
Isolated Dipolar ONN Schiff Base Regioisomers: Synthesis, Characterization and Crystallographic Study


Abstract
1. Introduction
2. Results and Discussion
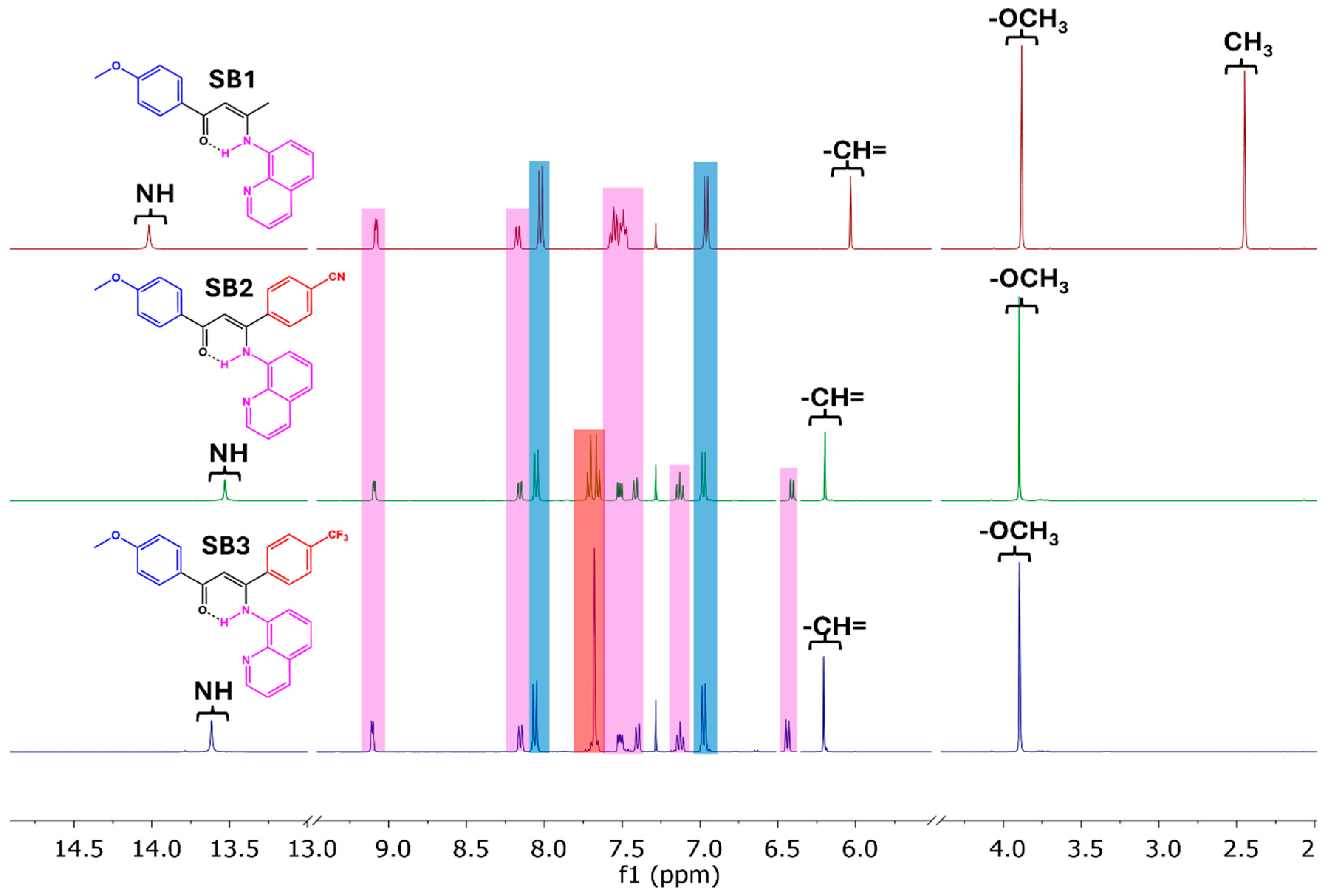
2.1. Synthesis and Characterization
2.2. Single-Crystal X-Ray Diffraction Analysis
2.3. Electronic Absorption Spectra of SB1–3
3. Materials and Methods
3.1. Materials and Physical Measurements
3.2. Synthesis of β-Diketone (β1–β3)
3.3. Synthesis of Schiff Bases (SB1–SB3)
3.4. Single-Crystal X-Ray Structure Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qin, W.; Long, S.; Panunzio, M.; Biondi, S. Schiff Bases: A Short Survey on an Evergreen Chemistry Tool. Molecules 2013, 18, 12264–12289. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizzi, L. Beauty in Chemistry: Making Artistic Molecules with Schiff Bases. J. Org. Chem. 2020, 85, 12212–12226. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Manzur, C.; Novoa, N.; Celedón, S.; Carrillo, D.; Hamon, J.R. Multidentate Unsymmetrically-Substituted Schiff Bases and Their Metal Complexes: Synthesis, Functional Materials Properties, and Applications to Catalysis. Coord. Chem. Rev. 2018, 357, 144–172. [Google Scholar] [CrossRef]
- Vigato, P.A.; Tamburini, S. The Challenge of Cyclic and Acyclic Schiff Bases and Related Derivatives. Coord. Chem. Rev. 2004, 248, 1717–2128. [Google Scholar] [CrossRef]
- Bargujar, S.; Ratnani, S.; Jain, R. Recent Advances in Microwave Assisted Synthesis of Schiff Base Metal Complexes. Inorg. Chem. Commun. 2024, 162, 112250. [Google Scholar] [CrossRef]
- Kargar, H.; Fallah-Mehrjardi, M.; Munawar, K.S. Metal Complexes Incorporating Tridentate ONO Pyridyl Hydrazone Schiff Base Ligands: Crystal Structure, Characterization and Applications. Coord. Chem. Rev. 2024, 501, 215587. [Google Scholar] [CrossRef]
- Costes, J.P. Synthetic Possibilities of the 2,4-Pentanedione-1,2-Diaminoethane System: An Overall View. Polyhedron 1987, 6, 2169–2175. [Google Scholar] [CrossRef]
- Fuentealba, M.; Trujillo, A.; Hamon, J.R.; Carrillo, D.; Manzur, C. Synthesis, Characterization and Crystal Structure of the Tridentate Metalloligand Formed from Mono-Condensation of Ferrocenoylacetone and 1,2-Phenylenediamine. J. Mol. Struct. 2008, 881, 76–82. [Google Scholar] [CrossRef]
- Novoa, N.; Roisnel, T.; Hamon, P.; Kahlal, S.; Manzur, C.; Ngo, H.M.; Ledoux-Rak, I.; Saillard, J.-Y.; Carrillo, D.; Hamon, J.-R. Four-Coordinate Nickel(II) and Copper(II) Complex Based ONO Tridentate Schiff Base Ligands: Synthesis, Molecular Structure, Electrochemical, Linear and Nonlinear Properties, and Computational Study. Dalton Trans. 2015, 44, 18019–18037. [Google Scholar] [CrossRef]
