrac, S.; Bisicchia, P.; Devine, K.M.; Msadek, T. A matter of life and death: Cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 2008, 70, 1307–1322. [Google Scholar] [CrossRef]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial peptides: Interaction with model and biological membranes and synergism with chemical antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef]
- Velikova, N.; Bem, A.E.; van Baarlen, P.; Wells, J.M.; Marina, A. WalK, the Path Towards New Antibacterials with Low Potential for Resistance Development. ACS Med. Chem. Lett. 2013, 4, 891–894. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Loura, L.M.S. Molecular Dynamics Simulations: Advances and Applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef] [PubMed]
- Venable, R.M.; Kramer, A.; Pastor, R.W. Molecular dynamics simulations of membrane permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef] [PubMed]
- Marzolini, C.; Telenti, A.; Decosterd, L.A.; Greub, G.; Biollaz, J.; Buclin, T. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. Aids 2001, 15, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- CLSI M100; Performance Standards for Antimicrobial Susceptibility Testing. CLSI: Berwyn, PA, USA, 2025.
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef]
- McNutt, A.T.; Li, Y.; Meli, R.; Aggarwal, R.; Koes, D.R. GNINA 1.3: The next increment in molecular docking with deep learning. J. Cheminform. 2025, 17, 1–8. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, L.; Liu, Y.; Qu, Y.; Li, Y.-Q.; Zhao, M.; Mu, Y.; Li, W. OnionNet-2: A convolutional neural network model for predicting protein-ligand binding affinity based on residue-atom contacting shells. Front. Chem. 2021, 9, 753002. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
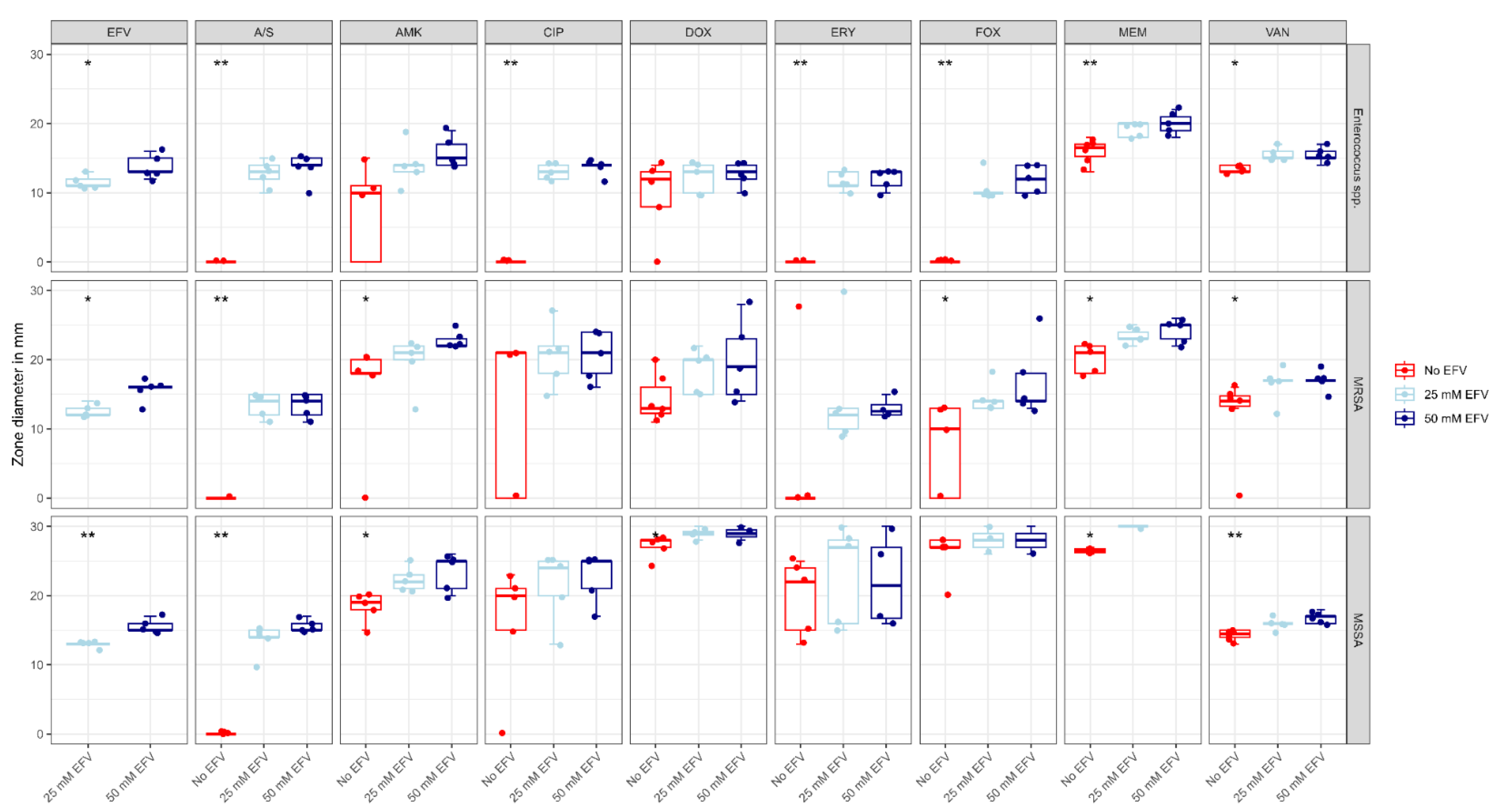
| Organism | Antibiotic | Zone Diameter | 25 mM Efavirenz | 50 mM Efavirenz | Control | p |
|---|---|---|---|---|---|---|
| Enterococci spp. | Amikacin | Median (IQR) | 14.0 (13.0 to 14.0) | 15.0 (14.0 to 17.0) | 10.0 (0.0 to 11.0) | 0.061 |
| Vancomycin | Median (IQR) | 15.0 (15.0 to 16.0) a | 15.0 (15.0 to 16.0) b | 13.0 (13.0 to 14.0) a,b | 0.01 * | |
| Ciprofloxacin | Median (IQR) | 13.0 (12.0 to 14.0) a | 14.0 (14.0 to 14.0) b | 0.0 (0.0 to 0.0) a,b | 0.004 * | |
| Meropenem | Median (IQR) | 20.0 (18.0 to 20.0) a | 20.0 (19.0 to 21.0) b | 17.0 (15.2 to 17.0) a,b | 0.006 * | |
| Ampicillin/Sulbactam | Median (IQR) | 13.0 (12.0 to 14.0) a | 14.0 (14.0 to 15.0) b | 0.0 (0.0 to 0.0) a,b | 0.006 * | |
| Doxycycline | Median (IQR) | 13.0 (10.0 to 14.0) | 13.0 (12.0 to 14.0) | 12.0 (8.0 to 13.0) | 0.555 | |