- Greenhill, J.V. Enaminones. Chem. Soc. Rev. 1977, 6, 277–294. [Google Scholar] [CrossRef]
- Duncan, N.C.; Garner, C.M.; Nguyen, T.; Hung, F.; Klausmeyer, K. Electronic Effects in the Reaction of 1,3-Diaryl-1,3-Diketones with Hydrazinopyridines. Tetrahedron Lett. 2008, 49, 5766–5769. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Gerling, K.A.; Rheingold, A.L.; Fritsch, J.M. Synthesis and Structures of Tridentate Ketoiminate Zinc Complexes Bearing Trifluoromethyl Substituents That Act as L-Lactide Ring Opening Polymerization Initiators. Dalton Trans. 2013, 42, 5573–5586. [Google Scholar] [CrossRef] [PubMed]
- Gerling, K.A.; Rezayee, N.M.; Rheingold, A.L.; Green, D.B.; Fritsch, J.M. Synthesis and Structures of Bis-Ligated Zinc Complexes Supported by Tridentate Ketoimines That Initiate L-Lactide Polymerization. Dalton Trans. 2014, 43, 16498–16508. [Google Scholar] [CrossRef] [PubMed]
- Slattery, R.M.; Stahl, A.E.; Brereton, K.R.; Rheingold, A.L.; Green, D.B.; Fritsch, J.M. Ring Opening Polymerization and Copolymerization of L-Lactide and ε-Caprolactone by Bis-Ligated Magnesium Complexes. J. Polym. Sci. A Polym. Chem. 2019, 57, 48–59. [Google Scholar] [CrossRef]
- Li, X.F.; Dai, K.; Ye, W.P.; Pan, L.; Li, Y.S. New Titanium Complexes with Two β-Enaminoketonato Chelate Ligands: Syntheses, Structures, and Olefin Polymerization Activities. Organometallics 2004, 23, 1223–1230. [Google Scholar] [CrossRef]
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The Chemistry of β-Diketiminatometal Complexes. Chem. Rev. 2002, 102, 3031–3065. [Google Scholar] [CrossRef]
- Muthu Tamizh, M.; Cooper, B.F.T.; MacDonald, C.L.B.; Karvembu, R. Palladium(II) Complexes with Salicylideneimine Based Tridentate Ligand and Triphenylphosphine: Synthesis, Structure and Catalytic Activity in Suzuki–Miyaura Cross Coupling Reactions. Inorganica Chim. Acta 2013, 394, 391–400. [Google Scholar] [CrossRef]
- Muthu Tamizh, M.; Mereiter, K.; Kirchner, K.; Karvembu, R. Ruthenium(II) Carbonyl Complexes Containing ‘Pincer like’ ONS Donor Schiff Base and Triphenylphosphine as Catalyst for Selective Oxidation of Alcohols at Room Temperature. J. Organomet. Chem. 2012, 700, 194–201. [Google Scholar] [CrossRef]
- Schmitz, L.A.; McCollum, A.M.; Rheingold, A.L.; Green, D.B.; Fritsch, J.M. Synthesis and Structures of Aluminum Ion-Pair Complexes That Act as L- and Racemic-Lactide Ring Opening Polymerization Initiators. Polyhedron 2018, 147, 94–105. [Google Scholar] [CrossRef]
- McCollum, A.M.; Longo, A.M.; Stahl, A.E.; Butler, A.S.; Rheingold, A.L.; Cundari, T.R.; Green, D.B.; Brereton, K.R.; Fritsch, J.M. Synthesis, Spectroscopy, and Crystallography of Mononuclear, Five-Coordinate Aluminum Complexes That Act as Cyclic Ester Polymerization Initiators. Polyhedron 2021, 204, 115233. [Google Scholar] [CrossRef]