| Cefoxitin | Median (IQR) | 10.0 (10.0 to 10.0) a | 12.0 (10.0 to 14.0) b | 0.0 (0.0 to 0.0) a,b | 0.004 * | |
| Erythromycin | Median (IQR) | 11.0 (11.0 to 13.0) a | 13.0 (11.0 to 13.0) b | 0.0 (0.0 to 0.0) a,b | 0.006 * | |
| EFV | Median (IQR) | 11.0 (11.0 to 12.0) | 13.0 (13.0 to 15.0) | - | 0.032 * | |
| MRSA | Amikacin | Median (IQR) | 21.0 (20.0 to 22.0) | 22.0 (22.0 to 23.0) a | 18.0 (18.0 to 20.0) a | 0.01 * |
| Vancomycin | Median (IQR) | 17.0 (17.0 to 17.0) | 17.0 (17.0 to 17.0) a | 14.0 (13.2 to 15.0) a | 0.031 * | |
| Ciprofloxacin | Median (IQR) | 21.0 (18.0 to 22.0) | 21.0 (18.0 to 24.0) | 21.0 (0.0 to 21.0) | 0.456 | |
| Meropenem | Median (IQR) | 23.0 (23.0 to 24.0) | 25.0 (23.0 to 25.0) a | 21.0 (18.0 to 22.0) a | 0.014 * | |
| Ampicillin/Sulbactam | Median (IQR) | 14.0 (12.0 to 15.0) a | 14.0 (12.0 to 15.0) b | 0.0 (0.0 to 0.0) a,b | 0.007 * | |
| Doxycycline | Median (IQR) | 20.0 (15.0 to 20.0) | 19.0 (15.0 to 23.0) | 13.0 (13.0 to 17.0) | 0.092 | |
| Cefoxitin | Median (IQR) | 14.0 (13.0 to 14.0) | 14.0 (14.0 to 18.0) a | 10.0 (0.0 to 13.0) a | 0.017 * | |
| Erythromycin | Median (IQR) | 12.0 (10.0 to 13.0) | 13.0 (12.0 to 15.0) | 0.0 (0.0 to 0.0) | 0.083 | |
| EFV | Median (IQR) | 12.0 (12.0 to 13.0) | 16.0 (16.0 to 16.0) | - | 0.018 * | |
| MSSA | Amikacin | Median (IQR) | 22.0 (21.0 to 23.0) a | 25.0 (21.0 to 25.0) b | 19.0 (18.0 to 20.0) a,b | 0.012 * |
| Vancomycin | Median (IQR) | 16.0 (16.0 to 16.0) | 17.0 (16.0 to 17.0) a | 15.0 (14.0 to 15.0) a | 0.004 * | |
| Ciprofloxacin | Median (IQR) | 24.0 (20.0 to 25.0) | 25.0 (21.0 to 25.0) | 20.0 (15.0 to 21.0) | 0.191 | |
| Meropenem | Median (IQR) | 35.0 (35.0 to 36.0) | 41.0 (36.0 to 42.0) a | 33.0 (27.0 to 33.0) a | 0.036 * | |
| Ampicillin/Sulbactam | Median (IQR) | 14.0 (14.0 to 15.0) a | 15.0 (15.0 to 16.0) b | 0.0 (0.0 to 0.0) a,b | 0.002 * | |
| Doxycycline | Median (IQR) | 29.0 (29.0 to 30.0) | 30.0 (29.0 to 35.0) a | 28.0 (27.0 to 28.0) a | 0.02 * | |
| Cefoxitin | Median (IQR) | 30.0 (28.0 to 31.0) | 31.0 (30.0 to 33.0) | 27.0 (27.0 to 28.0) | 0.109 | |
| Erythromycin | Median (IQR) | 27.0 (16.0 to 28.0) | 26.0 (17.0 to 30.0) | 22.0 (15.0 to 24.0) | 0.285 | |
| Efavirenz | Median (IQR) | 13.0 (13.0 to 13.0) | 15.0 (15.0 to 16.0) | - | 0.006 * |
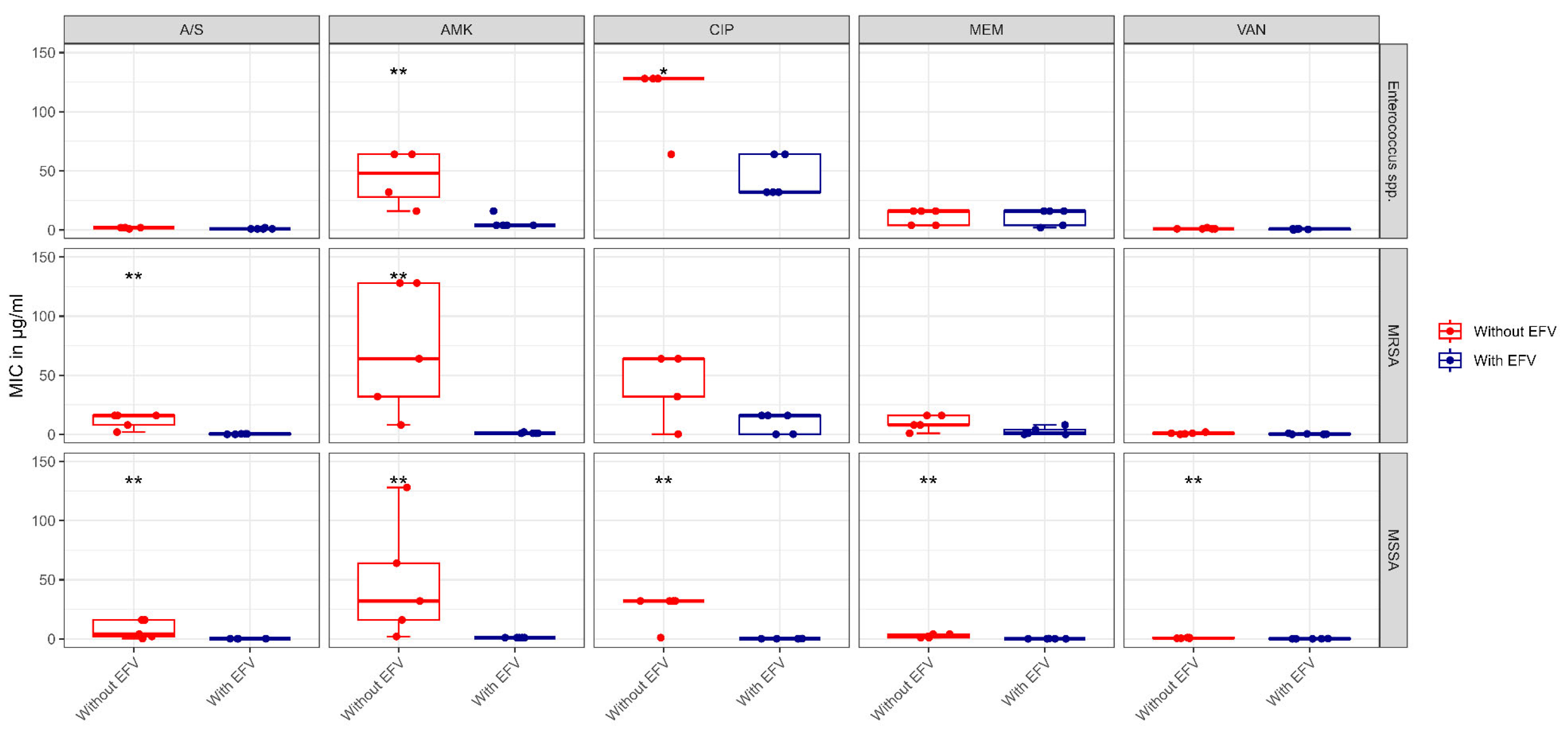
| Organism | Antibiotic | MIC in μg/mL | With Efavirenz ** | Control | p |
|---|---|---|---|---|---|
| Enterococcus spp. | Amikacin | Median (IQR) | 4.0 (4.0 to 4.0) | 64.0 (32.0 to 64.0) | 0.009 * |
| Vancomycin | Median (IQR) | 1.0 (0.5 to 1.0) | 1.0 (1.0 to 1.0) | 0.095 | |
| Ciprofloxacin | Median (IQR) | 32.0 (32.0 to 64.0) | 128.0 (128.0 to 128.0) | 0.011 * | |