- Ghorai, P.; Brandão, P.; Benmansour, S.; García, C.J.G.; Saha, A. Azido and Thiocyanato Bridged Dinuclear Ni(II) Complexes Involving 8-Aminoquinoline Based Schiff Base as Blocking Ligands: Crystal Structures, Ferromagnetic Properties and Magneto-Structural Correlations. Polyhedron 2020, 188, 114708. [Google Scholar] [CrossRef]
- Thakurta, S.; Maiti, M.; Butcher, R.J.; Gómez-García, C.J.; Tsaturyan, A.A. A Trinuclear Nickel(II) Schiff Base Complex with Phenoxido- and Acetato-Bridges: Combined Experimental and Theoretical Magneto-Structural Correlation. Dalton Trans. 2021, 50, 2200–2209. [Google Scholar] [CrossRef] [PubMed]
- Novoa, N.; Roisnel, T.; Dorcet, V.; Cador, O.; Manzur, C.; Carrillo, D.; Hamon, J.R. Efficient Preparation of Multimetallic ONO-Based Schiff Base Complexes of Nickel(II) and Copper(II). New J. Chem. 2016, 40, 5920–5929. [Google Scholar] [CrossRef]
- Novoa, N.; Manzur, C.; Roisnel, T.; Dorcet, V.; Cabon, N.; Robin-Le Guen, F.; Ledoux-Rak, I.; Kahlal, S.; Saillard, J.Y.; Carrillo, D.; et al. Redox-Switching of Ternary Ni(II) and Cu(II) Complexes: Synthesis, Experimental and Theoretical Studies along with Second-Order Nonlinear Optical Properties. New J. Chem. 2019, 43, 10468–10481. [Google Scholar] [CrossRef]
- González, D.M.; Hernández, L.A.; Oyarce, J.; Alfaro, A.; Novoa, N.; Cisterna, J.; Brito, I.; Carrillo, D.; Manzur, C. A new and efficient high-performance electrochemical glucose sensor based on a metallopolymer derived from a cobaltate(III) Schiff base complex. Synth. Met. 2021, 271, 116633. [Google Scholar] [CrossRef]
- Roberts, C.C.; Fritsch, J.M. Synthesis and Crystal Structures of Magnesium Complexes with NNO Schiff Base Ligands Bearing Quinolyl Pendant Donors. Polyhedron 2010, 29, 1271–1278. [Google Scholar] [CrossRef]
- Cisterna, J.; Artigas, V.; Fuentealba, M.; Hamon, P.; Manzur, C.; Dorcet, V.; Hamon, J.R.; Carrillo, D. Nickel(II) and Copper(II) Complexes of New Unsymmetrically-Substituted Tetradentate Schiff Base Ligands: Spectral, Structural, Electrochemical and Computational Studies. Inorganica Chim. Acta 2017, 462, 266–280. [Google Scholar] [CrossRef]
- Celedón, S.; Hamon, P.; Artigas, V.; Fuentealba, M.; Kahlal, S.; Carrillo, D.; Saillard, J.Y.; Hamon, J.R.; Manzur, C. Ferrocene Functionalized Enantiomerically Pure Schiff Bases and Their Zn(II) and Pd(II) Complexes: A Spectroscopic, Crystallographic, Electrochemical and Computational Investigation. New J. Chem. 2022, 46, 3948–3960. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Barbera, J.; Gimenez, R.; Serrano, J.L.; Alcala, R.; Villacampa, B.; Villalba, J.; Ledoux, I.; Zyss, J. Beta-Diketone, Pyrazole and Isoxazole Derivatives with Polar Groups: Liquid Crystalline and Non-Linear Optical Properties. Liq. Cryst. 1997, 22, 265–273. [Google Scholar] [CrossRef]
- Danilova, J.S.; Avdoshenko, S.M.; Karushev, M.P.; Timonov, A.M.; Dmitrieva, E. Infrared Spectroscopic Study of Nickel Complexes with Salen-Type Ligands and Their Polymers. J. Mol. Struct. 2021, 1241, 130668. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Elsevier: Amsterdam, The Netherlands, 1991; ISBN 978-0-12-451160-6. [Google Scholar]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for Intramolecular N-H···O Resonance-Assisted Hydrogen Bonding in β-Enaminones and Related Heterodienes. A Combined Crystal-Structural, IR and NMR Spectroscopic, and Quantum-Mechanical Investigation. J. Am. Chem. Soc. 2000, 122, 10405–10417. [Google Scholar] [CrossRef]
- Taylor, R.; Wood, P.A. A Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts. Chem. Rev. 2019, 119, 9427–9477. [Google Scholar] [CrossRef]
- Celedon, S.; Fuentealba, M.; Roisnel, T.; Hamon, J.R.; Carrillo, D.; Manzur, C. Stepwise Construction of a 4-Hydroxyphenyl Functionalized O,N,N-Tridentate Ferrocene-Containing Enaminone: Spectral, Analytical and Structural Studies. Inorganica Chim. Acta 2012, 390, 184–189. [Google Scholar] [CrossRef]
- González, D.; Arrué, R.; Matamala-Cea, E.; Arancibia, R.; Hamon, P.; Cador, O.; Roisnel, T.; Hamon, J.R.; Novoa, N. Homoleptic CoII, NiII, CuII, and ZnII Complexes Based on 8-Hydroxylquinoline Schiff Base Derivative: A Combined Synthetic, Spectral, Structural, and Magnetic Study. Eur. J. Inorg. Chem. 2018, 2018, 4720–4730. [Google Scholar] [CrossRef]
- Kalsi, P.S. Spectroscopy of Organic Compounds; New Age International: Mumbai, India, 2007; ISBN 8122415431. [Google Scholar]
- Bureš, F. Fundamental Aspects of Property Tuning in Push–Pull Molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Cha, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann Ltd.: London, UK, 2003; ISBN 9780750675710. [Google Scholar]
- Popic, V.V.; Korneev, S.M.; Nikolaev, V.A.; Korobitsyna, I.K. An Improved Synthesis of 2-Diazo-1,3-Diketones. Synthesis 1991, 1991, 195–198. [Google Scholar] [CrossRef]
- Rao, H.S.P.; Muthanna, N. Variations in the Blaise Reaction: Conceptually New Synthesis of 3-Amino Enones and 1,3-Diketones. Eur. J. Org. Chem. 2015, 2015, 1525–1532. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. IUCr Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. IUCr SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Novoa, N.; Manzur, C.; Roisnel, T.; Kahlal, S.; Saillard, J.Y.; Carrillo, D.; Hamon, J.R. Nickel(II)-Based Building Blocks with Schiff Base Derivatives: Experimental Insights and DFT Calculations. Molecules 2021, 26, 5316. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Distance (Å) | SB1 | SB2 | SB3 |