| Meropenem | Median (IQR) | 16.0 (4.0 to 16.0) | 16.0 (4.0 to 16.0) | 0.811 | |
| Ampicillin/sulbactam | Median (IQR) | 1.0 (1.0 to 1.0) | 2.0 (2.0 to 2.0) | 0.072 | |
| MRSA | Amikacin | Median (IQR) | 1.0 (1.0 to 1.0) | 64.0 (32.0 to 128.0) | 0.007 * |
| Vancomycin | Median (IQR) | 0.25 (0.062 to 0.5) | 1.0 (0.5 to 1.0) | 0.136 | |
| Ciprofloxacin | Median (IQR) | 16.0 (0.25 to 16.0) | 64.0 (32.0 to 64.0) | 0.053 | |
| Meropenem | Median (IQR) | 1.0 (0.062 to 4.0) | 8.0 (8.0 to 16.0) | 0.055 | |
| Ampicillin/sulbactam | Median (IQR) | 0.5 (0.125 to 0.5) | 16.0 (8.0 to 16.0) | 0.007 * | |
| MSSA | Amikacin | Median (IQR) | 1.0 (1.0 to 1.0) | 32.0 (16.0 to 64.0) | 0.005 * |
| Vancomycin | Median (IQR) | 0.062 (0.062 to 0.125) | 0.5 (0.5 to 1.0) | 0.007 * | |
| Ciprofloxacin | Median (IQR) | 0.125 (0.125 to 0.125) | 32.0 (32.0 to 32.0) | 0.005 * | |
| Meropenem | Median (IQR) | 0.062 (0.062 to 0.062) | 2.0 (1.0 to 4.0) | 0.007 * | |
| Ampicillin/sulbactam | Median (IQR) | 0.125 (0.125 to 0.125) | 4.0 (2.0 to 16.0) | 0.005 * |
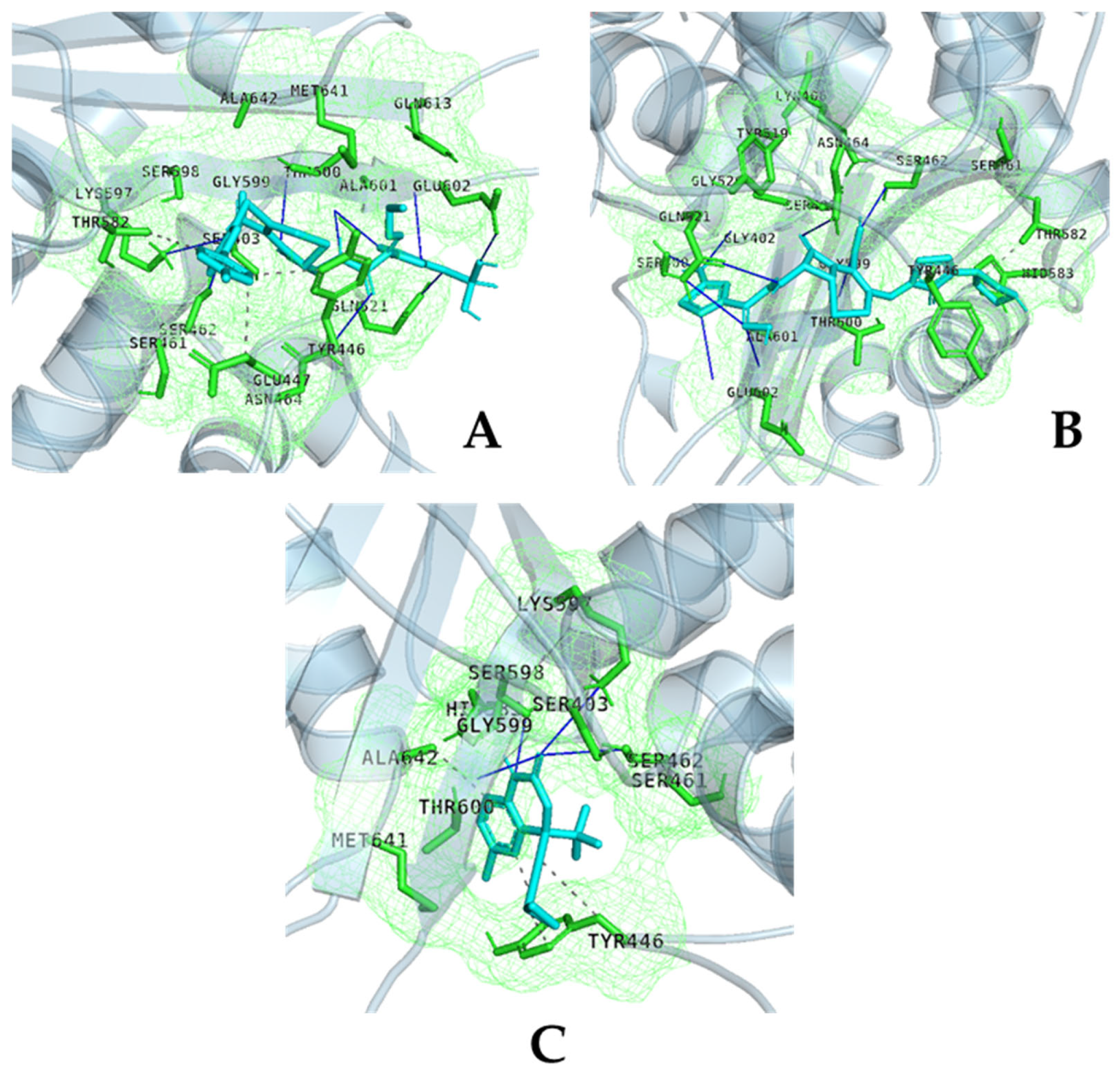
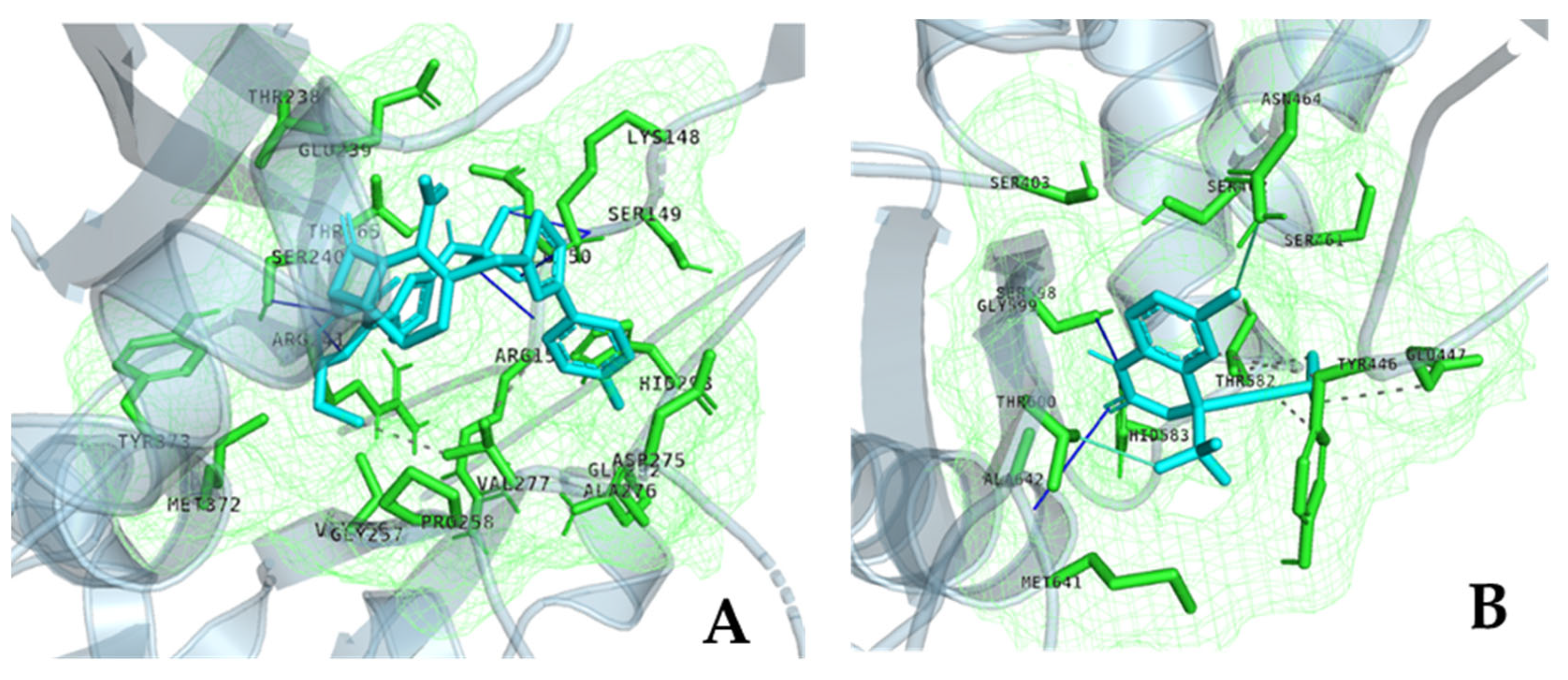
| Strain | Protein | Ligand | Vina Affinity (kcal/mol) | CNN-Score | CNN Affinity (kcal/mol) | OnionNet2 Predicted pKa |
|---|---|---|---|---|---|---|
| S. aureus/MRSA and MSSA | PBP1 | Amoxacillin | −8.21 | 0.3784 | 5.141 | 8.769 |