| C1-C10 | 1.4976 (14) | 1.484 (2) | 1.491 (3) |
| N1-C1 | 1.3418 (12) | 1.3563 (18) | 1.351 (3) |
| C1-C2 | 1.3839 (13) | 1.370 (2) | 1.369 (3) |
| C2-C3 | 1.4251 (14) | 1.436 (2) | 1.438 (3) |
| O2-C3 | 1.2547 (12) | 1.2480 (19) | 1.250 (3) |
| C3-C7 | 1.499 (14) | 1.488 (2) | 1.483 (3) |
| O3-C4 | 1.3687 (15) | 1.3703 (19) | 1.393 (3) |
| N3-C26 | 1.140 (2) | ||
| F1-C26 * | 1.3173 (7) | ||
| C21-C26 | 1.438 (2) | 1.464 (3) | |
| Bond Angle (°) | SB1 | SB2 | SB3 |
| O2-C3-C7 | 117.86 (9) | 118.29 (14) | 118.65 (19) |
| O2-C3-C2 | 123.04 (9) | 121.96 (14) | 121.60 (19) |
| C1-C2-C3 | 124.35 (9) | 123.63 (14) | 124.12 (19) |
| N1-C1-C2 | 120.60 (9) | 121.79 (14) | 121.38 (19) |
| C2-C1-C10 | 119.36 (9) | 119.15 (13) | 118.77 (18) |
| N1-C1-C10 | 119.98 (9) | 118.81 (12) | 119.62 (17) |
| C1-N1-C11 | 128.36 (9) | 129.12 (14) | 130.64 (18) |
| Compound | D-H···A | d(D-H)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° |
|---|---|---|---|---|---|
| SB1 | N1-H1···O2 | 0.86 | 1.97 | 2.666 (12) | 136.8 |
| C17H17···O2 i | 0.93 | 2.61 | 3.370 (13) | 139.3 | |
| C19H19···O2 ii | 0.93 | 2.83 | 3.480 (16) | 128.4 | |
| SB2 | N1-H1···O2 | 0.86 | 2.03 | 2.643 (17) | 127.7 |
| C5H5···O2 iv | 0.93 | 2.73 | 3.489 (2) | 139.4 | |
| C22-H22···O2 ii | 0.93 | 2.48 | 3.264 (2) | 142.7 | |
| C18-H18···O3 v | 0.93 | 2.92 | 3.496 (3) | 121.8 | |
| C19-H19···O3 vi | 0.93 | 2.73 | 3.353 (2) | 125.3 | |
| SB3 | N1-H1···O2 | 0.86 | 1.98 | 2.645 (2) | 133.7 |
| C15-H15···O2 vii | 0.93 | 2.76 | 3.350 (3) | 122.4 | |
| C6-H6···F3 viii | 0.93 | 2.91 | 3.490 (8) | 121.4 | |
| C5-H5···F3A viii | 0.93 | 2.61 | 3.293 (9) | 130.6 | |
| C25-H25···O2 ix | 0.93 | 2.70 | 3.354 (3) | 128.3 | |
| C25-H25···N2 ix | 0.93 | 2.63 | 3.370 (3) | 136.8 | |
| C24-H24···F1A x | 0.93 | 2.85 | 3.498 (7) | 127.9 | |
| Symmetry coordinates | |||||
| i 1 − x, 1 − y, 2 − z | ii −1 + x, +y, +z | iii 1 − x, 1 − y, −z | iv 2 − x, −y, 1 − z | ||
| v −1 + x, +y, −1 + z | vi 1−x, −y, 1−z | vii 1 − x, 1 − y, 1 − z | |||
| viii +x, +y, −1 + z | ix +x, 1/2 − y, 1/2 + z | x +x, 1/2 − y, −1/2 + z | |||
| Compound | Band | λ/nm (log ε) CH2Cl2 | λ/nm (log ε) DMSO | Solv. Shift (cm−1) |
|---|---|---|---|---|
| SB1 | A | 257 (4.67) | 260 (4.73) | 3 |
| B | 287 (4.26) | 289 (4.35) | 2 | |
| C | 391 (4.82) | 393 (4.91) | 2 | |
| SB2 | A | 253 (5.01) | 260 (4.86) | 7 |
| B | 301 (4.67) | 303 (4.64) | 2 | |
| C | 408 (4.90) | 411 (4.87) | 3 | |
| SB3 | A | 251 (4.84) | 260 (4.70) | 9 |
| B | 294 (4.45) | 293 (4.46) | −1 | |
| C | 405 (4.84) | 407 (4.84) | 2 |
| Compound | SB1 | SB2 | SB3 |
|---|---|---|---|
| Empirical formula | C20H18N2O2 | C26H19N3O2 | C26H19F3N2O2 |
| Formula mass, g mol−1 | 318.36 | 405.44 | 448.43 |
| Temperature, K | 296.15 | 296.15 | 296.15 |
| Crystal system | monoclinic | triclinic | monoclinic |
| Space group | P21/n | P | P21/c |
| a (Å) | 7.4516 (3) | 8.1445 (3) | 17.6152 (6) |
| b (Å) | 22.3305 (10) | 10.5294 (3) | 9.2876 (3) |
| c (Å) | 10.1970 (4) | 12.8440 (4) | 13.5040 (4) |
| α (°) | 90 | 98.0710 (10) | 90 |
| β (°) | 109.8860 (10) | 103.4230 (10) | 92.3570 (10) |
| γ (°) | 90 | 90.2180 (10) | 90 |
| Volume (Å3) | 1595.58 (12) | 1060.00 (6) | 2207.42 (12) |
| Z | 4 | 2 | 4 |
| Dcalc (g cm−3) | 1.325 | 1.270 | 1.349 |
| μ (mm−1) | 0.087 | 0.082 | 0.103 |
| F(000) | 672.0 | 424.0 | 928.0 |
| Crystal size (mm3) | 0.25 × 0.22 × 0.16 | 0.29 × 0.24 × 0.069 | 0.26 × 0.26 × 0.11 |
| 2Θ range (°) | 4.622–72.64 | 4.734–70.148 | 4.628–67.604 |
| Range, hkl | −12/12, −37/37, −16/16 | −13/13, −16/16, −20/20 | −27/27, −14/14, −21/21 |
| No. refl. collected | 47,985 | 51,112 | 59,859 |
| No. independent refl. | 7732 | 9346 | 8847 |
| Data/restraints/parameters | 7732/0/219 | 9346/0/281 | 8847/93/327 |
| Goodness-of-fit on F2 | 1.017 | 1.023 | 1.022 |
| Final R indexes [I >= 2σ (I)] | R1 = 0.0555, wR2 = 0.1539 | R1 = 0.0685, wR2 = 0.1955 | R1 = 0.0817, wR2 = 0.2406 |
| Final R indexes (all data) | R1 = 0.0908, wR2 = 0.1804 | R1 = 0.1392, wR2 = 0.2434 | R1 = 0.1385, wR2 = 0.2897 |
| Largest diff. peak and hole (e Å−3) | 0.39/−0.20 | 0.26/−0.16 | 0.66/−0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Tamay, P.; Villaman, D.; Hamon, J.-R.; Novoa, N. Isolated Dipolar ONN Schiff Base Regioisomers: Synthesis, Characterization and Crystallographic Study. Molecules 2024, 29, 5863. https://doi.org/10.3390/molecules29245863
Castro-Tamay P, Villaman D, Hamon J-R, Novoa N. Isolated Dipolar ONN Schiff Base Regioisomers: Synthesis, Characterization and Crystallographic Study. Molecules. 2024; 29(24):5863. https://doi.org/10.3390/molecules29245863
Chicago/Turabian StyleCastro-Tamay, Pablo, David Villaman, Jean-René Hamon, and Néstor Novoa. 2024. "Isolated Dipolar ONN Schiff Base Regioisomers: Synthesis, Characterization and Crystallographic Study" Molecules 29, no. 24: 5863. https://doi.org/10.3390/molecules29245863
APA StyleCastro-Tamay, P., Villaman, D., Hamon, J.-R., & Novoa, N. (2024). Isolated Dipolar ONN Schiff Base Regioisomers: Synthesis, Characterization and Crystallographic Study. Molecules, 29(24), 5863. https://doi.org/10.3390/molecules29245863