| PBP1 | Cefuroxime | −8.74 | 0.3964 | 5.074 | 7.090 | |
| PBP1 | Imipenem | −7.77 | 0.6219 | 4.950 | 7.066 | |
| PBP1 | penciling | −8.21 | 0.3784 | 5.141 | 8.769 | |
| PBP1 | Efavarinz | −7.60 | 0.5798 | 5.441 | 6.078 | |
| PBP2_Allostric Site | Ceftaroline | −8.03 | 0.2138 | 6.459 | 6.435 | |
| PBP2_Allostric Site | Efavarinz | −6.66 | 0.2110 | 4.377 | 5.877 | |
| PBP2_Catalytic Site | Ceftaroline | −8.92 | 0.2885 | 6.501 | 5.903 | |
| PBP2_Catalytic Site | Ceftobiprole | −10.37 | 0.1105 | 5.563 | 6.478 | |
| PBP2_Catalytic Site | Efavarinz | −7.23 | 0.1569 | 4.229 | 5.932 | |
| PBP3 | Aztronem | −8.47 | 0.1565 | 4.867 | 5.254 | |
| PBP3 | Cefepime | −8.79 | 0.1567 | 4.929 | 6.157 | |
| PBP3 | Cefiderocol | −10.27 | 0.0798 | 5.258 | 7.480 | |
| PBP3 | Cefotaxime | −8.45 | 0.3284 | 4.858 | 4.903 | |
| PBP3 | Ceftazidime | −9.25 | 0.4112 | 5.909 | 6.523 | |
| PBP3 | Meropnem | −8.20 | 0.4011 | 4.474 | 8.546 | |
| PBP3 | Efavarinz | −7.53 | 0.1576 | 4.385 | 5.741 | |
| PBP4 | Cefepime | −8.85 | 0.4317 | 5.467 | 4.829 | |
| PBP4 | Cefoxtitin | −8.51 | 0.2775 | 5.290 | 7.523 | |
| PBP4 | Piperacillin | −8.69 | 0.2913 | 5.740 | 6.796 | |
| PBP4 | Efavarinz | −7.07 | 0.2088 | 4.654 | 6.775 | |
| WalK | Efavarinz | −5.52 | 0.7506 | 5.217 | 5.349 | |
| Enterococcus spp. | PBP4 | Ceftaroline | −8.89 | 0.0503 | 5.651 | 6.771 |
| PBP4 | Imipenem | −7.07 | 0.1836 | 3.829 | 5.082 | |
| PBP4 | Efavarinz | −6.79 | 0.0874 | 3.243 | 6.775 | |
| PBP5 | Ceftaroline | −8.39 | 0.0463 | 5.214 | 6.489 | |
| PBP5 | Imipenem | −6.73 | 0.1726 | 3.427 | 7.654 | |
| PBP5 | Efavarinz | −6.86 | 0.2015 | 4.208 | 6.568 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, E.; Soliman, O.A.; Attia, N.; Rafaat, N.; Baecker, D.; Teleb, M.; Ghazal, A.; Amer, A.N. From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria. Molecules 2025, 30, 2925. https://doi.org/10.3390/molecules30142925
Saleh E, Soliman OA, Attia N, Rafaat N, Baecker D, Teleb M, Ghazal A, Amer AN. From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria. Molecules. 2025; 30(14):2925. https://doi.org/10.3390/molecules30142925
Chicago/Turabian StyleSaleh, Ezzeldin, Omar A. Soliman, Nancy Attia, Nouran Rafaat, Daniel Baecker, Mohamed Teleb, Abeer Ghazal, and Ahmed Noby Amer. 2025. "From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria" Molecules 30, no. 14: 2925. https://doi.org/10.3390/molecules30142925
APA StyleSaleh, E., Soliman, O. A., Attia, N., Rafaat, N., Baecker, D., Teleb, M., Ghazal, A., & Amer, A. N. (2025). From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria. Molecules, 30(14), 2925. https://doi.org/10.3390/molecules30142925